

Abstract
Beckwith-Wiedemann syndrome (BWS) is a rare disorder characterized by overgrowth and predisposition to embryonal tumors. BWS is caused by various epigenetic and/or genetic alterations that dysregulate the imprinted genes on chromosome region 11p15.5. Molecular analysis is required to reinforce the clinical diagnosis of BWS and to identify BWS patients with cancer susceptibility. This is particularly crucial prenatally because most signs of BWS cannot be recognized in utero. We established a reliable molecular assay by pyrosequencing to quantitatively evaluate the methylation profiles of ICR1 and ICR2. We explored epigenotype-phenotype correlations in 19 patients that fulfilled the clinical diagnostic criteria for BWS, 22 patients with suspected BWS, and three fetuses with omphalocele. Abnormal methylation was observed in one prenatal case and 19 postnatal cases, including seven suspected BWS. Seven cases showed ICR1 hypermethylation, five cases showed ICR2 hypomethylation, and eight cases showed abnormal methylation of ICR1 and ICR2 indicating paternal uniparental disomy (UPD). More cases of ICR1 alterations and UPD were found than expected. This is likely due to the sensitivity of this approach, which can detect slight deviations in methylation from normal levels. There was a significant correlation (p < 0.001) between the percentage of ICR1 methylation and BWS features: severe hypermethylation (range: 75–86%) was associated with macroglossia, macrosomia, and visceromegaly, whereas mild hypermethylation (range: 55–59%) was associated with umbilical hernia and diastasis recti. Evaluation of ICR1 and ICR2 methylation by pyrosequencing in BWS can improve epigenotype-phenotype correlations, detection of methylation alterations in suspected cases, and identification of UPD.
Keywords: BWS, genomic imprinting, DNA methylation, pyrosequencing, UPD
Introduction
Beckwith-Wiedemann syndrome (BWS; OMIM 130650) is a rare overgrowth syndrome that is characterized by prenatal and postnatal macrosomia, macroglossia, abdominal wall defects, and predisposition to embryonal tumors. Other signs are neonatal hypoglycemia, hemihyperplasia, ear anomalies, visceromegaly, renal abnormalities, adrenocortical cytomegaly, and an increased risk of embryonic tumors in childhood such as Wilms’ tumor. A family history of BWS is not frequently reported.1
BWS exhibits genotypic heterogeneity due to various genetic and/or epigenetic defects of the 11p15.5 region2 that harbors two imprinted domains, IGF2/H19 and KCNQ1/CDKN1C. Germline imprinting of IGF2/H19 and KCNQ1/CDKN1C is regulated by ICR1 (Imprinting Control Region 1) and ICR2, respectively. ICR1 is imprinted in the male germline and operates as an insulator. ICR2 is imprinted in the female germline and acts as a promoter for the regulatory non-coding RNA KCNQ1OT1. The pathogenetic abnormalities associated with BWS and their related frequencies are as follows: epigenetic defects of the 11p15.5 region (60%),2,3 point mutations of OCT4/SOX2-binding sites (10%),4 point mutations of CDKN1C (5–10%),3 anomalies of the trans-acting regulatory elements of the ICR2 domain (25%),2 and chromosomal rearrangements (2%).2–4 Epigenetic alterations are very common in BWS and include hypermethylation of ICR1 (5–10%) and/or hypomethylation of ICR2 (50–60%). These alterations can be primary or secondary to other genetic defects such as paternal uniparental disomy (UPD, 15–20%)5,6 and maternally-derived ICR1 microdeletions (10%).7 ICR1 defects (hypomethylation) are also associated with another genetically heterogeneous disorder, Silver-Russell syndrome (OMIM 180860), which is characterized by prenatal and postnatal growth restriction and mild dysmorphisms.8
Specific phenotype-(epi)genotype correlations have been described for BWS patients: macrosomia, macroglossia, and an increased risk of embryonic tumors are more frequently associated with ICR1 hypermethylation, while omphalocele is more common in individuals with ICR2 hypomethylation or CDKN1C point mutations.1–2,9
Although consensus diagnostic criteria for BWS have not been defined, the presence of three major features (e.g., prenatal and postnatal overgrowth, macroglossia, and abdominal wall defects) or two major features and one minor feature (e.g., ear anomalies, neonatal hypoglycemia, nephromegaly, and hemihyperplasia) is required for the postnatal clinical diagnosis of BWS.1 Molecular diagnosis is important to confirm the provisional BWS clinical diagnosis and to identify BWS patients with cancer susceptibility. Molecular diagnosis is particularly crucial for prenatal diagnosis because only a limited number of signs of BWS can be recognized in utero.
It has been suggested that BWS can be prenatally diagnosed in the mid-trimester of pregnancy by clinical findings of macrosomia, macroglossia, omphalocele, polyhydramnios, increased abdominal circumference, and renal or liver enlargement,9 and can be prenatally suspected by the identification of omphalocele in the first trimester. The prevalence of BWS in infants with omphalocele is reported to be 8–10%, and BWS is not prenatally suspected in the majority of these infants.10 Prenatal identification of BWS is important for pregnancy counseling, delivery planning, and postnatal management. Methylation defects in BWS can be identified by qualitative and quantitative methods. Quantitative methods are preferred11–13; however, clear indications for the use of these approaches have not been reported.
To establish a reliable molecular assay for prenatal and postnatal diagnosis of BWS, we quantitatively evaluated the methylation profiles of ICR1 and ICR2 using a pyrosequencing approach in cases that fulfilled all the clinical diagnostic criteria for BWS, cases with suspected BWS, and fetuses that exhibited signs of BWS. Genetic and epigenetic alterations can be in a mosaic condition (i.e., UPD) and can thereby lead to mild methylation defects. Therefore, we explored the relationship between the severity of methylation changes and BWS features to refine epigenotype-phenotype correlations.
Results
We performed pyrosequencing to investigate the methylation profiles of the imprinting control regions ICR1 and ICR2 located at 11p15.5. The following cases were eligible for analysis: postnatal cases in which BWS was clinically diagnosed, postnatal cases in which BWS was suspected due to the presence of at least one major feature of the disorder,1 and prenatal cases in which ultrasound (US) examinations revealed one or more features that were suggestive of BWS (i.e., abdominal wall defects, macrosomia, visceromegaly, and placentomegaly). None of the cases had a family history of BWS.
ICR1 epigenetic testing was designed to cover the CpG sites located at the sixth CTCF-binding site (Table S1) because we previously demonstrated that imprinting at this site is stable during development.13 For the ICR2 region, we established a new assay located within the KvDMR1 CpG island (Table S1) ~1 kb upstream of the NotI site, which is a locus that is extensively evaluated for diagnostic purposes by Southern blotting14 and pyrosequencing.11,12 The assay was not centered on the region defined by Bourque et al.11,12 because they reported higher mean methylation values (65–75%) than expected (~50%) in the control population. The authors hypothesized that this discrepancy was due to a PCR bias. We also evaluated the ICR2 locus including the NotI site by pyrosequencing and noticed unexpected methylation levels (hypomethylation) in both the prenatal and postnatal controls (mean: 24%, data not shown); consequently, this region was not considered an ideal target for diagnostic purposes. Therefore, we designed an assay located ~1 kb upstream of the NotI site. This site was considered useful for BWS testing since it showed hemimethylation in prenatal (mean: 45%) and postnatal (mean: 42%) tissues, as expected for an imprinted site.
Postnatal cases
The clinical features of the cases, which were described according to the criteria suggested by Weksberg et al.,1 and the methylation results are reported in Table 1. To define the methylation ranges in controls, we assessed the methylation values in peripheral blood lymphocytes (PBLs) of 103 healthy individuals (HIs) and found mean methylation levels of 46% (range: 40–52%; SD, 2.4%) and 45% (range: 39–50%; SD, 2.2%) at ICR1 and ICR2, respectively. For each sample, the methylation value represents the mean of at least two independent experiments with a standard deviation ≤ 3% (Fig. 1A). The methylation values resulted closely clustered (Fig. 1A), which allowed the threshold methylation levels of ICR1 and ICR2 to be identified. The methylation levels of ICR1 and ICR2 in each of the cases were then compared with these threshold and defined as normal or altered.
Table 1. Clinical features and methylation values of ICR1 and ICR2 in the postnatal cases.
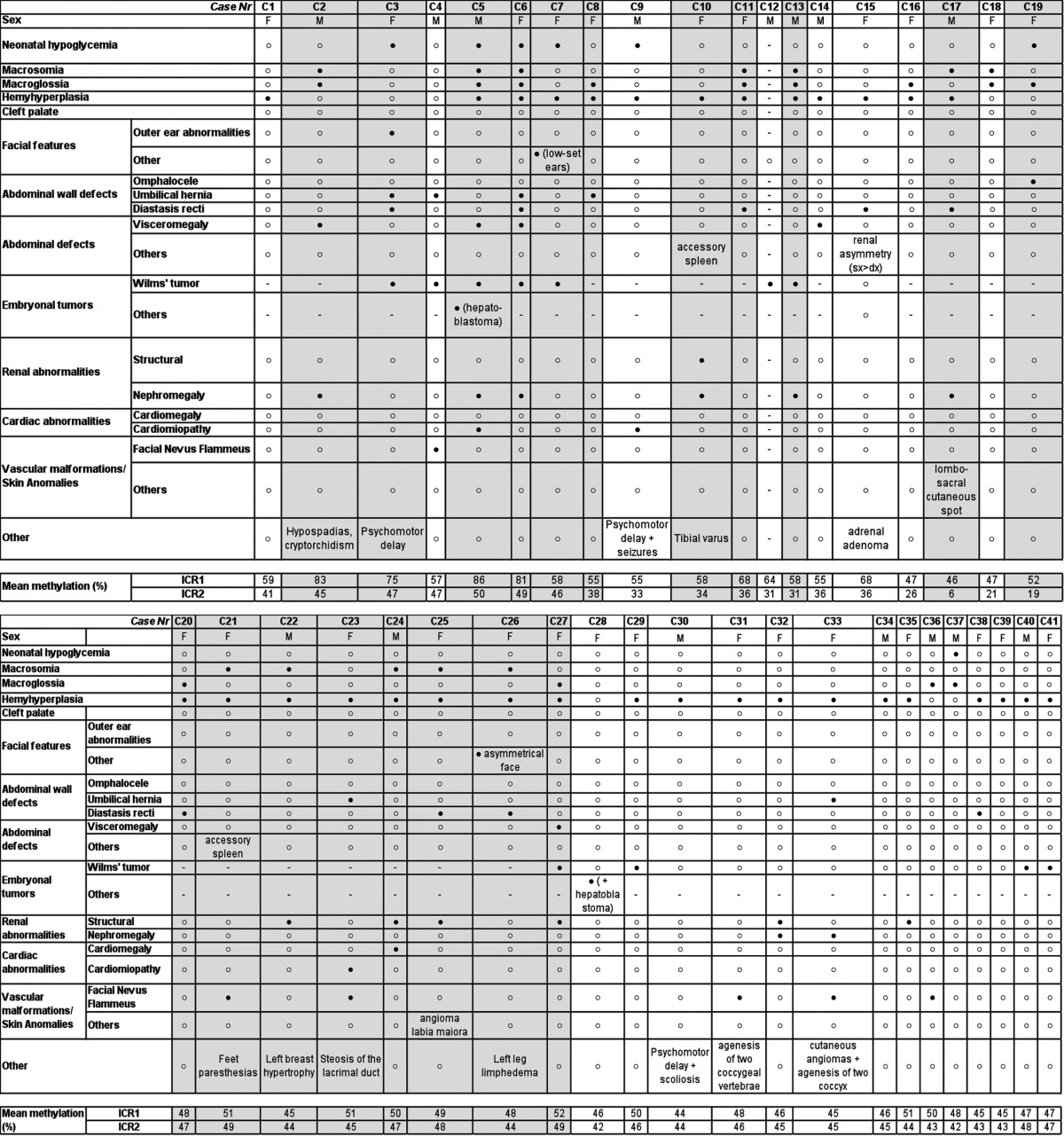
The cases that fulfilled all the clinical diagnostic criteria for BWS are shaded. Filled and unfilled circles denote the presence and absence of the feature, respectively. F, female; M, male.

Figure 1. Distribution of ICR1 and ICR2 methylation values in the control population (A) and postnatal BWS cases (B). Vertical (ICR1) and horizontal (ICR2) lines define the threshold levels of normal methylation values. The cases (▲) with mild ICR1 hypermethylation associated with abdominal wall defects (umbilical hernia or diastasis recti), but not with omphalocele, are encircled by a solid line. The cases (▲) with severe ICR1 hypermethylation associated with macroglossia, macrosomia, and visceromegaly are encircled by a dotted line. Cases with ICR2 hypomethylation (♦). Cases with both ICR1 and ICR2 methylation anomalies, indicating UPD ()
The postnatal study included 41 cases, of which 19 cases (13 females and 6 males, aged 1 mo–16 y) were clinically diagnosed with BWS and 22 cases (13 females and 9 males, aged 1 mo–14 y) presented with suspected BWS due to the presence of at least one major feature of the disorder.1 Abnormal methylation was observed in 19 of the 41 cases (Fig. 1B, Tables 1 and 2), and 11 of these 19 cases fulfilled all the criteria for the clinical diagnosis of BWS. In detail, of the 19 cases, seven showed ICR1 hypermethylation (mean: 71%; range: 57–86%; p < 0.001 vs. HI), four showed ICR2 hypomethylation (mean: 18%; range: 6–26%; p < 0.001 vs. HI), and eight showed both ICR1 hypermethylation (mean: 60%; range: 55–68%; p < 0.001 vs. HI) and ICR2 hypomethylation (mean: 34%; range: 31–36%; p < 0.001 vs. HI), which suggests paternal UPD spanning the 11p15.5 region. In three of these latter eight cases (C8, C9, and C12), microsatellite segregation analysis from parents to sibs confirmed paternal 11p15.5 UPD (data not shown). However, given that the concomitant presence of ICR1 and ICR2 methylation defects is the epigenetic manifestation of UPD,15 we considered all eight of these cases (C8-C15) to have UPD.
Table 2. : Percentage levels of ICR1 and ICR2 methylation in controls and cases.
| Population (number of subjects) |
Mean methylation, % (range) | |
|---|---|---|
| ICR1 | ICR2 | |
| Postnatal analyses | ||
| Controls (103) | 46 (40–52) | 45 (39–50) |
| Cases (41) | ||
| 7 | 71 (57–86)* | 47 (41–50) |
| 4 | 49 (46–53) | 18 (6–26)* |
| 8 | 59 (55–68)* | 34 (31–36)* |
| 22 | 48 (44–52) | 45 (42–49) |
| Prenatal analyses | ||
| Amniotic fluid controls (16) | 44 (38–48) | 42 (37–47) |
| Cases (3) | ||
| 1 | 48 | 30* |
| 2 | 46 (44–47) | 41 (37–45) |
Abnormal methylation.
When comparing the frequency of the different methylation defects of our cohort with those of the literature, we noticed higher levels of ICR1 alterations (37% vs. 7%) and UPD (42% vs. 27%) (Fig. 2). Although this could be related to the size of our population and to different inclusion criteria used in the literature studies, it may be because the pyrosequencing approach is more sensitive than other methods, as it allows slight deviations in methylation levels from normal values to be detected (see thresholds in Fig. 1B).
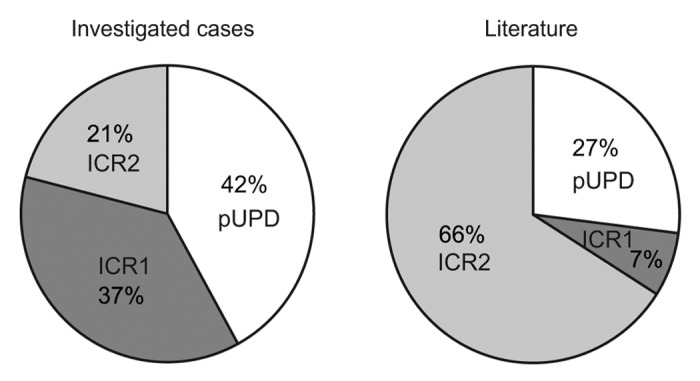
Figure 2. Frequencies of the different methylation defects (ICR1 hypermethylation, ICR2 hypomethylation, and both ICR1 and ICR2 methylation anomalies) in the cohort of the current study (left) and in cohorts of previous studies (http://www.ncbi.nlm.nih.gov/books/NBK1394/#bws.Summary) (right).
Epigenotype-phenotype correlations
A detailed description of the clinical features of the cases and the correlations of these features with the epigenetic defects are shown in Figure S1 and Table 1. We confirmed that omphalocele is associated with ICR2 hypomethylation and that an increased risk of embryonic tumors is associated with ICR1 hypermethylation, as previously reported.1,2,16 Regarding this latter point, it is remarkable that case C5, showing the highest ICR1 methylation values, developed two different embryonic tumors. This suggests a possible relationship between ICR1 hypermethylation grade and oncologic risk.
Finally, we found that all cases with alterations in both ICR1 and ICR2 methylation had hemihyperplasia (cases C8–15).
In the presence of hypermethylation at ICR1 (with normal ICR2 methylation), there was a correlation (p < 0.001) between the percentage of methylation and clinical BWS features (Table 1 and Fig. 1; Fig. S1). Two subgroups were identified (Fig. 1B): Group 1, in which ICR1 hypermethylation was severe (mean, 81%; range, 75–86%) and the cases had macroglossia, macrosomia, and visceromegaly; and Group 2, in which ICR1 hypermethylation was mild (mean: 57%; range: 55–59%) and the cases had abdominal wall defects comprising umbilical hernia or diastasis recti, but not omphalocele. No similar correlations between the percentage level of ICR2 methylation and BWS features were found.
Finally, seven of the eight cases with suspected BWS and epigenetic alterations coherently had a mild methylation defect: one case at ICR1 (57%), two cases at ICR2 (21% and 26%), and four cases with UPD (ICR1/ICR2: 55/38%, 55/33%, 64/31%, and 68/36%).
Prenatal cases
As controls, we analyzed amniotic fluid (AF) samples (14–18 weeks of gestation) from 16 pregnancies that underwent prenatal diagnosis due to advanced maternal age. From this, we defined the range of normal methylation levels (Table 2) of ICR1 (mean: 44%; range: 38–48%) and ICR2 (mean: 42%; range: 37–47%). Although we evaluated a small number of prenatal controls, the results are in agreement with those previously reported by our group on chorionic villus sampling CVS,13 suggesting that the imprinting marks at ICR1 and ICR2 are stable during development and in postnatal life.
We studied AF samples from three pregnancies with normal karyotypes, which were referred for molecular investigation following the detection of omphalocele by US imaging (Tables 2 and 3). Of these, one case displayed an abnormal methylation profile at ICR2 (mean methylation: 30%). The autopsy after therapeutic abortion confirmed the presence of omphalocele and revealed additional BWS features (Fig. 3) including macroglossia, ear anomalies, hepatomegaly, nephromegaly, ovarian hypertrophy, and adrenocortical citomegaly. Methylation analysis of DNA from paraffin-embedded CVS samples confirmed the hypomethylation at ICR2 (30%). The two other cases displayed normal methylation levels, and theautopsy after therapeutic abortion of one of these cases confirmed omphalocele but did not reveal any other BWS features.
Table 3. Descriptions of the three prenatal cases with suspected BWS.
| Sample ID | W.g. of AF sampling | Conception method | Features detected on ultrasound | Exitus | Karyotype | Clinical outcome |
Methylation level (%) ICR1 ICR2 |
|
|---|---|---|---|---|---|---|---|---|
| F1 | 18th+4 | Spontaneous | Omphalocele | Abortion at 20th w.g. | 46,XX | Omphalocele, visceromegaly, macroglossia, and peculiar craniofacial features | 48 | 30 |
| F2 | 16th+1 | Spontaneous | Omphalocele | Abortion at 20th w.g. | 46,XY | No additional signs of BWS | 47 | 45 |
| F3 | 16th+1 | Intra-cytoplasmic injection | Bi-amniotic bi-chorial twin pregnancy Omphalocele and a growth delay in the female fetus | Twins born at 37th w.g. | 46,XX (fetus with omphalocele) 46,XY (normal fetus) |
Omphalocele was not present at birth | 44 | 37 |
w.g, week of gestation

Figure 3. Autopsy of prenatal case F1. Hypertrophy of the labia majora (A), macroglossia (B), ear anomalies (C), visceromegaly with hepatomegaly, nephromegaly, and ovarian hypertrophy (D and E), and adrenocortical cytomegaly (F).
The other case was a twin pregnancy (bi-amniotic and bi-chorial fetuses). An umbilical hernia was present at birth and the suspicion of BWS was not confirmed.
Discussion
Accurate analysis of methylation at the imprinting control regions of 11p15.5 is an important tool in the molecular diagnosis of BWS. This is because deviations from the expected methylation levels are frequent in BWS2 and can result in the following genetic/epigenetic defects: loss of methylation at ICR2 and/or gain of methylation at ICR1 on the maternal chromosome; paternal UPD at 11p15.5; and duplication, inversion, or translocation of 11p15.5 leading to gene dosage alterations.
Sensitive methylation tests are also useful for the following reasons: (1) the clinical diagnostic criteria for BWS described by Weksberg1 help confirm suspected BWS; however, the diagnosis remains uncertain in cases that do not fulfill all these criteria, unless a molecular pathogenetic alteration is identified; (2) although correlations between genetic/epigenetic defects and BWS clinical features are known, specific associations based on the methylation percentages have not been delineated; and (3) BWS epigenetic defects are often mosaicisms (i.e., UPD), which suggests that even small variations in the methylation level can affect clinical features.
The complexities associated with methylation analysis, mainly in cases with low-level mosaicism, demand powerful analytical methods that can accurately quantify multiple CpG sites with high resolution and reproducibility. At present, pyrosequencing and mass spectrometry for nucleic acid analysis have unparalleled levels of accuracy to study methylation variation. In comparison, the accuracy of semi-quantitative (Southern blotting and methylation-sensitive multiplex ligation probe analysis) and qualitative (methylation-specific PCR) approaches is lower.17–19
We established a pyrosequencing approach to analyze methylation levels at the ICR1 and ICR2 loci of 11p15.5 with diagnostic purposes, in prenatal and postnatal cases, and investigated possible correlations between these methylation levels and the clinical features of BWS. The assays were validated using PBLs of HIs and AF of uncomplicated pregnancies.
Using this approach, we confirmed the diagnosis of BWS in ~60% of individuals that fulfilled all the clinical diagnostic criteria and in one-third of cases that did not fulfill these criteria. This indicates that this pyrosequencing approach is a reliable test to confirm clinically suspected BWS.
The high sensitivity of this test means it is particularly suitable to identify mosaic UPD, which can be underestimated by polymorphism analyses. Overlooking UPD might be an important limitation in the identification of BWS cases that have a high risk of cancer.20 Moreover, this approach allows UPD to be identified without the need to analyze the DNA of the parents, meaning the number of cases that can be tested is increased.
The pyrosequencing approach was also useful for prenatal diagnosis of BWS. This is particularly helpful as not all clinical diagnostic criteria of BWS can be evaluated in the fetus and this technique can be performed using fetal DNA, which is often of poor quality. Our results confirmed that imprinting at ICR113 and ICR2 is present and stable during development; therefore, prenatal diagnosis of BWS using this approach can be performed on both CVS and AF. Indeed, evidence of hypomethylation at ICR2 was found in the AF sample of one of the three prenatal cases examined, and the fetal autopsy and histological evaluations confirmed the diagnosis of BWS.
Another aim of this study was to investigate whether there is a correlation between the percentage of methylation at ICR1 and/or ICR2 and the features of BWS. ICR1 hypermethylation levels were shown to be suitable clinical markers of BWS. Indeed, there were significant correlations of severe ICR1 hypermethylation (range, 75–90%) with macroglossia, macrosomia, and visceromegaly, and of mild ICR1 hypermethylation (range, 55–70%) with abdominal wall defects comprising umbilical hernia or diastasis recti but not omphalocele. Interestingly, in almost all cases that had epigenetic alterations and in which BWS had not been clinically diagnosed (six of seven cases), only mild methylation defects were found. This suggests that there is a direct association between the methylation percentage and the severity of BWS.
Although methylation analysis is highly relevant to BWS molecular testing, the diagnostic procedures should also include additional analyses since ~30% of cases present other genetic abnormalities. In particular, the evaluation of CDKN1C point mutations and microdeletion/microduplication of chromosome 11p15.5 are required to identify families with a significantly increased recurrence risk.
In conclusion, we describe a fast, sensitive, and reliable pyrosequencing assay of ICR1 and ICR2 to detect methylation defects of the 11p15.5 imprinted region, which can be used to diagnose BWS in prenatal and postnatal cases. In addition, precise and quantitative identification of ICR1 and ICR2 methylation levels in BWS cases can be useful to improve epigenotype-phenotype correlations, although additional cases are needed to confirm our observations.
Material and Methods
Population
The study included 41 postnatal cases, of which 19 cases (13 females and 6 males, aged 1 mo–16 y) were clinically diagnosed with BWS and 21 cases presented with suspected BWS due to the presence of at least one major feature of the disorder as reported by Weksberg et al.1 Postnatal clinical diagnosis of BWS requires the presence of at least three major features of BWS (i.e., anterior abdominal wall defects, macroglossia, macrosomia, anterior ear lobe creases and/or posterior helical pits, visceromegaly, embryonal tumor in childhood, hemihyperplasia, cytomegaly of adrenal fetal cortex, renal abnormalities, positive family history of BWS, and cleft palate), or two major features and one minor feature of BWS (i.e., polyhydramnios, enlarged placenta and/or thickened umbilical cord, premature onset of labor and delivery, neonatal hypoglycemia, nevus flammeus, cardiomegaly/structural cardiac anomalies/cardiomyopathy, characteristic facies, diastasis recti, and advanced bone age). Descriptions of the prenatal clinical signs of BWS in the postnatal cases examined were not available.
The study also included three prenatal cases, which were selected because omphalocele was detected by US imaging. The cases were referred from various institutions (MBBM Foundation, San Gerardo Hospital, Monza; Istituto Auxologico Italiano; Fondazione IRCCS, Istituto Nazionale dei Tumori, Milano; and Fondazione IRCCS, Ca’ Granda Ospedale Maggiore Policlinico, Milano). Appropriate informed consent for the diagnostic molecular test was obtained from all parents. AF samples from pregnancies suspected of BWS and PBLs from patients with BWS were collected using standard procedures.
Standard cytogenetics procedures revealed all postnatal and prenatal cases had a normal karyotype. As controls, we analyzed 103 PBL samples from unaffected HI and 16 AF samples from uncomplicated pregnancies that underwent prenatal testing due to advanced maternal age.
DNA conversion and pyrosequencing
Genomic DNA was extracted from PBL samples, AF samples, and from the paraffin-embedded placental sample of case F1 using the QIAamp DNA Mini Kit (Qiagen). Sodium bisulphite conversion of DNA (500–700 ng) was performed using the EZ DNA Methylation-Gold Kit (Zymo Research Corporation,). PCR of ICR1 and ICR2 was performed using 20–100 ng of bisulphite-treated DNA and 10 pmol of the forward and reverse primers. Pyrosequencing assays were performed using the Pyro Mark ID instrument (Qiagen). Raw data were analyzed using the Q-CpG software v1.09 (Biotage AB), which calculates the ratio of converted C (T) to unconverted C at each CpG site, giving the percentage of methylation. Primer sequences and genomic positions are provided in Table S1.
Supplementary Material
Acknowledgments
This study was supported by the “Accordo Quadro Università-Regione Lombardia n. 17292” and by PRIN 2009 MBHZPR_003 (to LL). We thank Diatech Pharmacogenetics srl for the technical support for pyrosequencing.
Glossary
Abbreviations:
- AF
amniotic fluid
- BWS
Beckwith-Wiedemann syndrome
- HI
healthy individuals
- PBL
peripheral blood lymphocyte
- UPD
uniparental disomy
- US
ultrasound
Submitted
06/12/2013
Revised
07/16/13
Accepted
07/18/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/25812
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/25812
References
- 1.Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14. doi: 10.1038/ejhg.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demars J, Gicquel C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin Genet. 2012;81:350–61. doi: 10.1111/j.1399-0004.2011.01822.x. [DOI] [PubMed] [Google Scholar]
- 3.Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2013;163:131–40. doi: 10.1002/ajmg.c.31363. [DOI] [PubMed] [Google Scholar]
- 4.Poole RL, Leith DJ, Docherty LE, Shmela ME, Gicquel C, Splitt M, et al. Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur J Hum Genet. 2012;20:240–3. doi: 10.1038/ejhg.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henry I, Bonaiti-Pellié C, Chehensse V, Beldjord C, Schwartz C, Utermann G, et al. Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature. 1991;351:665–7. doi: 10.1038/351665a0. [DOI] [PubMed] [Google Scholar]
- 6.Dutly F, Baumer A, Kayserili H, Yüksel-Apak M, Zerova T, Hebisch G, et al. Seven cases of Wiedmann-Beckwith syndrome, including the first reported case of mosaic paternal isodisomy along the whole chromosome 11. Am J Med Genet. 1998;79:347–53. doi: 10.1002/(SICI)1096-8628(19981012)79:5<347::AID-AJMG4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Prawitt D, Enklaar T, Gärtner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, et al. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms’ tumor. Proc Natl Acad Sci U S A. 2005;102:4085–90. doi: 10.1073/pnas.0500037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartholdi D, Krajewska-Walasek M, Ounap K, Gaspar H, Chrzanowska KH, Ilyana H, et al. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet. 2009;46:192–7. doi: 10.1136/jmg.2008.061820. [DOI] [PubMed] [Google Scholar]
- 9.Williams DH, Gauthier DW, Maizels M. Prenatal diagnosis of Beckwith-Wiedemann syndrome. Prenat Diagn. 2005;25:879–84. doi: 10.1002/pd.1155. [DOI] [PubMed] [Google Scholar]
- 10.Cohen-Overbeek TE, Tong WH, Hatzmann TR, Wilms JF, Govaerts LC, Galjaard RJ, et al. Omphalocele: comparison of outcome following prenatal or postnatal diagnosis. Ultrasound Obstet Gynecol. 2010;36:687–92. doi: 10.1002/uog.7698. [DOI] [PubMed] [Google Scholar]
- 11.Bourque DK, Avila L, Peñaherrera M, von Dadelszen P, Robinson WP. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta. 2010;31:197–202. doi: 10.1016/j.placenta.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Bourque DK, Peñaherrera MS, Yuen RKC, Van Allen MI, McFadden DE, Robinson WP. The utility of quantitative methylation assays at imprinted genes for the diagnosis of fetal and placental disorders. Clin Genet. 2011;79:169–75. doi: 10.1111/j.1399-0004.2010.01443.x. [DOI] [PubMed] [Google Scholar]
- 13.Tabano S, Colapietro P, Cetin I, Grati FR, Zanutto S, Mandò C, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics. 2010;5:313–24. doi: 10.4161/epi.5.4.11637. [DOI] [PubMed] [Google Scholar]
- 14.Gaston V, Le Bouc Y, Soupre V, Burglen L, Donadieu J, Oro H, et al. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2001;9:409–18. doi: 10.1038/sj.ejhg.5200649. [DOI] [PubMed] [Google Scholar]
- 15.Romanelli V, Meneses HN, Fernández L, Martínez-Glez V, Gracia-Bouthelier R, F Fraga M, et al. Beckwith-Wiedemann syndrome and uniparental disomy 11p: fine mapping of the recombination breakpoints and evaluation of several techniques. Eur J Hum Genet. 2011;19:416–21. doi: 10.1038/ejhg.2010.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:343–54. doi: 10.1002/ajmg.c.30267. [DOI] [PubMed] [Google Scholar]
- 17.Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques. 2003;35:146–50. doi: 10.2144/03351md01. [DOI] [PubMed] [Google Scholar]
- 18.Tost J, El abdalaoui H, Gut IG. Serial pyrosequencing for quantitative DNA methylation analysis. Biotechniques. 2006;40:721–2, 724, 726. doi: 10.2144/000112190. [DOI] [PubMed] [Google Scholar]
- 19.Christians A, Hartmann C, Benner A, Meyer J, von Deimling A, Weller M, et al. Prognostic value of three different methods of MGMT promoter methylation analysis in a prospective trial on newly diagnosed glioblastoma. PLoS One. 2012;7:e33449. doi: 10.1371/journal.pone.0033449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalish JM, Conlin LK, Mostoufi-Moab S, Wilkens AB, Mulchandani S, Zelley K, et al. Bilateral pheochromocytomas, hemihyperplasia, and subtle somatic mosaicism: the importance of detecting low-level uniparental disomy. Am J Med Genet A. 2013;161A:993–1001. doi: 10.1002/ajmg.a.35831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.