

Abstract
Microgel multi-layer films assembled from anionic particles and linear polycation were prepared on elastomeric substrates and their self-healing properties studied. Dried films were imaged in situ during mechanical deformation and were determined to undergo plastic deformation in response to linear strain, leading to film buckling upon strain relaxation. Hydration leads to rapid reorganization of the film building blocks, permitting recovery of the film to the undamaged state. Additionally, films were determined to heal in the presence of high relative humidity environments, suggesting that film swelling and hydration is a major factor in the restoration of film integrity, and that full immersion in solvent is not required for healing. Films prepared from microgels with lower levels of acid content and/or polycation length, factors strongly connected to the charge density and presumably the connectivity of the film, also display self-healing characteristics.
Introduction
Self-healing materials are particularly desirable in applications where the integrity of a material or coating is crucial to its performance, especially if the material is to be used for long durations. Biological materials provide inspiration for design of self-healing structures because living systems have innate ability to sense and repair damage in order to restore functionality to the injured site. Accomplishing this feat with synthetic materials is a major challenge for current research, and requires thoughtful control over the underlying chemistry, configuration, and mechanical properties of the materials. Consequently, there is no “one-size-fits-all” approach to the development of self-healing materials, as different applications have very different material demands.1-5
Several distinctions can be made between types of self-healing materials. Autonomic self-healing materials are able to self-repair in the absence of an external trigger. Some of the best known examples feature encapsulated catalysts and monomers contained within a matrix, such that damage to the matrix leads to capsule rupture, catalyst and monomer mixing, polymerization, and thus restoration of matrix integrity.6 Taking a step towards bio-inspiration, this concept has been more recently extended such that two-phase microvasculature serves to deliver the necessary components for healing.7 Materials that require the input of energy, typically in the form of heat or light, in order to begin repair are referred to as “non-autonomic.”4 Their self-healing capabilities originate from the dynamic (and typically non-covalent) interactions that serve to reversibly connect their substituents. The external trigger often serves to fluidize or decrease the viscosity of the system or alter the equilibrium towards reactive, unconnected species. Removal of the stimulus resets the connected, original state. Some examples of reversible chemistries that have been used in self-healing systems include Diels-Alder reactions,8-10 π-π interactions,11, 12 disulfide linkages,13, 14 trithiocarbonates,15, 16 hydrogen bonding,17-20 metal complexes,21, 22 ion-pairing23, 24 and even systems using a combinations of strategies.25
Our group has previously described thin films composed of anionic hydrogel microparticles (microgels) cross-linked by linear polycations and assembled using layer-by-layer techniques.26, 27 An intriguing finding is that when such films are deposited on elastomeric substrates, they undergo autonomic self-healing in aqueous media in response to damage events such as bending, stretching, or mechanical “poking”, leading to restoration of the initial film integrity.28 Further investigation of such films found that their mechanical properties could be reinforced by gold nanoparticles.29 The initial discovery of this self-healing behavior was a serendipitous result of studying the deposition of microgel-based films onto elastomeric substrates.28 However, none of the studies so far have offered a great deal of insight into the mechanism of film damage or what drives restoration of film integrity following damaging events. In this work, we elucidate key factors in understanding the mechanical origins of this damage, as well as the drivers that promote rapid self-healing. In addition, we study the applicability of this healing effect to a range of film compositions.
Experimental
Materials
All reagents were purchased from Sigma Aldrich (St Louis, MO) and used as received unless otherwise noted. The monomer N-isopropylacrylamide (NIPAm) was recrystallized from hexane (VWR International, West Chester, PA) and dried under vacuum before use. Reagents acrylic acid (AAc), N,N′-methylenebisacrylamide (BIS), sodium dodecyl sulfate (SDS), ammonium persulfate (APS), sodium phosphate, monobasic (NaH2PO4), sodium chloride, magnesium nitrate hexahydrate, potassium chloride, (3-aminopropyl)trimethoxysilane (APTMS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and 2-(N-morpholino)ethanesulfonic acid (MES) were used as received. Solutions of 20% w/w poly(diallyldimethylammonium chloride) (PDADMAC) in water with molecular weights of 100-200 kDa and 400-500 kDa were used as received. Water used in all reactions, particle purifications, and buffer preparations was purified to a resistance of 18 MΩ (Barnstead E-Pure System), and filtered through a 0.2 μm filter to remove particulate matter.
Microgel Synthesis
Microgels were synthesized using previously described aqueous precipitation polymerization methods.30 Anionic microgels were prepared by dissolving NIPAm, BIS, and SDS in water. The solution was filtered through a 0.2 μm syringe filter, heated to 70 °C, and purged with N2 for one hour. Acrylic acid (AAc) was added 10 minutes before initiation of the reaction, which was achieved with 1 mM APS. The reaction was carried out under a N2 atmosphere for 4 hours with constant stirring. All syntheses were filtered through glass wool to remove coagulum and then subjected to at least 4 rounds of centrifugation and resuspension in distilled water. Finally, all syntheses were lyophilized for 72 hours before use.
Microgel Characterization
Diffusion coefficients were determined by dynamic light scattering (DLS) using a Protein Solutions DynaPro DLS (Wyatt Technology Corporation, Santa Barbara, CA) equipped with temperature control. Hydrodynamic radii (RH), autocorrelation functions, and polydispersities were calculated using Dynamics software for each particle type used in these experiments. All DLS measurements were carried out at 20 °C in either 10 mM PBS at pH 7.4 and 15 mM ionic strength or 10 mM formate buffer at pH 3.0 with 15 mM ionic strength to confirm pH responsivity. Particle sizes are presented in the supplementary information, Table S1.
Substrate Preparation
Films were made using layer-by-layer techniques (LbL) previously described by our group.28 Briefly, poly(dimethylsiloxane) (PDMS) was mixed in a 10:1 ratio by weight of elastomer to curing agent (Sylgard 184 Silicone Elastomer Kit, Dow Corning), degassed for 30 minutes, and cured at 50 °C overnight. Cured PDMS was cut into 9 × 18 mm pieces, equilibrated in hexane for 2 hours, and again incubated at 50 °C in an oven for 2 hours to remove hexane. Pieces were stored in individual Eppendorf tubes filled with water until further use. Prior to use, the water was removed and replaced with 1.2 M HCl for approximately sixteen hours. The pieces were then removed, washed 3 times with water, washed 2 times with absolute ethanol, and shaken in absolute ethanol for 30 minutes. Finally, the pieces were removed and placed in a separate solution of 1% APTMS in absolute ethanol for 2 hours while shaking. Once removed, the pieces were rinsed with water, at which point they were ready for LbL coating.
Film Construction
Films were constructed using an LbL approach described previously.26, 28 Briefly, prepared substrates were placed in well plates and covered with a 0.1 mg/mL solution of microgels in pH 7.4 phosphate buffered saline (PBS). The plates were centrifuged with an Eppendorf 5804 R centrifuge in a microwell plate rotor at 2250 × g for 10 minutes. The substrates were then removed and rinsed with water. The microgels were covalently coupled to the substrate using a carbodiimide coupling reaction using 2 mM EDC and 5 mM NHS in 10 mM MES pH 5.7 buffer for 2 hours. The microgel-modified substrates were then shaken in a solution of polycation for 30 min in either 100-200 kDa or 400-500 kDa Mw PDADMAC at a concentration of 0.1 monoM in PBS, and the layer-by-layer process was repeated up to 8 layers of microgels.
Film Stretching
Films were loaded into a homemade stretching apparatus that allowed precise mechanical control over the degree of strain applied to the substrate. Coated PDMS substrates were clamped at both ends between glass plates connected to a micrometer-controlled, single-axis translation stage. The film was parallel to the floor with the coated side up. Before applying a stress, the exposed film area was wet with distilled water for at least 1 minute to heal any damage associated with handling, dried with N2, and measured along the stretching axis.
Film Characterization and Analysis
Brightfield microscopy images at 40× or 100× magnification were captured on an Olympus IX-70 inverted microscope equipped with a PixelFly black and white CCD camera. Atomic force microscopy images were captured on an Asylum Research (Santa Barbara, CA) MFP-3D AFM in AC-mode. The resultant images were analyzed using the MFP-3D software written in the IgorPro software environment (WaveMetrics, Inc, Lake Oswego, OR). Cantilevers for the Asylum instrument were operated in tapping mode and were Si-SPM with Al reflex coating, 42 N/m force constant, model NCHR purchased from NANOWORLD (Neuchâtel, Switzerland). Atomic force microscopy images of films under stress were captured using a Nanosurf (Boston, MA) EasyScan 2 AFM. Cantilevers used with the Nanosurf were operated in tapping mode and were Si, N-type with Al reflex coating, 45 N/m nominal force constant, model ACLA purchased from APPNANO (Santa Clara, CA). The Nanosurf scan head was positioned on top of the stretching apparatus to suspend the tip over a mounted sample. The scan head imaged at an angle 45 degrees relative to the axis of stretching. Collected images were analyzed using analysis tools in the Nanosurf AFM software (v. 3.0.2.4) to determine root-mean-square (RMS) roughness, with values reported as the average ± SD of four 20 × 20 μm regions.
Steam Healing
A 200-mL beaker was filled with approximately 170 mL of DI water. The water was heated until steam was visibly rising from the surface. Microgel films were damaged by pressing a plastic pipette tip against the film and gently dragging the tip across the coating, such that a mark was clearly visible. The sample was then suspended above the steaming beaker, film side down, for a period of five seconds.
Healing Driven by Relative Humidity
To prepare the high relative humidity environment, two petri dishes were filled with DI water, placed within slightly larger petri dishes to minimize spills, and placed within a sealed 2-gallon plastic bag. This bag was sealed within a second bag, and allowed to equilibrate. An Acu-Rite relative humidity sensor was sealed along with the petri dishes allowed monitoring of the humidity within the inner bag. The inner bag was allowed to equilibrate until humidity measured within the bag exceeded 80%. To observe intermediate humidity states, saturated solutions were prepared by adding the following masses to approximately 80 mL of deionized water in a 100-mL beaker, such that undissolved solids coated the bottom of the beakers: 71 g magnesium nitrate hexahydrate (55% relative humidity), 30 g sodium chloride (73% relative humidity), 36 g potassium chloride (78% relative humidity). These were again sealed within two bags and allowed to equilibrate to the steady state humidity value. Films that were damaged either through controlled stretching or pipette scratching (as described above) were placed in open-topped 6-well plates, then placed within the inner bag of the humidity chamber. Films could be monitored visually during healing, and were removed after one hour for further characterization.
Film Thickness
Cleaned glass coverslips were functionalized using 1% v/v APTMS in absolute ethanol, then incubated overnight. Following rinsing and incubation in PBS for 30 minutes, film assembly proceeded as described for the PDMS substrates above, creating films with identical composition. A clean razor blade was used to scratch the surface of the films, exposing the glass substrate. The region of the scratch was imaged using the Asylum MFP-3D AFM. Film thicknesses for hydrated films were determined using the iDrive accessory for the MFP3D after incubating the scratched area in deionized water for a minimum of thirty minutes. The iDrive cantilevers were silicon nitride coated with Cr/Au with nominal spring constant of 0.02 N/m (Asylum Research, Santa Barbara, CA). All images were collected at 512 × 512 scan resolution. The resultant images of the film scratches were analyzed by averaging the results from 150 line traces wherein the surface of the film and the glass substrate were clearly discernible, then the height difference between the substrate and film used to determine the thickness.
Results and Discussion
Film Damage
Stretching microgel multi-layered films on elastomeric substrates had previously been shown to result in a series of parallel cracks perpendicular to the axis of applied stress. These cracks subsequently disappear upon immersion in water.28, 29 Though these results demonstrate the reversibility of even large-scale damage, the origin of these parallel cracks was unclear, with two prevailing hypotheses being that they arose due to cracking of the microgel film under the applied stress (e.g. the films are brittle), or that the films underwent plastic deformation as the degree of strain was increased (e.g. the films are deformable). The Nanosurf's compact scanning head allowed it to be stationed atop the stretching apparatus and thus allowed the surface topography of the films to be monitored in situ under applied stress in order to ascertain the origin of the cracking or wrinkling pattern. A photograph of the setup for measuring the film under applied stress at a variety of strain magnitudes is available in the supplementary information.
As shown in Fig. 1, the RMS roughness of the film as a function of applied stress remains relatively constant as the degree of strain is increased. As the stress is released, however, the parallel cracking pattern appears and results in an increase in the RMS roughness. Roughness continues to increase as stress is removed, until restoration of the zero-strain state. At that point, wetting and subsequent drying of the film leads to the characteristic healing response, and the RMS roughness returns to its baseline level as the wrinkled pattern disappears. The same pattern is observed even when lower degrees of maximal strain are applied (Fig. 1f, g), as the roughness begins to increase only when the strain is removed. Additionally, stretching the film to 30% strain, then exposing to water, as shown in Fig. 2, revealed minimal changes in surface topography. Relaxation of the stress from this state leads to the same wrinkling pattern as that observed without the intermediate wetting step.
Fig. 1.

a, f, g) RMS roughness as a function of applied strain for 8-layer 30% AAc microgels/400-500 kDa PDADMAC films. Maximal strain a) 30%, f) 20%, g) 10%. b-e) Representative AFM images obtained during in situ stretching of the films. Film roughness remains relatively constant as the degree of strain increases (b, c), then buckling occurs as the stress is removed (c, d). Wetting of the films leads to restoration of the initial roughness (d, e). All AFM images are 40 μm × 40 μm.
Fig. 2.
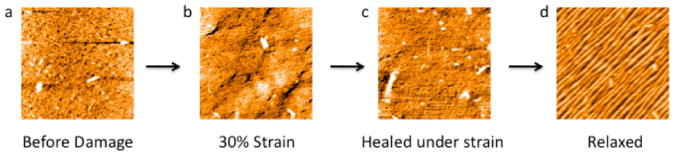
Film healed under stress. a) Film before damage displays a smooth surface with low roughness. b) Application of 30% strain does not result in a large change in surface topography. c) Film exposed to water while under strain also has a smooth surface with low roughness. d) When stress is removed from the film, the characteristic buckling behavior results. All AFM images are 40 μm × 40 μm.
Further in situ investigation of the stretched film revealed additional evidence that the wrinkling pattern is the result of a plastic deformation. Fig. 3 reveals that as an undamaged film is subjected to 30% strain and then relaxed, the characteristic buckling pattern appears. Returning that film to 30% strain without healing leaves wrinkles behind, with the wrinkling pattern lying orthogonal to the original pattern. The wrinkles seen upon initial relaxation of the film are always observed to lie on the diagonal from the bottom left of the image to the top right, due to the Nanosurf's 45 degree angle of imaging (see Fig. 1, for example). This corresponds to wrinkles that are perpendicular to the stretching direction, suggesting buckling behavior. However, when a film was re-stretched to 30% strain without a healing step, the lines were observed to lie along the opposite diagonal (Fig. 3d), indicative that the wrinkles are now parallel to the axis of stretching. We attribute this change in direction to the elongation and compression forces that the film experiences during stretching on an elastomeric substrate. As PDMS is stretched, it elongates along one axis and compresses along the perpendicular axes (Poisson ratio = 0.5).31 Therefore, if we assume that the entire film undergoes some deformation during the initial stretching event, the in-plane film axis perpendicular to the initial stretching axis is actually under compression during a second stretching event, thereby resulting in a new wrinkling pattern perpendicular to the axis of the second stretch. As seen in Fig. 3e, this pattern persists following multiple rounds of film stretching and relaxation.
Fig. 3.
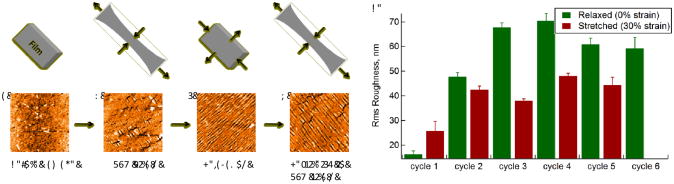
Cycles of stretching of microgel films leads to sensitivity to perpendicular compression. a) Film under 0% strain, before cycling, b) 30% strain leads to compression along the short axis, perpendicular to the applied stress, c) the film is relaxed back to 0% strain; during relaxation, the film is effectively being compressed along the long axis, d) 30% elongation is reapplied with compression again introduced along the short axis. e) This pattern persists through multiple cycles of stretching when the film is not healed. All AFM images are 40 μm × 40 μm.
The above evidence supports the conclusion that the observed damage pattern arises from plastic deformation (stretching) when the film is under linear stress, followed by film wrinkling as the stress is removed. Upon stretching, the total area of the film increases to accommodate the stretching of the elastomeric substrate. Removing the stress reduces the effective area of the substrate, while the film lacks the elasticity needed to return to its original area, thus resulting in wrinkling. In contrast, observation of cracking in the films as stress was applied would have indicated strain-induced cracking as the origin of the observed damage pattern.
It is illustrative to think of the microparticulate structure of the films when considering the origin of the differential responses to elongation versus compression. The multilayered film consists of anionic polymer nanoparticles cross-linked Coulombically with highly-charged, linear polycation chains. Consequently, the individual particles have their own elastic properties, and connectivity to neighboring particles in the film is through a strong, though non-covalent (charge-based) interaction. As stress is applied, the Coulombic interactions between discrete acid sites on the microgels and individual monomer units of PDADMAC would seem to be the weakest link, and these interactions may be sacrificed in favor of an altered ion pairing structure that allows for an increase in film dimension along the stretching axis. Upon removal of the elongational stress, the original ion pairing arrangement does not recover, and the now elongated film simply wrinkles to accommodate the elastic recovery of the substrate. The polymer and ion mobility that occurs during hydration of the film then allows restoration of the smooth, low-energy confirmation.
Film Healing
The rapidity with which damage to microgel multi-layered films is resolved upon immersion in water has limited our ability to observe the stages of the healing process, and thus limited our understanding what drives self-healing. In an attempt to reduce the influence of passing the film across the air/water interface during immersion, film healing was attempted by exposure to the high relative humidity above a steaming beaker of water. As can be seen in Fig. 4, this too leads to the complete restoration of film integrity following damage by a plastic pipette tip. Once again, this response is rapid, requiring less than five seconds of exposure to steam (see video available in supplementary information). This finding suggests that immersion is unnecessary, as is the need for bulk water; surface hydration is sufficient to effect healing.
Fig. 4.

Damage induced by scratching an 8-layer 30% AAc microgels/400-500 kDa PDADMAC film rapidly heals in response to steam. a) Scratched film before healing b) Film exposed to steam for 5 seconds. Insets: microscopy of the scratched region before and after. Scale bars are 10 μm.
Having ascertained that water vapor was sufficient to heal film damage, we next sought to determine what level of ambient humidity was necessary to drive fully autonomous film healing. We explored the effects of intermediate humidity on films that had been stretched by 20%, thereby damaging the entire film area. These films were then placed them into sealed environments with controlled relative humidity, ranging from 55-84% by Rh sensor. As shown in Fig. 5e, little effect is observed in films at 55% relative humidity. At 73% humidity, in Fig. 5f, the films seem to have almost completely healed – the wrinkled pattern has nearly disappeared, but small parallel lines are still discernible. When exposed to higher relative humidity, as in Fig. 5g, h, the films do not show any remnant wrinkling. This suggests that the films are sufficiently hygroscopic to imbibe enough ambient water under high humidity conditions to effect the requisite polymer mobility for healing to occur. This hydration of the film leads to swelling, which in and of itself necessitates a rearrangement of the microgels and polycation in the film. The intermediate state visible in Fig. 5f suggests that healing proceeds first in the dimension normal to the plane of the film, since the intensity of the wrinkles diminishes as the film progresses from a wrinkled to a smooth state, as opposed to changes in the periodicity or width of the wrinkled features. This ability to drive film healing based on ambient humidity levels suggests that these coatings may be autonomous in high humidity environments, making them “equatorial-” or “rainy-day-” autonomous.
Fig. 5.

a – d) Damage caused by applying 20% strain to 8-layer 30% AAc microgels/400-500 kDa PDADMAC films. Film recovery following one hour exposure to e) 55% relative humidity, f) 73% relative humidity, g) 78% relative humidity, h) 84% relative humidity. Little change is noted at 55%, while slight remnant damage is visible at 73%. Complete recovery proceeds at 78% or higher. Scale bar in all images is 10 μm.
Hydration and swelling of an individual microgel is very rapid because of its relatively small dimensions, occurring within fractions of a second.32, 33 Swelling therefore may provide the driving force responsible for the rapidity with which film integrity is restored. Films composed of 30% acrylic acid microgels cross-linked with 400-500 kDa PDADMAC (8 microgel layers) have a typical dry thickness of approximately 75 nm, while film swelling leads to a greater than four-fold increase in film thickness to approximately 350 nm (Supporting Information). Note that this dimension is thinner than might be expected from swollen microgel diameters. This discrepancy arises from microgel condensation that occurs during polycation complexation. As observed by AFM, and shown in Fig. 6, wrinkling damage induced by pipette damage to the films leads to a feature size of the damage on the order of 100 nm. As a result, swelling of the film leads to a greater overall change in the film thickness than that which is introduced by damage. The rearrangement of polymer chains in the microgels and polycation within the film accompanying swelling are likely responsible for restoration of the overall film integrity.
Fig. 6.

a) Damage induced by pipette in a 8-layer 30% AAc/400-500 kDA PDADMAC film. b) After being exposed to an 83% relative humidity environment for one hour. Both AFM images are 20 × 20 μm. c) Line traces across the two images reveal the characteristic roughness associated with damaged and healed films.
Film Composition
To test the generalizability of the self-healing phenomenon in microgel multi-layered films, we explored changes in the charge density and distribution in the films. Recently other examples of self-healing polyelectrolyte multilayer assemblies driven by water addition have been demonstrated,23, 24 suggesting charge pairing and ion mobility are an important component of the self-healing characteristics. We explored the effects of particle charge density by reducing the acrylic acid content of pNIPAm-AAc microgels from 30 to 10%. Additionally, film connectivity can be manipulated by changing the length of the PDADMAC used during assembly. The longer polymer chains associated with higher molecular weight polycation results in limited penetration into the microgel network,34 as well as serving to cross-link neighboring microgels. As such, one would also anticipate that higher molecular weights would tend to lead to higher levels of microgel-to-microgel cross-linking within the films; shorter chains should result in poorer connectivity. The molecular weights of PDADMAC used in this work, 100-200 kDa and 400-500 kDa, would have estimated end-to-end distances ranging from 15 - 21 nm and 31 - 34 nm, respectively. As shown in Fig. 7, using lower AAc content microgels and shorter PDADMAC still leads to the formation of self-healing microgel multilayers, as neither the reduced acid content nor the shorter polycation disrupts connectivity sufficiently for the self-healing characteristic to be suppressed.
Fig. 7.

a, b) 8-layer 30% AAc microgel films cross-linked with 100-200 kDa PDADMAC. c, d) 10% AAc microgel films cross-linked with 100-200 kDa PDADMAC. e, f) 10% AAc microgel films cross-linked with 400-500 kDa PDADMAC. a, c, e) Films subjected to 20% strain, then relaxed. b, d, f) Films heal after being exposed to 84% relative humidity for one hour. Scale bars in all images are 10 μm.
Conclusions
In this work, we have demonstrated that microgel multi-layered films undergo plastic deformation in response to elongation, as evidenced by the onset of a characteristic buckling pattern upon removal of the linear stress. Additionally, the rapid healing response of the microgel films does not require immersion in water as exposure to a relative humidity greater than 75% is sufficient to promote healing. This suggests that film swelling is a key driving force in the rearrangement of polymer within the film, and hence healing. Finally, the generality of the self-healing phenomenon for microgel multilayer films has been extended to lower acid content and shorter polycation, both of which reduce film connectivity. Taken together, it is clear that microgel multi-layered films represent a dynamic and intriguing approach to self-healing materials, and that better understanding of the fundamental parameters of these assemblies will aid in their future applications.
Supplementary Material
Acknowledgments
Funding for JCG was provided by the National Institutes of Health (1 R01 GM088291-01) and training grant: GTBioMAT Graduate Training for Rationally Designed, Integrative Biomaterials (T32 EB 006343), U.S. Department of Education GAANN award, the Georgia Tech Center for Drug Design, Development and Delivery, and the Georgia Tech TI:GER® program. Funding for MWS was provided by the Georgia Tech Center for Drug Design, Development, and Delivery GAANN Fellowship.
Footnotes
Electronic Supplementary Information (ESI) available: Supplemental figures and schemes for the experimental design, microgel sizes, and AFM images from thickness and swelling determination are available. Additionally, a video depicting the healing of a film upon exposure to steam is presented. See DOI: 10.1039/b000000x/
References
- 1.Bergman SD, Wudl F. J Mater Chem. 2008;18:41–62. [Google Scholar]
- 2.Burattini S, Greenland BW, Chappell D, Colquhoun HM, Hayes W. Chem Soc Rev. 2010;39:1973–1985. doi: 10.1039/b904502n. [DOI] [PubMed] [Google Scholar]
- 3.Syrett JA, Becer CR, Haddleton DM. Polym Chem. 2010;1:978–987. [Google Scholar]
- 4.Hager MD, Greil P, Leyens C, van der Zwaag S, Schubert US. Adv Mater. 2010;22:5424–5430. doi: 10.1002/adma.201003036. [DOI] [PubMed] [Google Scholar]
- 5.van Gemert GML, Peeters JW, Sontjens SHM, Janssen HM, Bosman AW. Macromol Chem Phys. 2012;213:234–242. [Google Scholar]
- 6.White SR, Sottos NR, Geubelle PH, Moore JS, Kessler MR, Sriram SR, Brown EN, Viswanathan S. Nature. 2001;409:794–797. doi: 10.1038/35057232. [DOI] [PubMed] [Google Scholar]
- 7.Toohey KS, Sottos NR, Lewis JA, Moore JS, White SR. Nat Mater. 2007;6:581–585. doi: 10.1038/nmat1934. [DOI] [PubMed] [Google Scholar]
- 8.XX Chen F, Wudl F, Mal AK, Shen HB, Nutt SR. Macromolecules. 2003;36:1802–1807. [Google Scholar]
- 9.Reutenauer P, Buhler E, Boul PJ, Candau SJ, Lehn JM. Chem Eur J. 2009;15:1893–1900. doi: 10.1002/chem.200802145. [DOI] [PubMed] [Google Scholar]
- 10.Syrett JA, Mantovani G, Barton WRS, Price D, Haddleton DM. Polym Chem. 2010;1:102–106. [Google Scholar]
- 11.Burattini S, Colquhoun HM, Greenland BW, Hayes W. Faraday Discuss. 2009;143:251–264. doi: 10.1039/b900859d. [DOI] [PubMed] [Google Scholar]
- 12.Burattini S, Colquhoun HM, Fox JD, Friedmann D, Greenland BW, Harris PJF, Hayes W, Mackay ME, Rowan SJ. Chem Commun. 2009:6717–6719. doi: 10.1039/b910648k. [DOI] [PubMed] [Google Scholar]
- 13.Canadell J, Goossens H, Klumperman B. Macromolecules. 2011;44:2536–2541. [Google Scholar]
- 14.Yoon JA, Kamada J, Koynov K, Mohin J, Nicolay R, Zhang YZ, Balazs AC, Kowalewski T, Matyjaszewski K. Macromolecules. 2012;45:142–149. [Google Scholar]
- 15.Nicolay R, Kamada J, Van Wassen A, Matyjaszewski K. Macromolecules. 2010;43:4355–4361. [Google Scholar]
- 16.Amamoto Y, Kamada J, Otsuka H, Takahara A, Matyjaszewski K. Angew Chem, Int Ed. 2011;50:1660–1663. doi: 10.1002/anie.201003888. [DOI] [PubMed] [Google Scholar]
- 17.Cordier P, Tournilhac F, Soulie-Ziakovic C, Leibler L. Nature. 2008;451:977–980. doi: 10.1038/nature06669. [DOI] [PubMed] [Google Scholar]
- 18.Chen YL, Kushner AM, Williams GA, Guan ZB. Nature Chem. 2012;4:467–472. doi: 10.1038/nchem.1314. [DOI] [PubMed] [Google Scholar]
- 19.Cui JX, del Campo A. Chem Commun. 2012;48:9302–9304. doi: 10.1039/c2cc34701f. [DOI] [PubMed] [Google Scholar]
- 20.Phadke A, Zhang C, Arman B, Hsu CC, Mashelkar RA, Lele AK, Tauber MJ, Arya G, Varghese S. Proc Natl Acad Sci U S A. 2012;109:4383–4388. doi: 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burnworth M, Tang LM, Kumpfer JR, Duncan AJ, Beyer FL, Fiore GL, Rowan SJ, Weder C. Nature. 2011;472:334–U230. doi: 10.1038/nature09963. [DOI] [PubMed] [Google Scholar]
- 22.Yuan JC, Fang XL, Zhang LX, Hong GN, Lin YJ, Zheng QF, Xu YZ, Ruan YH, Weng WG, Xia HP, Chen GH. J Mater Chem. 2012;22:11515–11522. [Google Scholar]
- 23.Wang X, Liu F, Zheng XW, Sun JQ. Angew Chem, Int Ed. 2011;50:11378–11381. doi: 10.1002/anie.201105822. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Chen SS, Wu MC, Sun JQ. Adv Mater. 2012;24:4578–4582. doi: 10.1002/adma.201201306. [DOI] [PubMed] [Google Scholar]
- 25.Deng GH, Li FY, Yu HX, Liu FY, Liu CY, Sun WX, Jiang HF, Chen YM. ACS Macro Letters. 2012;1:275–279. doi: 10.1021/mz200195n. [DOI] [PubMed] [Google Scholar]
- 26.Serpe MJ, Jones CD, Lyon LA. Langmuir. 2003;19:8759–8764. [Google Scholar]
- 27.South AB, Whitmire RE, Garcia AJ, Lyon LA. ACS Appl Mater Interf. 2009;1:2747–2754. doi: 10.1021/am9005435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.South AB, Lyon LA. Chem, Int Ed. 2010;49:767–771. doi: 10.1002/anie.200906040. [DOI] [PubMed] [Google Scholar]
- 29.Park CW, South AB, Hu XB, Verdes C, Kim JD, Lyon LA. Colloid Polym Sci. 2011;289:583–590. [Google Scholar]
- 30.Hendrickson GR, Smith MH, South AB, Lyon LA. Adv Funct Mater. 2010;20:1697–1712. [Google Scholar]
- 31.Mark JE. Polymer data handbook. Oxford University Press,; Oxford; New York: 1998. [Google Scholar]
- 32.Suarez IJ, Fernandez-Nieves A, Marquez M. J Phys Chem B. 2006;110:25729–25733. doi: 10.1021/jp0643754. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka T, Fillmore DJ. J Chem Phys. 1979;70:1214–1218. [Google Scholar]
- 34.Kleinen J, Klee A, Richtering W. Langmuir. 2010;26:11258–11265. doi: 10.1021/la100579b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.