

Abstract
Genome-wide disruption of the epigenetic code is a hallmark of malignancy that encompasses many distinct, highly interactive modifications. Delineating the aberrant epigenome produced during toxicant-mediated malignant transformation will help identify the underlying epigenetic drivers of environmental toxicant-induced carcinogenesis. Gene promoter DNA methylation and gene expression profiling of arsenite-transformed prostate epithelial cells showed a negative correlation between gene expression changes and DNA methylation changes; however, less than 10% of the genes with increased promoter methylation were downregulated. Studies described herein confirm that a majority of the DNA hypermethylation events occur at H3K27me3 marked genes that were already transcriptionally repressed. In contrast to aberrant DNA methylation targeting H3K27me3 pre-marked silent genes, we found that actively expressed C2H2 zinc finger genes (ZNFs) marked with H3K9me3 on their 3′ ends, were the favored targets of DNA methylation linked gene silencing. DNA methylation coupled, H3K9me3 mediated gene silencing of ZNF genes was widespread, occurring at individual ZNF genes on multiple chromosomes and across ZNF gene family clusters. At ZNF gene promoters, H3K9me3 and DNA hypermethylation replaced H3K4me3, resulting in a widespread downregulation of ZNF gene expression, which accounted for 8% of all the downregulated genes in the arsenical-transformed cells. In summary, these studies associate toxicant exposure with widespread silencing of ZNF genes by DNA hypermethylation-linked H3K9me3 spreading, further implicating epigenetic dysfunction as a driver of toxicant associated carcinogenesis.
Keywords: C2H2 zinc finger genes, DNA methylation, H3K27me3, H3K4me3, H3K9me3, arsenic, epigenetics, malignant transformation
Introduction
One important function of epigenetics is to provide a mechanism of genomic plasticity so that cells may respond to a changing and potentially harmful environment. Global disruption of the epigenetic code is a hallmark of malignancy that encompasses many distinct modifications that are highly interactive. For instance, during normal differentiation, histone H3 lysine 9 trimethylation (H3K9me3) acts as a substrate for heterochromatin binding proteins that recruit DNA methyltransferases thereby directing DNA methylation to specific genomic elements.1,2 Another repressive histone modification, H3 lysine 27 trimethylation (H3K27me3), predetermines many of the aberrant de novo DNA methylation sites in cancer most likely through protein-protein interactions which recruit DNA methyltransferases.3,4 On the other hand, trimethylation of histone H3 lysine 4 (H3K4me3) is found at the promoters of actively transcribed genes where it aids in the formation of an active chromatin conformation and prevents DNA methylation.5 Although some may function as individual enzymes, most epigenetic modifying proteins work as members of higher order protein complexes which often contain several epigenetic modifying enzymes, collectively controlling multiple distinct epigenetic marks. Generally, when one epigenetic mark is pathologically disrupted, several others are likely to be altered as a result. By characterizing the interplay between epigenetic modifications in toxicant-associated malignant transformation we can begin to uncover the underlying pathways frequently disrupted in cancer that drive widespread epigenetic malfunction.
The C2H2 zinc finger genes (ZNFs) make up one of the largest families of sequence specific DNA binding transcriptional repressors. At least a third of ZNF genes contain the KRAB domain (KRAB-ZNFs), which is known to interact with TRIM28 (also known as KAP-1) and indirectly recruit histone and DNA methyltransferases to specific genomic sequences that become marked with H3K9me3 and DNA methylation as a result.6-8 In non-malignant cells, this targeted repressive activity of ZNFs participates in important anti-cancer defenses including regulation of cell proliferation and silencing of endogenous retroviruses.6,9,10 Because ZNFs are indirectly responsible for recruitment of distinct epigenetic modifying enzymes they represent a potential common pathway that when disrupted would alter multiple epigenetic marks at specific genomic loci.
Our past studies have identified global DNA methylation and histone modification changes that occurred in toxicant-treated, malignant cell lines relative to their untreated, immortal counterparts.11-13 These models suggested that non-mutagenic environmental toxicants, such as arsenic, could induce aberrant agglomerative DNA methylation.14 We also found that in all the malignant cell lines and tumor biopsies tested, DNA hypermethylation occurred disproportionately within human embryonic stem cell (hESC) H3K27me3 and H3K9me3 domains with a striking correlation between densely packed hESC H3K9me3 domains and aberrantly methylated gene family clusters.14 Finding that DNA hypermethylation occurred disproportionately within H3K27me3 and H3K9me3 domains was not surprising, since these histone modifications are known to be linked to DNA methylation in certain scenarios.1,3 In the context of gene promoters, H3K27me3, H3K9me3 and DNA methylation are each known to participate in transcriptional repression, which led us to investigate the relationships between H3K27me3, H3K9me3, aberrant DNA hypermethylation and gene expression changes in environmental toxicant-associated malignant transformation.
In this pursuit, we have expanded on earlier studies by incorporating gene expression and selected chromatin immunoprecipitation analyses of the immortal prostate epithelial cell line RWPE-1 and its arsenic-exposed malignant variant, CAsE-PE, to better understand the relationships between aberrant DNA methylation, H3K9me3, H3K27me3 and gene expression of individual genes and gene family clusters in toxicant-associated malignant transformation. Our results confirm that H3K27me3 marked genes are targeted for aberrant DNA methylation and remain inactive. Contrarily, in immortalized cells we found that ZNFs in particular are frequently marked with H3K9me3 on their 3′ ends and H3K4me3 at the 5′ ends while actively transcribed.15 However, after malignant transformation, H3K9me3 had replaced H3K4me3 at the promoters of ZNFs and was accompanied by aberrant DNA hypermethylation, resulting in a widespread downregulation of ZNF gene expression.
Results
To interpret global DNA methylation changes associated with malignant transformation we performed MeDIP-on-Chip on 2 replicates of CAsE-PE and 2 replicates of RWPE-1 using Agilent Human promoter microarrays. This microarray probes the genomic regions between 5.5 kb upstream and 2.5 kb downstream of the transcription start sites (TSSs) of approximately 17,000 genes (Fig. S1A). Therefore, the probes are well positioned to detect DNA methylation changes that occur in close proximity to TSSs of protein coding genes but any genomic elements which are far from gene promoters have minimal coverage. With this probe distribution, the microarray is more likely to detect hypermethylation events associated with malignant transformation, since the DNA of normal cells is generally unmethylated near TSSs. In agreement with the array distribution and the general profile of DNA methylation at gene promoters in cancer, our final MeDIP-on-Chip data set of CAsE-PE relative to RWPE-1 contained predominately DNA hypermethylation events occurring near TSSs, while DNA hypomethylation events were fewer and more evenly distributed through the regions analyzed (Fig. S1A).
Hypermethylated genes share common ontologies
Overall, the MeDIP-on-Chip detected 869 hypermethylated and 166 hypomethylated genes in CAsE-PE cells (Supplemental Data Set 1). Most of the genes hypermethylated in CAsE-PE are H3K27me3 marked in human embryonic stem cells (hESC), consistent with data published previously (Fig. S1B).14 Several of the aberrant DNA methylation events detected by MeDIP-on-Chip were confirmed by MassARRAY technology (Fig. S2). To determine if particular classes of genes were targeted for hypermethylation in CAsE-PE, we performed a gene set enrichment analysis. Results showed that genes involved in developmental processes, and genes that are DNA or chromatin binding transcription factors were targeted for DNA hypermethylation (Table S1). This term enrichment could mean that these genes were selected for hypermethylation by nature of their function, which when silenced by methylation conferred some growth advantage to the cells. When taking into account that H3K27me3 domains of stem cells are targeted for aberrant DNA methylation in cancer, a more likely explanation is that these functional categories of genes are aberrantly methylated because they are normally regulated by the same epigenetic mechanisms, including H3K27me3 (Fig. S1B).16
Widespread downregulation of ZNFs in CAsE-PE
In addition to DNA methylation profiling, we also performed gene expression profiling to identify changes that were associated with the malignant phenotype of CAsE-PE. We measured global gene expression levels in 3 replicates of CAsE-PE and 3 replicates of RWPE-1 using the Affymetrix Human Gene ST 1.0 microarray platform. Based on the criteria of fold change ≥ 2 and adjusted p-value ≤ 0.05, we found 428 downregulated and 514 upregulated genes in the malignant CAsE-PE cells relative to the non-malignant RWPE-1 (Supplemental Data Set 2). We found several gene expression changes that have been linked to cancer progression of the prostate and other tissues and we used QRT-PCR to confirm a small representative set (Fig. S3). Surprisingly, there was a highly significant downregulation of ZNF and KRAB-ZNF genes, comprising almost 8% of all the downregulated genes. Thirty-four out of the total 519 ZNF genes covered by the expression array were significantly downregulated and 26 of these were KRAB-ZNF genes (p < 1 × 10−12, hypergeometric test of ZNFs). Over-representation of the ZNFs and particularly the KRAB-ZNFs among the downregulated genes suggests that they were silenced in a coordinated fashion which may be driven by a common epigenetic mechanism and that their broad silencing could have a substantial impact on the phenotype of CAsE-PE cells.
A negative correlation between DNA methylation and gene expression is driven by hypermethylation of ZNF gene promoters
Cancer-associated, DNA hypermethylation occurs disproportionately in H3K27me34 and H3K9me3 domains,14 leading us to question how each of these potential targeting mechanisms would affect cellular phenotype since both these histone modifications and DNA methylation at gene promoters repress transcription. To begin to address this question we determined to what degree the DNA methylation changes correlated with gene expression changes. We merged the MeDIP-on-Chip data with the expression data by calculating the methylation changes within 2 kb windows centered on TSSs. Plotting the DNA methylation changes relative to the gene expression changes showed that they were negatively correlated which indicates that the hypermethylation did not only occur at genes that were already silenced by H3K27me3 or H3K9me3 (Fig. 1). Forty-six genes were significantly downregulated and had a log2 fold DNA methylation increase greater than 0.5 (Fig. 1, blue text). Some of the genes that met these criteria are LAMB2, ANXA6, SLC4A11, UCHL1, FGF5, PTGS2, SPARC, and BDNF. Upregulation of only three genes was linked to decreased methylation of the TSS (S100P, INSL6, and ADRB2, Fig. 1). In all, about 9% of the observed gene expression changes were linked to DNA methylation changes, with almost all the changes being repressive.

Figure 1. Expression by methylation plot. The blue-shaded background density cloud shows the distribution of all the genes covered by both arrays. Differentially expressed genes (553 genes, represented as black dots) were used to calculate a correlation between methylation change and expression change. Spearman rank correlation coefficient = -0.37. Genes labeled with blue text had a significant decrease in expression and a log2 increase in methylation greater than 0.5. Not all the genes that meet these criteria are labeled due to space limitations. Selected genes labeled with green text had minimal expression change and a log2 increase in methylation greater than 0.5. Genes labeled with red text had a significant increase in expression and a log2 decrease in methylation greater than 0.5. Selected genes labeled with black text were also analyzed by QRT-PCR or MassARRAYTM (Figs. S2 and S3). CASP14 is labeled due to its high expression in CAsE-PE relative to RWPE-1.
It was readily apparent that many ZNFs were among the genes that were both hypermethylated and downregulated. Twelve ZNF genes (ZNF542, ZNF300, ZNF560, ZNF454, ZNF471, ZNF667, ZFP28, ZFP42, ZSCAN18, ZNF677, ZIK1, and ZNF582), 9 of which are KRAB-ZNFs, demonstrated significant downregulation and increased DNA methylation (> 0.5 log2 fold change). Overall, silencing of ZNF genes represented a substantial portion (12 of 46, ~26%) of all the significantly downregulated genes in CAsE-PE that were correlated with increased DNA methylation.
H3K27me3 domains are targets of aberrant DNA methylation
After merging the expression and methylation data sets it was apparent that many of the DNA hypermethylation events were not associated with changes in gene expression, since the target genes themselves were not expressed in parental RWPE-1 cells. In fact, genes associated with some of the most extensive DNA hypermethylation events, such as PHOX2B and DBC1, experienced no expression change (Fig. 1, green text). VSX1, SLITRK3, HOXC12, and PAX3 are other examples of substantial DNA hypermethylation events with no expression change, which is understandable since this group of genes is expressed at low levels in the parental RWPE-1 cells (Supplemental Data Set 2). Chromatin immunoprecipitations determined that VSX1, HOXC12 and PHOX2B were marked with H3K27me3 in RWPE-1 and CAsE-PE (Fig. 2). Overall, 92% of the genes with increased promoter methylation were not downregulated. Of these, 61% correspond to hESC H3K27me3 domains and 11% correspond to hESC H3K9me3 domains. While only a select few genes were verified as H3K27me3 targets in our models, based on the large overlap between hESC H3K27me3 domains and CAsE-PE hypermethylated genes (Fig. S1B) it is likely that a high proportion of the hypermethylated genes are H3K27me3 marked in the parental RWPE-1. These data further support that H3K27me3 is a major factor in directing aberrant DNA methylation during malignant progression, but the phenotypic effects of this phenomenon are unknown since it has not been associated with gene expression changes.
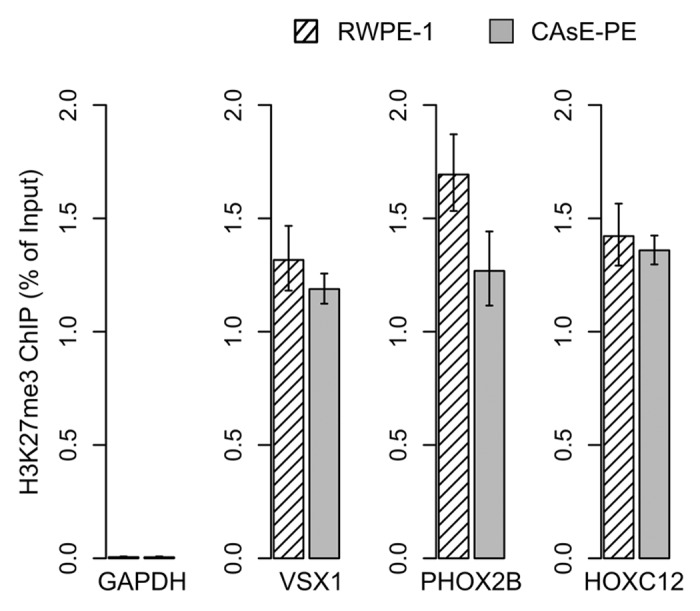
Figure 2. H3K27me3 target genes. H3K27me3 ChIPs were quantified by QRT-PCR. Error bars represent the SEM of three experiments. GAPDH primers were used as a control to show ChIP levels of a H3K27me3 negative region.
Zinc finger gene clusters associate with hESC H3K9me3 domains and are silenced by DNA methylation
In contrast to aberrant DNA methylation targeting H3K27me3 pre-marked silent genes, we found that actively expressed ZNF genes that overlap hESC H3K9me3 domains are preferred targets of DNA methylation linked gene silencing. We found a major overlap of hESC H3K9me3 domains with ZNF genes, many of which were downregulated and DNA hypermethylated. Most of the human ZNF genes reside within one of the 81 ZNF gene clusters across the human genome that are known to associate with H3K9me3 and heterochromatin binding proteins.17,18 Chromosome 19 is particularly rich with ZNF genes, accounting for about 44% of all the human ZNF genes including the largest ZNF gene cluster (19.13) with 76 ZNF genes. We found that 9 of the ZNF genes in cluster 19.13 were significantly hypermethylated while 10 were significantly downregulated in CAsE-PE relative to RWPE-1 (Fig. 3). Of the downregulated ZNFs in this cluster, 5 are hypermethylated, while ZFP28 and ZNF667 have increased promoter DNA methylation that did not reach the strict DMR requirement (Fig. 3). ZNF cluster 19.13 and most other ZNF gene clusters are hESC H3K9me3 domains (Fig. 4A). ZNF gene cluster 19.7 was also significantly downregulated and associated with H3K9me3 stem cell domains, but the methylation data for this cluster is limited because it was poorly covered by the MeDIP-on-Chip microarray (Fig. 4A). Four other ZNF clusters on chromosome 19 (19.12, 19.11, 19.6, and 19.5) also had downregulated and hypermethylated ZNFs. Downregulated, hypermethylated ZNFs were also found on other chromosomes, for example ZNF454, which is part of ZNF gene cluster 5.1 on chromosome 5, was hypermethylated, downregulated and associated with H3K9me3 stem cell domains (Fig. 4B). Likewise, ZNF300 on chromosome 5 was hypermethylated and downregulated, but does not belong to a ZNF gene cluster (Fig. 4B). In summary, while the data are limited, they suggest that ZNFs with unique connections to hESC H3K9me3 domains, are widely targeted for aberrant DNA hypermethylation and downregulation during toxicant-induced malignant transformation.
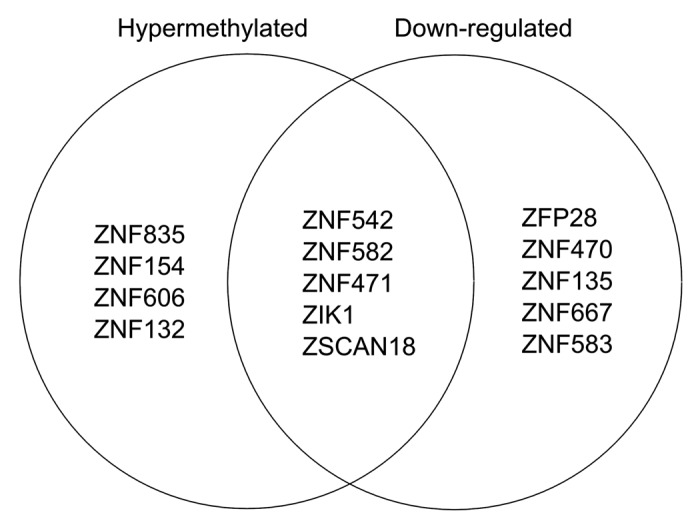
Figure 3. Venn diagram of ZNF genes in ZNF gene cluster 19.13 that are hypermethylated (i.e., contain a hyper DMR) or are downregulated.

Figure 4. Expression and Methylation Changes in CAsE-PE. Significantly upregulated genes or downregulated genes are plotted in red or blue respectively while other genes are shown in gray. Purple stars show the methylation change at differentially expressed genes where available, measured within a 2kb window centered on the TSS. Grey boxes mark the ZNF gene family clusters and green boxes mark H3K9me3 stem cell domains. (A) Chromosome 19. (B) Portion of Chromosome 5q.
Aberrant spreading of H3K9me3 is linked to DNA methylation and gene silencing
One would expect little to no transcription from genes associated with H3K9me3 since this histone modification induces heterochromatin formation and gene silencing. Therefore, it would seem contradictory for ZNF genes to be expressed in RWPE-1 cells and associated with domains commonly marked with H3K9me3 in other cell types. As a resolution to this contradiction, work by Blahnik et al. showed that actively expressed ZNF genes are marked by H3K9me3 primarily at their 3′ exons rather than through the entire gene.15 Based on this information we analyzed the histone methylation patterns at the 5′ and 3′ ends of several ZNF genes that were silenced by DNA methylation in CAsE-PE. Histone methylation levels were measured by ChIP at ZNF542, ZNF471 and ZFP28, all of which pertain to ZNF gene cluster 19.13 (Fig. 5). In parental RWPE-1 cells we found that the 3′ end of each of these genes was marked with H3K9me3 whereas at the 5′ ends of the genes H3K4me3 was present and H3K9me3 was absent. In the malignantly transformed CAsE-PE cells, H3K4me3 had been replaced by H3K9me3 at the 5′ ends of all three genes (Fig. 5). This same pattern was found at the ends of ZNF560 in ZNF cluster 19.5 (Fig. 6). This pattern of histone methylation suggests that during arsenic induced malignant transformation, H3K9me3 spreads from the 3′ exons of expressed ZNF genes to the 5′ end near the promoter leading to gene silencing and DNA methylation. Similar changes to histone methylation patterns were also observed at ZNF genes on chromosome 5 which experienced a replacement of H3K4me3 with H3K9me3 at their 5′ ends, DNA hypermethylation and downregulation in CAsE-PE (Fig. 6). Together, these data show that in malignant cells, individual ZNF genes and entire ZNF gene clusters across different chromosomes are silenced by spreading of H3K9me3 and DNA hypermethylation.

Figure 5. ZNF cluster 19.13 ChIPs. ChIPs of 5′ and 3′ ends of three ZNF genes in cluster 19.13 were quantified by QRT-PCR. (Top) H3K4me3. (Middle) H3K9me3. Error bars represent SEM of three experiments (*p = 0.05, Wilcoxon rank sum test). (Bottom) Locations of the ChIP amplicons.
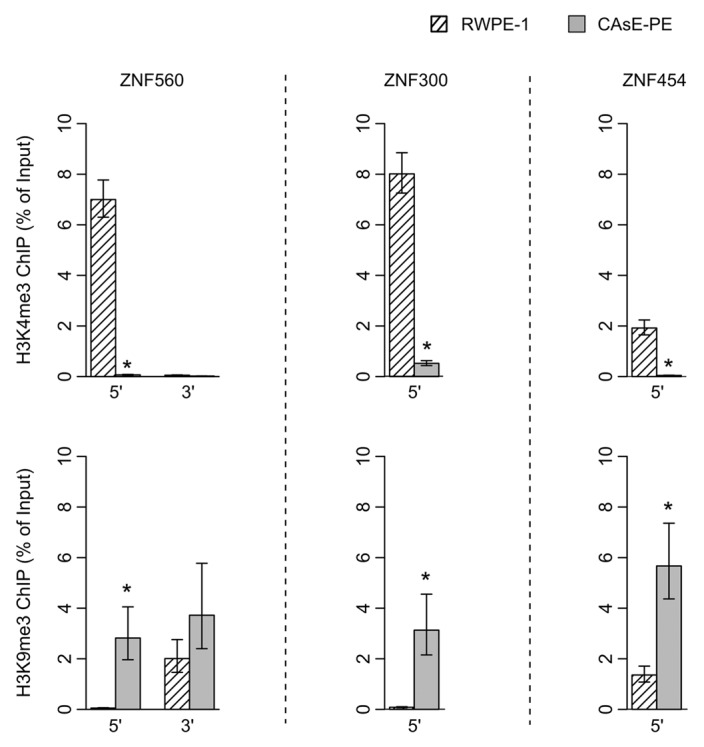
Figure 6. ChIPs of 5′ and 3′ ends of ZNF560 and 5′ ends of ZNF300 and ZNF454 were quantified by QRT-PCR. (Top) H3K4me3. (Bottom) H3K9me3. Error bars represent the SEM of three experiments (*p = 0.05, Wilcoxon rank sum test)
Discussion
In this study we measured 1035 differentially methylated and 942 differentially expressed genes in a prostate epithelial model of malignant transformation by an environmental toxicant. Most of the hypermethylated genes had no gene expression change and this is most likely a result of H3K27me3 targeted aberrant DNA methylation. In the malignant prostate epithelial cells there was widespread downregulation of ZNF genes that was mediated by H3K9me3 spreading from the 3′ ends of ZNF genes to the ZNF gene promoters. Loss of expression of ZNF genes was associated with a replacement of H3K4me3 with H3K9me3 and DNA methylation at the ZNF gene promoters. These findings suggest a mechanism in which the boundaries of H3K9me3 domains are disrupted, allowing H3K9me3 to spread and direct de novo DNA methylation to the promoters of ZNF genes in particular.
While H3K27me3 may be linked to more DNA hypermethylation events than H3K9me3 (Fig. S1B), the effect of aberrant DNA hypermethylation at H3K27me3 marked genes is unclear. One reasonable possibility is that aberrant DNA hypermethylation adds a more permanent level of repression to already silent genes, causing them to become non-inducible in cancer. In contrast to H3K27me3, H3K9me3 linked aberrant DNA methylation is clearly connected with gene expression changes and therefore could be an important contributor to the malignant phenotype by reducing global expression of ZNF genes.
Individual ZNFs and even some clusters of ZNFs have often been found hypermethylated and silenced in several tumor types, suggesting that our findings represent a commonly disrupted epigenetic pathway in cancer progression that is not a rare event.19-27 Perhaps the most compelling evidence that silencing of ZNFs in the arsenite-transformed prostate epithelial model is a relevant event in human carcinomas are the publicly available TCGA gene expression and DNA methylation data sets from normal prostate and prostate adenocarcinoma tissues (http://cancergenome.nih.gov/). The TCGA data show that DNA methylation-linked silencing of ZNFs is common (Fig. S4). In addition, large deletions along chromosome 19 are common in several malignancies including cervical cancer, esophageal squamous cell carcinoma and nasopharyngeal carcinoma.28-30 Together, these data suggest that silencing of ZNFs is an important event in the progression of various cancers and is not an event unique to arsenic-associated malignancies.
In the present study we identified 34 downregulated ZNFs and therefore we can only speculate on what the consequences of such a broad silencing event would be. Several studies have identified individual ZNFs as potential tumor suppressors (including ZNFs 382, 569, 383, 649, 540, 446, 411, 418, and 322) that regulate cellular proliferation by inhibiting MAPK signaling and repressing various oncogenes such as MYC, MITF, HMGA2, and CDK6.9,19,31-37 Together, these studies suggest that widespread silencing of ZNFs could lead to increases in MAPK signaling and proliferation. Another interesting possibility arises from recent work which has shown that ZNF genes co-evolved with human endogenous retroviruses (HERVs) and that one of their functions is to recruit epigenetic machinery to HERV sequences in the genome, which leads to HERV silencing by histone and DNA methylation.6,38-40 This suggests that one possible effect of widespread ZNF gene silencing could be hypomethylation and de-repression of HERVs, events that are commonly found in tumors (reviewed in ref. 41).
Finally, results from our ChIP assays indicated that in malignant cells, H3K9me3 had spread into previously unoccupied genomic regions. One potential mechanism underlying aberrant H3K9me3 spreading in malignant transformation involves the CTCF/PARP-1 insulator elements that are found at the boundaries between inactive and active genomic regions. Previous studies have shown that CTCF and PARP-1 together, protect certain genomic regions from acquiring repressive epigenetic marks.42,43 Oxidative stress, a common effect of arsenic exposure, disrupts the CTCF/PARP-1 interaction and may lead to increases in DNA methylation and other repressive epigenetic marks at the corresponding genomic loci.44,45 Therefore, disruption of CTCF/PARP-1 insulator elements by arsenical induced ROS is one possible explanation of how H3K9me3 could spread from the 3′UTR of ZNF genes to the promoter regions, but future studies will have to show whether this hypothesis holds any merit.
Materials and Methods
Cell culture and sample acquisition
The samples used in this study along with their culture conditions were described previously.14 The non-tumorigenic RWPE-1 cell line was derived from prostate epithelial cells that were infected with human papillomavirus 18 to induce immortalization.46 CAsE-PE (arsenite transformed RWPE-1) cells were derived and cultured as described previously.47,48 STR profiling confirmed that RWPE-1 and CAsE-PE are from the same individual.
Nucleic acid isolation
Genomic DNA was isolated using the DNeasy Blood and Tissue Kit according to the manufacturer’s protocol (Qiagen). Total RNA was harvested using TRIzol and purified using the miRNeasy Kit (Qiagen). The quantity of each sample was assessed using absorbance at 260 nm on the NanoDrop 1000 Spectrophotometer.
MeDIP coupled to microarray (MeDIP-on-Chip)
Methyl-cytosine DNA Immunoprecipitations (MeDIP) were performed as previously described.49 MeDIP and input samples were amplified, labeled, co-hybridized to Human Promoter ChIP-on-Chip Microarrays Set 244K (Agilent Technologies, G4489A) and washed as previously described.14
MeDIP-on-Chip data analysis
Microarray data were imported to R50 and normalized as previously described.51 M values (log2 ratios of immunoprecipitated and input channel) were used for further analysis as a measure of enrichment of a region centered on individual probes. For calculation of differentially methylated regions (DMRs), M values were analyzed in a sliding window of 1200 bp, with a step of one probe. DMRs were defined as regions of at least three consecutive probes less than 600 bp apart where the mean difference of ratios was at least 1.5 fold. A p-value cut was selected to maintain the false discovery rate (FDR) ≤ 5%. The FDR was determined by analysis of permutated data. DMRs were calculated between RWPE-1 and CAsE-PE samples. The coverage of the Human Promoter ChIP-on-Chip Microarray Set 244K was annotated to individual genes and a gene was considered differentially methylated when its annotated region contained a DMR. Gene set enrichment analyses of hypermethylated genes were performed using the GOstats package in R.52
Analysis of gene expression change
Three biological replicates of RWPE-1 and 3 replicates of CAsE-PE were grown until they reached approximately 80% confluence in 25 cm2 flasks. Total RNA was isolated from each flask and concentrations and quality were determined using absorbance at A260 and A280 followed by analysis on an Agilent Nanochip (Agilent Technologies). RNA samples were used to produce labeled target, hybridized to Affymetrix GeneChip® Human Gene 1.0 ST Arrays, and read using an Affymetrix scanner according to the manufacturer’s protocols. Raw data (CEL files) were normalized and summarized according to Irizarry et al.,53 as implemented in the package aroma.affymetrics.54 Differential expression was tested using the package LIMMA in the R programming environment.55 All p-values were adjusted according to Benjamini and Hochberg’s method to control the false discovery rate using a global method. Differential gene expression was defined as a fold change ≥ 2 and adjusted p-value ≤ 0.05. Several gene expression changes were confirmed by quantitative real-time (QRT)-PCR as described previously.11
Merging DNA methylation data with gene expression data
MeDIP-on-Chip data were merged with gene expression microarray data by calculating the average methylation differences within 2 kb windows centered on TSSs of genes that had 5 or more MeDIP-on-Chip probes within the 2 kb window.
Chromatin immunoprecipitation
Chromatin immunoprecipitations (ChIPs) were performed using the MAGnify Chromatin Immunoprecipitation system (Life Technologies, 49-2024) according to the manufacturer’s protocol with the following adjustments. Antibodies against H3K4me3 (Millipore, 05-745R), H3K27me3 (Cell Signaling, C36B11) and H3K9me3 (Diagenode, mAb-146-050) were used for chromatin immunoprecipitation. Cells were grown in 10-cm dishes, washed with Hank’s Balanced Salt Solution (HBSS) and then crosslinked by adding 5 mL of HBSS with 1% formaldehyde for 8 min at room temperature. The crosslinking reaction was stopped by adding 500 μL of 1.25 M glycine and incubating 5 min. Dishes were washed 2× with 5 mL HBSS and then the cells were scraped from the dishes in two steps using 2.5 mL of HBSS with 2% FBS, 0.1% EDTA and protease inhibitors for each scraping. Chromatin was sheared by sonication in a chilled Bioruptor (Diagenode) set to high for 42 cycles of 30 sec on/off. Approximately 200,000 cells were used for each ChIP. Input DNA was purified from a sample aliquot equal to 10% of the total cells used for each ChIP. Equal volumes of ChIP and input DNA were quantified by QRT-PCR using the ABI 7500 Real-Time Detection System. Primer sequences are provided (Table S2).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (P42 ES04940, ES006694, CA23074, 5T32ES16652-3 and 2T32ES007091-31 to PLS). This article may be the work product of an employee or group of employees of the National Institute of Environmental health Sciences (NIEHS), National Institutes of Health (NIH), however, the statements, opinions or conclusions contained herein do not necessarily represent the statements, opinions or conclusions of NIEHS, NIH or the US government.
Glossary
Abbreviations:
- ZNF
C2H2 zinc finger gene
- DMR
differentially methylated region
- H3K9me3
histone H3 lysine 9 trimethylation
- H3K27me3
histone H3 lysine 27 trimethylation
- H3K4me3
histone H3 lysine 4 trimethylation
- hESC
human embryonic stem cells
Data access
Microarray data were deposited in NCBI’s Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo) under the accession numbers GSE38930 and GSE47047.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/25936
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/25926
References
- 1.Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15:1176–83. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13:1192–200. doi: 10.1016/S0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 3.Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 4.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 5.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, et al. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development. 2013;140:519–29. doi: 10.1242/dev.087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quenneville S, Turelli P, Bojkowska K, Raclot C, Offner S, Kapopoulou A, et al. The KRAB-ZFP/KAP1 system contributes to the early embryonic establishment of site-specific DNA methylation patterns maintained during development. Cell Rep. 2012;2:766–73. doi: 10.1016/j.celrep.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Dénervaud N, et al. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010;6:e1000869. doi: 10.1371/journal.pgen.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X, Yuan W, Huang W, Bai Y, Deng Y, Zhu C, et al. ZNF569, a novel KRAB-containing zinc finger protein, suppresses MAPK signaling pathway. Biochem Biophys Res Commun. 2006;346:621–8. doi: 10.1016/j.bbrc.2006.05.109. [DOI] [PubMed] [Google Scholar]
- 10.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–40. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 11.Jensen TJ, Novak P, Eblin KE, Gandolfi AJ, Futscher BW. Epigenetic remodeling during arsenical-induced malignant transformation. Carcinogenesis. 2008;29:1500–8. doi: 10.1093/carcin/bgn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen TJ, Novak P, Wnek SM, Gandolfi AJ, Futscher BW. Arsenicals produce stable progressive changes in DNA methylation patterns that are linked to malignant transformation of immortalized urothelial cells. Toxicol Appl Pharmacol. 2009;241:221–9. doi: 10.1016/j.taap.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wnek SM, Jensen TJ, Severson PL, Futscher BW, Gandolfi AJ. Monomethylarsonous acid produces irreversible events resulting in malignant transformation of a human bladder cell line following 12 weeks of low-level exposure. Toxicol Sci. 2010;116:44–57. doi: 10.1093/toxsci/kfq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Severson PL, Tokar EJ, Vrba L, Waalkes MP, Futscher BW. Agglomerates of aberrant DNA methylation are associated with toxicant-induced malignant transformation. Epigenetics. 2012;7:1238–48. doi: 10.4161/epi.22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blahnik KR, Dou L, Echipare L, Iyengar S, O’Geen H, Sanchez E, et al. Characterization of the contradictory chromatin signatures at the 3′ exons of zinc finger genes. PLoS One. 2011;6:e17121. doi: 10.1371/journal.pone.0017121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 17.O’Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, et al. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Lodén M, Talhout W, et al. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 2006;16:1493–504. doi: 10.1101/gr.5391806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J, et al. KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer Res. 2010;70:6516–26. doi: 10.1158/0008-5472.CAN-09-4566. [DOI] [PubMed] [Google Scholar]
- 20.Omura N, Li CP, Li A, Hong SM, Walter K, Jimeno A, et al. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146–56. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita S, Tsujino Y, Moriguchi K, Tatematsu M, Ushijima T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2′-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006;97:64–71. doi: 10.1111/j.1349-7006.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mori Y, Olaru AV, Cheng Y, Agarwal R, Yang J, Luvsanjav D, et al. Novel candidate colorectal cancer biomarkers identified by methylation microarray-based scanning. Endocr Relat Cancer. 2011;18:465–78. doi: 10.1530/ERC-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lleras RA, Adrien LR, Smith RV, Brown B, Jivraj N, Keller C, et al. Hypermethylation of a cluster of Krüppel-type zinc finger protein genes on chromosome 19q13 in oropharyngeal squamous cell carcinoma. Am J Pathol. 2011;178:1965–74. doi: 10.1016/j.ajpath.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andresen K, Boberg KM, Vedeld HM, Honne H, Hektoen M, Wadsworth CA, et al. Novel target genes and a valid biomarker panel identified for cholangiocarcinoma. Epigenetics. 2012;7:1249–57. doi: 10.4161/epi.22191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30:1390–401. doi: 10.1038/onc.2010.525. [DOI] [PubMed] [Google Scholar]
- 26.Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, et al. The presence of aberrant DNA methylation in noncancerous esophageal mucosae in association with smoking history: a target for risk diagnosis and prevention of esophageal cancers. Cancer. 2009;115:3412–26. doi: 10.1002/cncr.24394. [DOI] [PubMed] [Google Scholar]
- 27.Mihara M, Yoshida Y, Tsukamoto T, Inada K, Nakanishi Y, Yagi Y, et al. Methylation of multiple genes in gastric glands with intestinal metaplasia: A disorder with polyclonal origins. Am J Pathol. 2006;169:1643–51. doi: 10.2353/ajpath.2006.060552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du Plessis L, Dietzsch E, Van Gele M, Van Roy N, Van Helden P, Parker MI, et al. Mapping of novel regions of DNA gain and loss by comparative genomic hybridization in esophageal carcinoma in the Black and Colored populations of South Africa. Cancer Res. 1999;59:1877–83. [PubMed] [Google Scholar]
- 29.Shao JY, Huang XM, Yu XJ, Huang LX, Wu QL, Xia JC, et al. Loss of heterozygosity and its correlation with clinical outcome and Epstein-Barr virus infection in nasopharyngeal carcinoma. Anticancer Res. 2001;21(4B):3021–9. [PubMed] [Google Scholar]
- 30.Tsuda H, Takarabe T, Okada S, Uchida H, Kasamatsu T, Yamada T, et al. Different pattern of loss of heterozygosity among endocervical-type adenocarcinoma, endometrioid-type adenocarcinoma and adenoma malignum of the uterine cervix. Int J Cancer. 2002;98:713–7. doi: 10.1002/ijc.10228. [DOI] [PubMed] [Google Scholar]
- 31.Xiang Z, Yuan W, Luo N, Wang Y, Tan K, Deng Y, et al. A novel human zinc finger protein ZNF540 interacts with MVP and inhibits transcriptional activities of the ERK signal pathway. Biochem Biophys Res Commun. 2006;347:288–96. doi: 10.1016/j.bbrc.2006.06.076. [DOI] [PubMed] [Google Scholar]
- 32.Cao L, Wang Z, Zhu C, Zhao Y, Yuan W, Li J, et al. ZNF383, a novel KRAB-containing zinc finger protein, suppresses MAPK signaling pathway. Biochem Biophys Res Commun. 2005;333:1050–9. doi: 10.1016/j.bbrc.2005.05.193. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Wang Y, Zhang C, Yuan W, Wang J, Zhu C, et al. ZNF322, a novel human C2H2 Kruppel-like zinc-finger protein, regulates transcriptional activation in MAPK signaling pathways. Biochem Biophys Res Commun. 2004;325:1383–92. doi: 10.1016/j.bbrc.2004.10.183. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Yang D, Bai Y, Mo X, Huang W, Yuan W, et al. ZNF418, a novel human KRAB/C2H2 zinc finger protein, suppresses MAPK signaling pathway. Mol Cell Biochem. 2008;310:141–51. doi: 10.1007/s11010-007-9674-4. [DOI] [PubMed] [Google Scholar]
- 35.Liu F, Zhu C, Xiao J, Wang Y, Tang W, Yuan W, et al. A novel human KRAB-containing zinc-finger gene ZNF446 inhibits transcriptional activities of SRE and AP-1. Biochem Biophys Res Commun. 2005;333:5–13. doi: 10.1016/j.bbrc.2005.05.069. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Zhu C, Luo J, Wang Y, Li D, Li Y, et al. ZNF411, a novel KRAB-containing zinc-finger protein, suppresses MAP kinase signaling pathway. Biochem Biophys Res Commun. 2004;320:45–53. doi: 10.1016/j.bbrc.2004.05.130. [DOI] [PubMed] [Google Scholar]
- 37.Yang H, Yuan W, Wang Y, Zhu C, Liu B, Wang Y, et al. ZNF649, a novel Kruppel type zinc-finger protein, functions as a transcriptional suppressor. Biochem Biophys Res Commun. 2005;333:206–15. doi: 10.1016/j.bbrc.2005.05.101. [DOI] [PubMed] [Google Scholar]
- 38.Guallar D, Pérez-Palacios R, Climent M, Martínez-Abadía I, Larraga A, Fernández-Juan M, et al. Expression of endogenous retroviruses is negatively regulated by the pluripotency marker Rex1/Zfp42. Nucleic Acids Res. 2012;40:8993–9007. doi: 10.1093/nar/gks686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rowe HM, Trono D. Dynamic control of endogenous retroviruses during development. Virology. 2011;411:273–87. doi: 10.1016/j.virol.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Thomas JH, Schneider S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011;21:1800–12. doi: 10.1101/gr.121749.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romanish MT, Cohen CJ, Mager DL. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin Cancer Biol. 2010;20:246–53. doi: 10.1016/j.semcancer.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Zampieri M, Guastafierro T, Calabrese R, Ciccarone F, Bacalini MG, Reale A, et al. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem J. 2012;441:645–52. doi: 10.1042/BJ20111417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell. 2009;34:271–84. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang F, Zhou X, Liu W, Sun X, Chen C, Hudson LG, et al. Arsenite-induced ROS/RNS generation causes zinc loss and inhibits the activity of poly(ADP-ribose) polymerase-1. Free Radic Biol Med. 2013;61C:249–56. doi: 10.1016/j.freeradbiomed.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guastafierro T, Catizone A, Calabrese R, Zampieri M, Martella O, Bacalini MG, et al. ADP-ribose polymer depletion leads to nuclear Ctcf re-localization and chromatin rearrangement(1) Biochem J. 2013;449:623–30. doi: 10.1042/BJ20121429. [DOI] [PubMed] [Google Scholar]
- 46.Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis. 1997;18:1215–23. doi: 10.1093/carcin/18.6.1215. [DOI] [PubMed] [Google Scholar]
- 47.Achanzar WE, Brambila EM, Diwan BA, Webber MM, Waalkes MP. Inorganic arsenite-induced malignant transformation of human prostate epithelial cells. J Natl Cancer Inst. 2002;94:1888–91. doi: 10.1093/jnci/94.24.1888. [DOI] [PubMed] [Google Scholar]
- 48.Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Webber MM, Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol Appl Pharmacol. 2005;206:288–98. doi: 10.1016/j.taap.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 49.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–62. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 50.R_Development_Core_Team. R: A language and environment for statistical computing. 2010.
- 51.Vrba L, Garbe JC, Stampfer MR, Futscher BW. Epigenetic regulation of normal human mammary cell type-specific miRNAs. Genome Res. 2011;21:2026–37. doi: 10.1101/gr.123935.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–8. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 53.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bengtsson H, Simpson K, Bullard J, Hansen K. Aroma.affymetrix: A generic framework in R for analyzing small to very large affymetrix data sets in bounded memory. 2008. [Google Scholar]
- 55.Smyth GK. Limma: Linear models for microarray data. 2005:397-420. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.