

Abstract
Measles virus (MV) vectors are promising candidates for designing new recombinant vaccines since the parental live vaccines have a well-known safety and efficacy record. Like all viral vectors, the MV vector efficacy in inducing a protecting immune answer could be affected by the pre-existing immunity among the human population. In order to determine the optimal immunization route and regimen, we mimicked a MV pre-immunity by passively administrating MV neutralizing antibodies (MV-nAb) prior intramuscular (i.m.) and/or intranasal (i.n.) immunization with recombinant MV expressing the SIV-gag antigen (rMV-SIVgag). Our results revealed that 500 mIU of MV-nAb allowed the induction of a humoral and cellular immune response against the vector and the transgene, while higher titers of the MV-nAb were significantly inhibitory. In a prime-boost regimen, in the presence of MV-nAb, the intranasal-intramuscular (i.n.-i.m.) or intramuscular-intramuscular (i.m.-i.m.) routes induced higher humoral immune responses against the vector and the transgene (SIV-gag). In naive animals, cellular immune response was significantly higher by i.m. immunization; however, MV pre-immunity did not seem to affect the cellular immune response after an i.n. immunization.
In summary, we show that a pre-existing immunity of up to 500 mIU anti-MV neutralizing antibodies had little effect on the replication of rMV and did not inhibit the induction of significant humoral and cellular immune responses in immune-competent mice.
Keywords: vaccine, pre-existing immunity, immunization route, measles, SIV, recombinant virus, reverse genetics, immune competent mice
Introduction
Novel technologies in vaccinology are necessary to tackle today's major diseases, such as AIDS, malaria and tuberculosis. Reverse genetics permits the designing of viruses, to be used as vectors, in order to deliver foreign antigens to the body.1-3 Due to its size, Measles virus (MV) is a an excellent vector candidate since its genome allows the incorporation of up to 40% of additional foreign nucleotides.4 The live MV vaccine has a long record of safety and efficacy, especially since the reversion to pathogenicity has never been observed with this vaccine. The live vaccine is injected subcutaneously or dispensed as an aerosol for adults, children and infants.2,10-12,22 Since the manufacturing and production of MV vaccine is well established, an MV-based vaccine vector would be easily manufactured and thus affordable. These characteristics prompted us and others to design and produce several MV vectors carrying diverse antigens derived from human pathogens, including Hepatitis B,5 Dengue,6 HIV-1,4,7,28 SARS8 and Malaria.9 For the rescue of recombinant measles viruses (rMV), a helper-cell based system had to be developed. The uniqueness of the rescue of these rMV viruses, using a human cell line, represents an additional advantage in terms of vaccine safety.
The measles vaccine has also been extensively used as an aerosol vaccine in children.2,9-12 Generally, aerosolized measles vaccine appeared to be more immunogenic than subcutaneous measles vaccines in children aged 10 mo and older. Aerosol applications have the advantage to be distributed over a significant surface, e.g., humans lungs represent around 50m2.1,2 In addition, the pulmonary, nasal and oral immune system comprise the mucosal-associated lymphoid tissues, which are responsible for the activation of approximately 80% of all immunocytes.13 Mucosal immunity is often the first line of defense against pathogens and is therefore of utmost importance. Many studies indicate that systemic immunization produce strong anti-viral systemic responses, while mucosal immunization can stimulate both the mucosal and systemic response and confer long-term immunological memory. However, the magnitude of these immune responses is often somewhat reduced, as seen with immunizations using adenovirus vectors.14-16,26
We constructed a MV-based vector containing various HIV-1 and SIV antigens with the ultimate goal of using it as pediatric vaccine against MV and HIV-1 infections. The possibility to dispense it as an aerosol is ideal for resource-limited countries where refrigeration and needle use are problematic. However, adults could also benefit of such an rMV-HIV-1 vaccine, since it could be used as an additional tool among HIV-1 therapeutics.
Certain questions arose as to whether such a vaccine would still be efficacious in the presence of a MV pre-immunity, which can be expected in a large number of an adult population. As seen in the STEP and Phambili study, the presence of an anti-vector pre-immunity can have a detrimental impact on the development of an immune response and on the potential of a vaccine to protect against new infections.17-19 However, other studies showed that Ad5-based vaccines given by mucosal routes can circumvent the effect of pre-existing immunity and induce significant immune response against the transgene.20 Similarly, other studies described the possibility to circumvent a measles pre-immunity using immunization by aerosol.21,22
We studied the effect of pre-existing anti-MV immunity on the magnitude of immune response against MV and transgene after immunization with a rMV vector expressing SIV-gag in immune competent hCD46 transgene mice.31 We show that at a limited MV pre-immunity (up to 500mIU), a prime-boost regimen can circumvent and maximize the induction of a specific humoral and cellular immune response against MV and the transgene.
Results
Passive infusion of MV neutralizing antibodies and its effect on the induction of humoral immune responses against MV and the SIVgag transgene
In order to investigate the impact of a MV pre-existing immunity prior immunization with a recombinant measles vector (rMV); 500, 2,000 or 5,000 mIU of MV neutralizing antibodies (nAb), respectively, were administered intravenously (i.v.) to mice. The following day, blood samples were taken and mice were immunized either intramuscularly (i.m.) or intranasaly (i.n.) with 1 × 105 pfu rMV-SIVgag, and the development of the humoral immune response against MV and the transgene was monitored.
Humoral IgG responses against MV
Figure 1 shows the fold increases of median MV-IgG titers between day 1 and week 2 after i.m. immunization. As expected, the group of animals that did not receive any MV-nAb induced the highest fold increase of all groups, which implies that even the lower dose (500 mIU) of MV-nAb had some inhibitory effect on the development of the anti-MV humoral immune response. Interestingly, the group of mice that received 5,000 mIU of MV-nAb had a fold increase similar to the group that was immunized with UV-inactivated rMV. These results suggest that the reduced antibody response in these animals was due to an inhibition of viral replication. Indeed, the median fold increase between day one and week 2 was inversely correlated with the amount of MV-nAb received, with R2 equal to 0.86.

Figure 1. hCD46tg mice were given 0, 500, 2,000 or 5,000 mIU MV-nAb i.v., respectively, one day prior i.m. immunization with 1 × 105 pfu rMV-SIVgag. UV-inactivated virus and mice that did not receive measles nAb prior immunization served as controls. Measles antibody titers were determined by endpoint titration at day one and week 2. Fold increase of the median titers between these two time-points are shown in the different groups.
The experiment was then repeated using either an i.m. or an i.n. prime, and the results are shown in Figure 2. After 4 weeks, mice that received 500 mIU of MV-nAb followed by an i.m. immunization showed an increase of anti MV-IgG titers 2 weeks post immunization (panel A). The i.n. group showed a somewhat delayed response with increased anti MV-IgG titers at week 4 (Fig. 2B). The groups that were given 2,000 or 5,000 mIU MV-nAb prior immunization did not show any increase in the development of anti-MV antibody (not shown), and were therefore excluded from further experiments. We conclude that mice pretreated with 500 mIU MV-nAb still experience viral replication but at a reduced degree after either i.m. or i.n. prime.
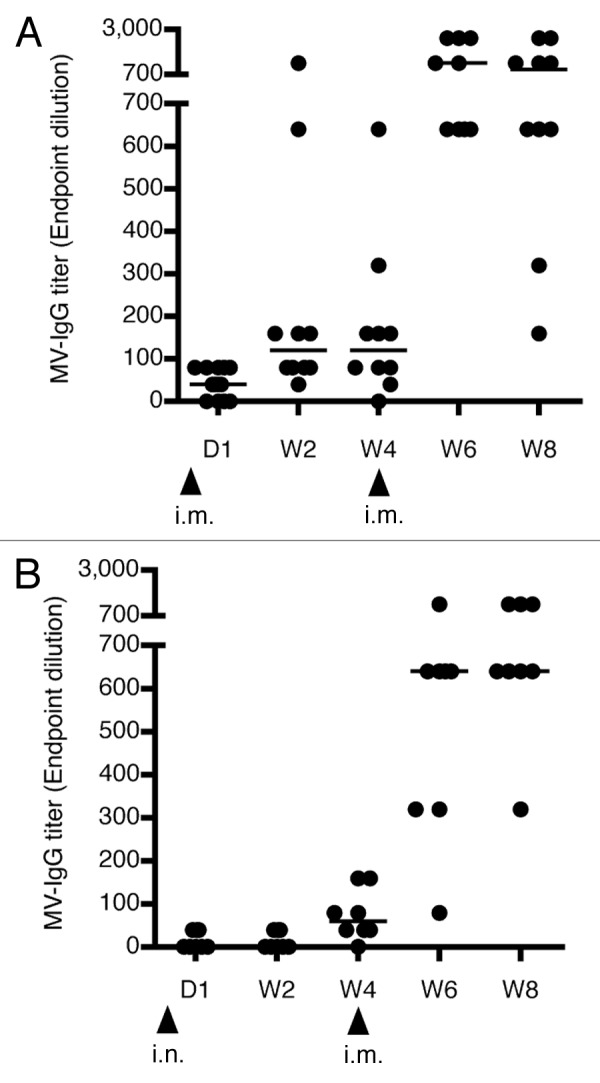
Figure 2. Humoral response against MV. hCD46tg mice were given 500 mIU MV-nAb one day prior to immunization i.m. (panel A) or i.n. (panel B) with 1 × 105 pfu rMV-SIVgag. Development of MV antibody titers over time is shown as median values in lines. The i.m. boost was performed at week 4 with 1 × 105 pfu rMV-SIVgag. Arrows show the time points of immunization.
To determine whether the specific humoral immune response could be improved even in the presence of 500 mIU of MV-nAb, the same animals were then boosted i.m. at week 4 with 1 × 105 pfu of rMV-SIVgag. The results show that the i.m-i.m. immunization route significantly increased anti-MV antibodies by 8-fold (median titer from 1:120 to 1:960, Fig. 2A). On the other hand, mice immunized by an i.n.-i.m. regimen showed an 11-fold increase of anti-MV antibody titers, with maximal titers reaching 1:1280 (Fig. 2B). Control animals (naive without MV-nAb) induced also quite a homogeneous MV antibody titers which increased steadily after the boost, up to median titers of 1:5,120 for i.m. prime-boost and 1:1,280 for i.n.-i.m. prime-boost at week eight.
Next, we assessed the capacity of the induced humoral response to neutralize the cognate antigen by determining MV-nAb. Pooled sera were used for this purpose. A positive development of MV-nAb could only be seen in the control group of mice and in the mice pre-treated to 500 mIU of MV-nAb (i.m. or i.n. prime). The naive control group had a mean MV-nAb titer of 1,153 mIU at 4 weeks post i.m. prime, which was increased to reach a mean titer of 15,124 mIU after boost (Fig. 3A, white bars). The pre-immune mice (with 500 mIU of MV-nAb) showed generally less MV-nAb, but a boost could triple the amount of MV-nAb to reach 600 mIU (Fig. 3A, gray bars). In comparison, the control mice primed via i.n. route generated less MV-nAb, reaching only 2,900 mIU after boost (Fig. 3B, white bars). However, when pre immune mice (having 100 mIU or 500 mIU of MV-nAb) were subject to i.n.-i.m. prime-boost, a marked increase in the anti MV-nAb, reaching 2,400 mIU and 770 mIU, respectively was observed (Fig. 3B, gray bars). Mice that were given 2,000 mIU or 5,000 mIU of MV-nAb prior to i.m. or i.n. immunization did not show any increase in MV-nAb after the boost (not shown).
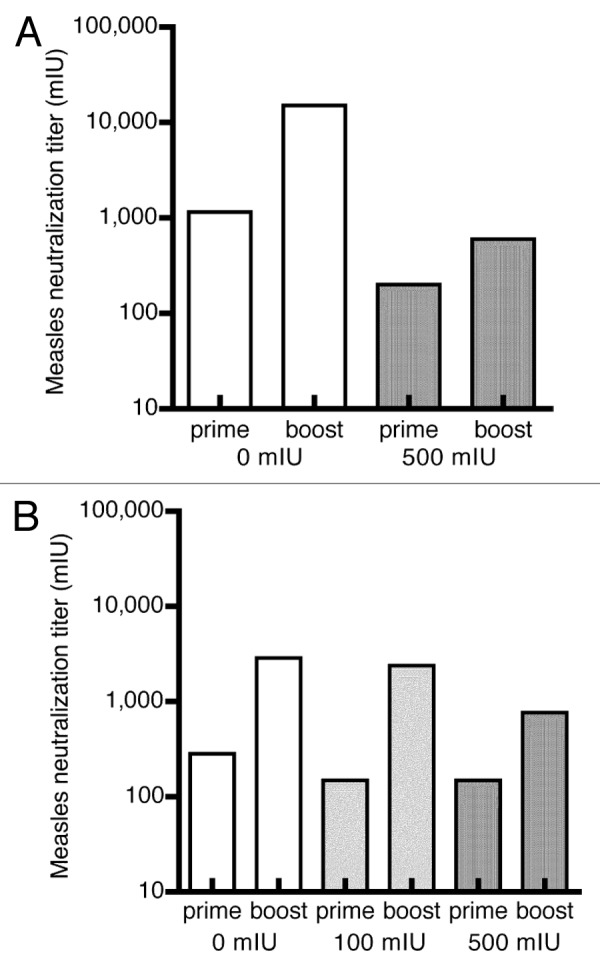
Figure 3. Induction of neutralization antibodies against MV in pre-immune hCD46tg mice by an i.m.-i.m or i.n.-i.m. prime-boost scenario. Transgenic mice were given MV-nAb (i.v.) one day prior to immunization with 1 × 105 pfu rMV-SIVgag. The concentration of antibodies are shown on panel (A) i.m.-i.m., and panel (B) i.n.-i.m. The i.m. boost of all mice groups was performed at 4 weeks post the prime. Neutralization anti MV antibodies were determined at week 4 (prior to boost) and 8 (after boost) are shown.
Taken together, these results indicate that a persistent MV IgG and neutralizing antibodies (MV-nAb) could be induced in the presence of up to 500 mIU of MV-nAb by i.m. or i.n. immunization. In addition, we show that a prime-boost schedule, i.n.-i.m or i.m.-i.m. could significantly increase the magnitude of MV antibody responses.
Humoral immune responses against SIVgag
The humoral immune response against the transgene, SIVgag, was determined under the same conditions mentioned above (i.e., either an i.m. or i.n. prime, followed by an i.m. boost at week 4) (Fig. 4). The presence of 500 mIU MV-nAb did not affect the induction of anti-SIVgag antibodies, whether immunized by i.m. or i.n. Similar endpoint anti-SIVgag antibodies were measured in both control animals and pre-immune animals, upon i.m.-i.m. prime-boost (Fig. 4A). However, we observed a delayed induction of antibodies against SIVgag upon an i.n. prime (Fig, 4B) in the absence or presence of a MV pre-immune status. At week 4, only few animals generated SIVgag antibodies with titers not exceeding 1:100 after immunization by the i.n. route. Interestingly, 4 weeks after the i.m. boost, both i.n.-i.m. groups, with or without anti measles preimmunity, showed a significant increase of anti SIVgag antibodies. No significant differences were detected between the naive control animals and the pre-immune animals. However, the i.n.-i.m. prime-boost group induced significantly more anti-SIVgag antibodies (p = 0.008) as compared with the i.m.-i.m. group, with titers reaching up to 1:1,280.
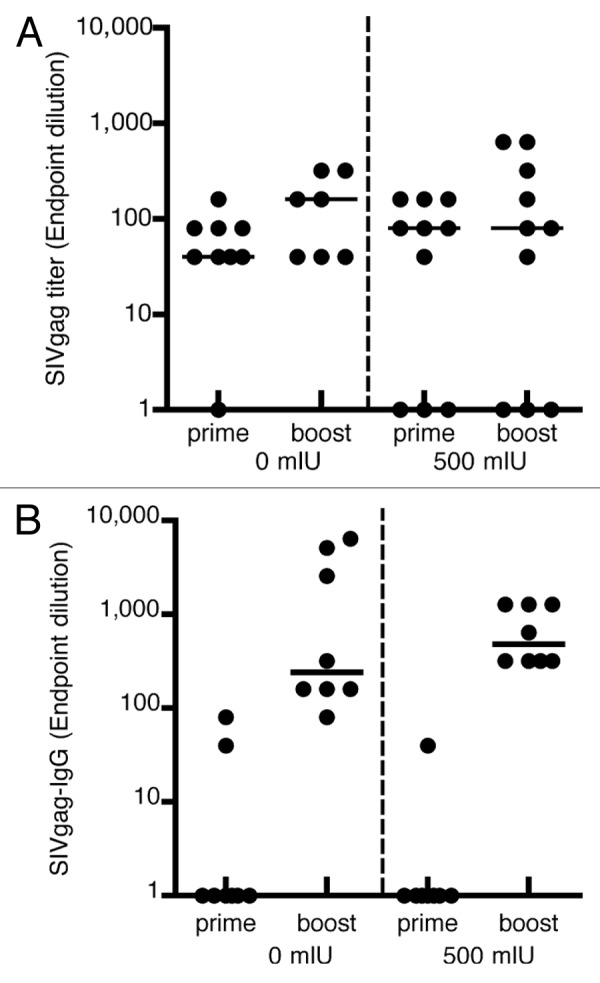
Figure 4. Induction of humoral immune responses against SIVgag. hCD46tg mice were given 0 or 500 mIU MV-nAb prior to i.m. or i.n. immunization with 1 × 105 pfu rMVSIVgag. Samples were collected at 4 weeks post prime i.m. (panel A) or i.n. (panel B) and 4 weeks post i.m. boost with 1 × 105 pfu rMV-SIVgag. The median values are shown as lines.
Effect of MV neutralizing antibodies on the induction of cellular immune response against MV and the SIVgag transgene
To assess the impact of pre-existing anti-MV antibodies on the induction of cellular immune response against the vector (MV) or the transgene (SIVgag), transgenic hCD46 mice were given 500 mIU of MV-nAb i.v. prior to i.m. or i.n. immunization with rMVSIVgag.
ELISpots were performed using 106 splenocytes of hCD46tg mice extracted 2 weeks post i.m. or i.n. immunization. The results are shown in Figure 5. By the i.m. immunization route, naive control animals reached median values of 440 anti-measles specific INFgamma secreting spot forming cells (SFC), whereas animals that received 500 mIU MV-nAb had median values of 180 anti-measles SFC (p = 0.018, Fig. 5A). Similarly, SIVgag-specific IFNγ-secreting cells were reduced from a median value of around 1,100 SFC, in naive animals, to 380 SFC in animals that received 500 mIU of MV-nAb (p = 0.0089, Fig. 5B). As expected, the group of animals immunized by UV-inactivated rMV-SIVgag did not induce cellular immune responses as measured by IFNγ Elispot assay (Fig. 5A and B).

Figure 5. Cellular immune response against MV and the transgene, SIVgag. hCD46tg mice were given 0 or 500 mIU MV-nAb, one day prior to either i.m. or i.n. immunization with 1 × 105 pfu rMV-SIVgag. ELISPOTs were performed 2 weeks post immunization. Panels A and B represent the intramuscular (i.m.) immunization and the measurement of cellular immune response against MV-N (Panel A) and SIVgag (Panel B). Panels C and D represent the intranasal (i.n.) immunization and the measurement of cellular immune responses against MV-N (Panel C) and SIVgag (Panel D). The lines represent the median values.
In the group of mice immunized i.n., we could observe that the cellular response against MV or SIVgag was generally lower as compared with i.m. immunization. The administration of 500 mIU of MV-nAb prior to i.n. immunization, however, did not significantly affect the magnitude of cellular immune responses. An equivalent cellular response against MV-N or SIVgag as compared with untreated mice was observed (Fig. 5C and D).
In conclusion, the ability to mount a cellular immune response against MV and the transgene was negatively affected by the pre-existing anti MV-nAb only when immunization was performed i.m. In contrast, no inhibitory effect was observed upon i.n. immunization, although the magnitudes of cellular immune responses were relatively lower as compared with i.m. immunization.
Discussion
New concepts and strategies are nowadays necessary for finding solutions toward a vaccine against Human Immunodeficiency Virus 1 (HIV-1), malaria and tuberculosis. Ideally, a vaccine should be safe, efficacious, and affordable; conditions that are all fulfilled in the commercial measles vaccine. In the case of HIV-1, the correlates of protection are not known; therefore, a protective vaccine may need to follow multiple strategies to optimize the breadth, quality, magnitude and effectiveness of the immune response,26 e.g., working with new antigen concepts,23 heterologous prime-boost regimens and/or heterologous delivery systems. One of these options is antigen delivery through viral vectors, such as measles, adenovirus, vaccinia, and canarypox. The disadvantage using viral vectors lies often in a pre-existing immunity status against the vector that can hamper the efficacy of these vectors.
Recent clinical studies demonstrated the negative impact of a pre-existing immunity against the vector used for immunization. In the STEP trial, that used a recombinant Adenovirus 5 (rAd5) vector-based vaccine, the vaccine failed to protect against HIV-1 or to reduce viral loads after infection because of the pre-existing immunity against the natural adenovirus.17-19
A similar situation may be expected with rMV vectors, thus it was pertinent to examine the effect of MV pre-immunity on the immune response against the vector as well as against the transgene. In humans, MV-nAb titers of > 200 mIU are considered protective;27 persons vaccinated against measles have usually MV-nAb titers of around 300 mIU, and the naturally infected persons induce higher MV-nAb titers of up to 3,000 mIU. To mimic such MV pre-immunity, we chose to work with an immune competent mice model expressing the human CD46 receptor, and which reflect more closely a natural immune response. In the context of a MV pre-immunity, the optimal route of immunization had to be defined and its effect on the humoral and cellular immune response had to be determined. We therefore administered MV-nAb intravenously at different concentrations prior to i.m. or i.n. immunization with rMV-SIVgag. The presence of MV-nAb had a direct impact on the replication of the recombinant measles viruses that were administered i.m. as seen by the inverse correlation between quantity of injected MV-nAb and measles antibody induction, as well as the reduced MV-nAb or cellular immune responses. The administration of 500 mIU MV-nAb prior immunization seemed to minimally affect mounting a humoral or cellular immune response against MV or SIVgag. This group had the highest capacity to induce antibodies against MV, the antibodies titers were persistent, and the increase in titers after a boost was homogeneous. A single intramuscular immunization, however, failed at inducing a strong humoral immunity against the transgene. Similarly, a single intranasal immunization was not able to induce a systemic immune response in the presence of MV-nAb. Therefore, we explored the effect of a homologous and heterologous prime-boost immunization using the intramuscular and the intranasal routes. Significantly higher titers of SIV antibodies were induced when the mice were primed intranasally and boosted intramuscularly. In contrast, the cellular response against MV and SIVgag was quite well developed with i.m. immunization and less developed with the i.n. immunization. Interestingly, when 500 mIU MV-nAb were injected prior i.m. or i.n. immunization, the cellular response was affected in the i.m. but not i.n. immunization route, although the magnitude of the responses were higher by the i.m. route. However, it appears that an intranasal immunization route has some advantages over the intramuscular route since higher levels of SIVgag antibodies could be induced with an i.n.-i.m. prime-boost regimen as compared with a i.m.-i.m. regimen. We did not measure any loss of cellular responses even in the presence of 500 mIU MV-nAb. Previous studies proposed that aerosol vaccines could circumvent a pre-existing immunity against the vector.21,22 Our results corroborate that a MV pre-immunity can be circumvented by an i.n. or aerosol immunization, although only in a heterologous immunization regimen. In contrast, the cellular immune response was less developed after an i.n. immunization, but can probably be optimized by an additional boost. We also performed broncho-alveolar lavages (BAL) after i.m. or i.n. immunization, and the highest amount of IFNγ-producing T-cells were observed after an i.m-i.n. prime-boost regimen, with 550 SFC per 106 cells for MV and 125 SFC per 106 cells for SIVgag (data not shown). As compared with an i.m. prime (which was undetectable in BAL), the i.m.-i.n. regimen seem to be optimal for a local cellular response induction, but not for a systemic response. Damjanovic et al. showed that after i.n. immunization with rAd5, the transgene was expressed after 1–3 d in the trachea and lungs.29 In addition, Bolton et al., showed that after an aerosol administration of rAd5 in rhesus macaques, local immune responses were persistent and of high quality, but the systemic responses were transient.30
In summary, this study provides evidence that MV pre-immunity with neutralization titers up to 500 mIU does not preclude MV replication and can induce significant humoral and cellular immune responses against the vector and the transgene. Applying an i.n.-i.m. prime-boost regiment increases the humoral and cellular immune responses specifically against the transgene. By extrapolation of the results, it seems possible to use a recombinant measles virus vector even in persons who were vaccinated against MV. However, the use of MV vectors in a population where naturally occurring infections are dominant does not seem to be very promising. However, further tests have to be performed, perhaps also in different animal models such as non-human primates to draw definitive conclusions concerning this issue.
Material and Methods
Virus and recombinant measles vector (rMV)
The commercial MV vaccine strain was used to construct the MV vector used in this study.3 Different sequences were introduced in the vector to enable the expression of SIVgag, HIVenv, or others.4 All viruses have been produced on human fetal lung fibroblasts called MRC5 cells and reached titers of 5 × 105 to 2 × 106 pfu. Most experiments used the original vaccine or the empty vector as control.
rMV virus batches were controlled for correct sequences of the vector and the transgene. Stability of transgene expression and the replication performance was monitored by serial cell culture passages of the virus. For these analyses, methods such as western blots, immunofluorescence, growth curve, sequencing of the passaged viruses were applied.
Antigens
The SIVgag was kindly provided by M. Feinberg, Emory University, Atlanta, USA and is not optimized for human codon usage.
Mice
Many studies on measles virus have used immunodeficient mice (IFNARhCD46tg) to control for the immune response potential of the vaccine. We chose to use immune competent mice that express human CD46 - the measles receptor-, named hCD46tg [B6.FVB-Tg(CD46)2Gsv]24,31 in order to ensure a more natural immune response.
The animals were kept at the Animal Facility under specified pathogen-free conditions during the experiment. The Swiss Federal Veterinary Office for Animal Experimentation reviewed and approved the mouse experiments.
Mice were immunized intramuscularly (i.m.) or intranasally (i.n.) at the age of 6 to 12 weeks with parental or recombinant measles virus (rMV) expressing the Simian Immunodeficiency Virus (SIV) gag protein (rMVb-SIVgag). The immunization dosage was mostly of 1 × 105 pfu or as indicated. Venous blood was collected via the retroorbital venous sinus at different time points.
For producing measles neutralizing antibodies, we immunized IFNARCD46tg mice several times with our rMV empty vector. The animals were euthanized, and sera were collected and pooled. Before determining the neutralization titers, the sera were concentrated using a Vivaspin column (Sartorius Biotech). The sera were then diluted in PBS to reach the desired concentration before intravenous injection in the mice.
ELISA assays
Anti-MV antibody IgG titers were determined according to a standard ELISA protocol.3 SIVgag IgG analysis was performed using the Bio Rad Genetic System HIV-2 ELISA detection kit using HRP anti-mouse detection antibodies (BioConcept) and OPD as substrate (Fluka). Cut-offs were set as three times the mean of negative sera. Titers are represented as log10 of the reciprocal end-dilution.
Measles neutralization antibody assay
Plaque neutralization assays were performed to determine the amount of measles-specific neutralizing antibodies. Sera were pre-diluted 1:32 in culture medium (MEM with 5% FBS, L-glutamine and gentamycin) before heat-inactivation at 56°C for 30 min. As controls, a WHO serum standard, a measles-positive serum and a measles-negative serum were taken with each experiment. Sera were serially diluted in culture medium and incubated with measles virus for one hour before transferring them to a Vero cell monolayer. The cells were then kept at room temperature for 1h in a humid chamber and a pre-warmed overlay medium (MEM with 1% methylcellulose, 5% FBS, L-glutamine, gentamycin) was added for an incubation period of 1 week. Plates were washed twice with PBS before staining with crystal violet. Neutralization antibody titers were calculated by the method of Reed and Muench.25
ELISpot assays
Spleens were collected two weeks post immunization and splenocytes were extracted. Shortly, spleens were meshed through a 70µm cell strainer (BD Falcon) in PBS 2% FCS. The erythrocytes were lyzed using 1.5ml BD Pharm Lyse (BD Biosciences PharMingen) for SIVgag. IFN-γ ELISPOT assay was performed according to manufacturer’s instructions (BD PharMingen). Duplicates of 0.2 × 106 or 0.5 × 106 splenocytes per 96 well plate were subsequently re-stimulated for 17–18 h at 37°C in 5% CO2 with either an equal volume of medium (negative control), medium containing peptide pools, or medium containing 20ng/ml PMA and 1µM Ionomycine (both Sigma) as positive control. Peptides for measles re-stimulation covered the whole N-protein region and were synthesized by Anawa. Peptides for HIVenv or SIVgag re-stimulation were obtained from the NIH AIDS Research and Reference Reagent Program and covered the whole HIVenv or SIVgag region, respectively.
ELISPOT development was performed according to AEC Substrate Reagent Set (BD Biosciences PharMingen). ELISpot plates were evaluated on an AID Viruspot Reader (Pharma Consulting). Values three times above background were considered positive. Test was considered valid with medium negative controls lower than < 50 spots per million splenocytes.
Acknowledgments
We thank A. Brogli and Y. Zeller for excellent animal care, and K. Scherler and T. Steiner for skilled technical assistance. This study was performed at Berna Biotech-Crucell with the financial support of NIH grant AI46007 and the NIH contract No. N01-AI-60018 to HYN. We would like to thank Dr Fabian Wild for his generous gift of anti MV antibodies and the NIH AIDS research and reference reagent program for supplying SIV reagents.
Glossary
Abbreviations:
- MV
measles virus
- rMVb
recombinant measles virus
- SIV
simian immunodeficiency virus
- hCD46tg
immune competent transgenic mice bearing the human CD46 measles receptor
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/23241
References
- 1.Hasleton PS. The internal surface area of the adult human lung. J Anat. 1972;112:391–400. [PMC free article] [PubMed] [Google Scholar]
- 2.Low N, Kraemer S, Schneider M, Restrepo AM. Immunogenicity and safety of aerosolized measles vaccine: systematic review and meta-analysis. Vaccine. 2008;26:383–98. doi: 10.1016/j.vaccine.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Zuniga A, Wang Z, Liniger M, Hangartner L, Caballero M, Pavlovic J, et al. Attenuated measles virus as a vaccine vector. Vaccine. 2007;25:2974–83. doi: 10.1016/j.vaccine.2007.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liniger M, Zuniga A, Morin TN, Combardiere B, Marty R, Wiegand M, et al. Recombinant measles viruses expressing single or multiple antigens of human immunodeficiency virus (HIV-1) induce cellular and humoral immune responses. Vaccine. 2009;27:3299–305. doi: 10.1016/j.vaccine.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol. 2007;81:10597–605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandler S, Lucas-Hourani M, Moris A, Frenkiel MP, Combredet C, Février M, et al. Pediatric measles vaccine expressing a dengue antigen induces durable serotype-specific neutralizing antibodies to dengue virus. PLoS Negl Trop Dis. 2007;1:e96. doi: 10.1371/journal.pntd.0000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorin C, Mollet L, Delebecque F, Combredet C, Hurtrel B, Charneau P, et al. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol. 2004;78:146–57. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liniger M, Zuniga A, Tamin A, Azzouz-Morin TN, Knuchel M, Marty RR, et al. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine. 2008;26:2164–74. doi: 10.1016/j.vaccine.2008.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Locke E, Bruder J, Clarke D, Doolan DL, Havenga MJ, et al. Viral vectors for malaria vaccine development. Vaccine. 2007;25:2567–74. doi: 10.1016/j.vaccine.2006.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dilraj A, Cutts FT, de Castro JF, Wheeler JG, Brown D, Roth C, et al. Response to different measles vaccine strains given by aerosol and subcutaneous routes to schoolchildren: a randomised trial. Lancet. 2000;355:798–803. doi: 10.1016/S0140-6736(99)95140-1. [DOI] [PubMed] [Google Scholar]
- 11.Dilraj A, Cutts FT, Bennett JV, Fernandez de Castro J, Cohen B, Coovadia HM. Persistence of measles antibody two years after revaccination by aerosol or subcutaneous routes. Pediatr Infect Dis J. 2000;19:1211–3. doi: 10.1097/00006454-200012000-00021. [DOI] [PubMed] [Google Scholar]
- 12.Dilraj A, Sukhoo R, Cutts FT, Bennett JV. Aerosol and subcutaneous measles vaccine: measles antibody responses 6 years after re-vaccination. Vaccine. 2007;25:4170–4. doi: 10.1016/j.vaccine.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Janeway CA, Travers P, Walport M, Schlomchik MJ. Antigen presentation to T Lymphocytes. In: The immune system in health and disease. Garland Science 2008;169-201. [Google Scholar]
- 14.Patel A, Zhang Y, Croyle M, Tran K, Gray M, Strong J, et al. Mucosal delivery of adenovirus-based vaccine protects against Ebola virus infection in mice. J Infect Dis. 2007;196(Suppl 2):S413–20. doi: 10.1086/520603. [DOI] [PubMed] [Google Scholar]
- 15.Kiyono H, Fukuyama S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat Rev Immunol. 2004;4:699–710. doi: 10.1038/nri1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–58. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 17.STEP study: disappointing, but not a failure. Lancet. 2007;370:1665. doi: 10.1016/S0140-6736(07)61702-4. [DOI] [PubMed] [Google Scholar]
- 18.Fauci AS, Johnston MI, Dieffenbach CW, Burton DR, Hammer SM, Hoxie JA, et al. HIV vaccine research: the way forward. Science. 2008;321:530–2. doi: 10.1126/science.1161000. [DOI] [PubMed] [Google Scholar]
- 19.Sekaly RP. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? J Exp Med. 2008;205:7–12. doi: 10.1084/jem.20072681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, et al. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One. 2008;3:e3548. doi: 10.1371/journal.pone.0003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cutts FT, Clements CJ, Bennett JV. Alternative routes of measles immunization: a review. Biologicals. 1997;25:323–38. doi: 10.1006/biol.1997.0103. [DOI] [PubMed] [Google Scholar]
- 22.Wong-Chew RM, Islas-Romero R, García-García MdeL, Beeler JA, Audet S, Santos-Preciado JI, et al. Induction of cellular and humoral immunity after aerosol or subcutaneous administration of Edmonston-Zagreb measles vaccine as a primary dose to 12-month-old children. J Infect Dis. 2004;189:254–7. doi: 10.1086/380565. [DOI] [PubMed] [Google Scholar]
- 23.Graham BS, Koup RA, Roederer M, Bailer RT, Enama ME, Moodie Z, et al. Vaccine Research Center 004 Study Team Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 DNA candidate vaccine. J Infect Dis. 2006;194:1650–60. doi: 10.1086/509259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marty RR, Knuchel MC, Azzouz-Morin TN, Zuniga A, Ilter O, Wiegand M, et al. An immune competent mouse model for the characterization of recombinant measles vaccines. doi: 10.4161/hv.34358. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–7. [Google Scholar]
- 26.Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613–9. doi: 10.1038/nature07352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cutts F. The immunological basis for immunization series, module 7: Measles. World Health Organization 1993; 1-19. [Google Scholar]
- 28.Guerbois M, Moris A, Combredet C, Najburg V, Ruffié C, Février M, et al. Live attenuated measles vaccine expressing HIV-1 Gag virus like particles covered with gp160DeltaV1V2 is strongly immunogenic. Virology. 2009;388:191–203. doi: 10.1016/j.virol.2009.02.047. [DOI] [PubMed] [Google Scholar]
- 29.Damjanovic D, Zhang X, Mu J, Medina MF, Xing Z. Organ distribution of transgene expression following intranasal mucosal delivery of recombinant replication-defective adenovirus gene transfer vector. Genet Vaccines Ther. 2008;6:5. doi: 10.1186/1479-0556-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolton DL, Kozlowski P, Rao S, Roederer M, Song K. Fine aerosol adenovirus vaccination against SIV in rhesus macaque. AIDS Res Hum Retroviruses. 2008;24:14. [Google Scholar]
- 31.Mrkic B, Pavlovic J, Rülicke T, Volpe P, Buchholz CJ, Hourcade D, et al. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72:7420–7. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]