

Abstract
Cardiovascular diseases claim more lives worldwide than any other. Etiologically, the dominant trajectory involves atherosclerosis, a chronic inflammatory process of lipid-rich lesion growth in the vascular wall that can cause life-threatening myocardial infarction (MI). Those who survive MI can develop congestive heart failure, a chronic condition of inadequate pump activity that is frequently fatal. Leukocytes – white blood cells – are important participants at the various stages of cardiovascular disease progression and complication. This review will discuss leukocyte function in atherosclerosis, myocardial infarction, and heart failure.
INTRODUCTION
The immune system protects us against pathogens such as viruses, bacteria, and parasitic worms, but its influence is much broader. The system recognizes and responds to divergent environmental and endogenous stimuli, and every known disease is at least partially associated with or dependent on immune function. Cardiovascular disease is no exception.
Over the last half century, advances in public health (emphasis on healthy diet, exercise, smoking cessation), clinical cardiology (chest pain units, coronary stenting, cardiac defibrillation), and scientific discovery (lipid-lowering 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, angiotensin-converting-enzyme (ACE) inhibitors) have contributed to a steady decline in deaths from cardiovascular disease (1). Despite these improvements, the disease is still responsible for 30% of deaths worldwide, surpassing all others including cancer, and costing the global economy (in 2010) an estimated US $863 billion. Even after surviving myocardial infarction or stroke, the likelihood of developing secondary complications, such as reinfarction or heart failure, is high, which increases costs through hospitalizations and follow-up clincal care. The world population is rising, and it is estimated that by 2030 cardiovascular disease costs could increase to US $1,044 billion (2). Aside from addressing unmet medical needs at the public health policy and clinical care levels, a deep and nuanced understanding of the underlying biological processes should enable development of safe and effective treatments.
Atherosclerosis is the pathology that leads to myocardial infarction and stroke. For many years after its recognition, atherosclerosis was thought to involve passive lipid deposition in the vessel wall. Today we understand that atherosclerosis is a chronic inflammatory disease driven by lipids, specifically low density lipoproteins (LDL) and leukocytes. Neither atherosclerosis nor its complications adhere to a simple arithmetic of dietary lipid imbalance, but rather comprise a syndrome in which environmental and genetic inputs disrupt biological systems. In other words, lifestyle, age, hereditary factors, and co-morbidities disturb immune, digestive, endocrine, circulatory, and nervous systems, thereby altering immune function, metabolism, and many other processes, while eliciting inflammation, hypercholesterolemia, and hypertension. Atherosclerosis develops and causes myocardial infarction or stroke when many things go wrong in many different ways.
Leukocytes are keepers of the immune system. The various classes of myeloid and lymphoid cells that encompass the leukocyte repertoire recognize and eliminate pathogens and molecular patterns perceived to be dangerous. Working together, leukocytes can engender protective immunity and keep the host from harm. Yet they can also contribute to disease. Virtually every leukocyte class has been implicated in atherosclerosis and its complications, and their action is neither uniform nor hierarchical. Some leukocytes are atherogenic whereas others are atheroprotective; some sustain inflammation after myocardial infarction while others resolve it. This functional heterogeneity is a challenge but also an opportunity for therapeutic intervention because it suggests that specific disease-promoting functions can be targeted and those required for normal homeostasis can be spared.
LEUKOCYTES IN ATHEROSCLEROSIS
The natural progression of atherosclerosis in the human involves the acquisition of specific features in the growing lesion. A key initiating process of atherosclerosis is the intimal retention of apolipoprotein (apo) B-containing lipoproteins in regions of disturbed blood flow and low shear stress (3). Lipid-rich macrophages – otherwise known as foam cells – appear early in the intima and can be identified in nearly 40% of newborns; they typically regress before the age of 2. The existence of small pools of extracellular lipids in the intima is a feature of a preatheroma, whereas an easily discernible core of extracellular lipid marks an atheroma. Increasingly complicated lesions are defined by fibrous thickening; the appearance of fissures, hematoma, and thrombi; and calcification. By the age of 40, 95% of people have some type of lesion. Problems occur if a lesion interferes with tissue oxygenation when either the lesion’s size reduces blood flow or the lesion ruptures and occludes the vessel altogether. Of the two, lesion rupture is far more dangerous. Myocardial infarction and stroke are sudden events that result from occlusion of vessels that oxygenate the heart and brain, respectively (4).
Macrophage as protagonist
Macrophages are most numerous among leukocytes in any type of lesion and, with the possible exception of smooth muscle cells, the most prominent cellular contributors to the lesion’s physical bulk (5). In response to intimal lipid accumulation, disturbed blood flow, low shear stress, and other stimuli, endothelial cells permit monocytes – major precursors of macrophages – passage across the endothelium (6, 7). Newly-infiltrated monocyte-derived macrophages recognize and ingest lipids that have accrued in the intima as a consequence of hypercholesterolemia. Macrophages are specialized phagocytes that rely on different strategies to sense, internalize, and process the diverse lipid moieties they encounter. Pattern recognition receptors (PRR) expressed on the plasma membrane recognize various native and oxidized lipoproteins and facilitate their uptake for lysosomal degradation (8). Likewise, cytoplasmic sensors such as the NLRP3 inflammasomes respond to cholesterol crystals and release IL-1β, a major inflammatory cytokine (9, 10). The relative importance of specific sensors to the generation of foam cells and development of atherosclerosis continues to be debated: PRRs such as SR-A and CD36 are dispensable to foam cell formation (11), whereas the NLRP3 inflammasome influences atherosclerosis in one mouse model (9) but not another (12).
Macrophage lipid uptake may be viewed as a protective response that backfires. Ingesting oxidized lipoproteins, like ingesting microbes, involves the scavenging of substances perceived to be dangerous. Lipoprotein recognition and consequent ingestion morphs macrophages into foam cells, many of which eventually die and contribute to a large lipid core, a characteristic of lesions most vulnerable to rupture (5). Surprisingly, foam cell formation suppresses, rather than promotes, inflammatory gene expression (13). Leukocytes may fuel an inflammatory cycle, but they also, in some incarnations, quench it.
Hypothetically, macrophages can prevent atherosclerosis by scavenging and removing surplus lipoproteins from the artery wall, without becoming foam cells and without escalating inflammation. Can such a scenario exist? Several lines of investigation on the production, migration, and function of macrophages provide clues.
Leukocytosis predicts cardiovascular events
Human and mouse blood contains at least two distinct monocyte subsets with differential migratory properties (14). Mouse Ly-6Chigh monocytes share several properties with human CD16− CD14+ monocytes, and are inflammatory. In response to hypercholesterolemia, the bone marrow and spleen overproduce Ly-6Chigh monocytes that enter the circulation, contribute to excessive monocytosis, preferentially accumulate in lesions, and differentiate to macrophages (15–17). Ly-6Clow monocytes, which are thought to be primarily reparative, are similar to CD16+CD14dim human monocytes, patrol the vasculature, and infiltrate atheromata less frequently. If an overzealous immune system generates too many cells of the wrong type which contribute to pathology, then targeting the culprit subset may be atheroprotective.
Genetically-engineered disruption of cholesterol metabolism defines the two major models of atherosclerosis: apoE−/− and LDL receptor (R)−/− mice develop atherosclerotic lesions with striking resemblance to those that evolve in people. Between the two, apoE−/− mice develop more severe blood leukocytosis and monocytosis. Recent studies, utilizing apoE−/− and other mice deficient in lipid-efflux proteins such as the ATP-binding cassette transporter 1 (ABCA1) and ATP-binding cassette sub-family G member 1 (ABCG1), have forged a mechanistic link between leukocyte and lipid biology. Trapped cholesterol in hematopoietic stem and progenitor cells (HSPCs) that lack the crucial cholesterol efflux machinery leads to the expression of the GM-CSF and IL-3 common beta chain receptor on the plasma membrane, and contributes to excessive proliferation (18, 19). In other words, cholesterol efflux suppresses proliferation. Concurrently, lipid-rich splenic phagocytes release IL-23 which induces a cascade that eventually liberates HSPCs from their medullary niches (20). When HSPCs seed extramedullary sites, they encounter GM-CSF and IL-3 (17). The net effect is HSPC proliferation, extramedullary hematopoiesis, leukocytosis, and accelerated atherosclerosis.
Most humans are not apoE, LDLR, ABCA1, or ABCG1 deficient; they are not impaired in handling cholesterol. But human cardiovascular disease does associate with leukocytosis (21). In comparison with the mouse model, where a defect in cholesterol clearance leads to severe systemic inflammation, the human leukocytosis is almost always mild. The mouse phenotype may represent an extreme version of a process that merely simmers in people, perhaps a version of insufficient cholesterol handling rather than its complete breakdown. If this is so, then precise targeting of leukocyte-centric cholesterol pathways will be therapeutically desirable.
Macrophage flux and function
Macrophage flux involves monocyte migration across the endothelium, monocyte differentiation to macrophages, macrophage retention in the atheromata, exit, or death. The preferential accumulation of Ly-6Chigh monocytes in the growing atheromata relies on the CCR2-CCL2, CX3CR1-CX3CL1, and CCR5-CCL5 (16), and neutralizing these axes in mice almost abolishes atherosclerosis (22, 23). Upon accumulation and lipid ingestion, macrophages release netrin-1, a guidance molecule that binds to UNC5b on the plasma membrane and blocks the directed migration of macrophages out of the lesion (24). In the absence of netrin-1, lesions are smaller. The relative contribution of macrophage exit and/or death may indeed be critical to how disease progresses. On the one hand, efficient macrophage exit (25) – or reduced monocyte influx (26) – are central to regression; when lesions regress, macrophage numbers diminish. On the other hand, limited macrophage death, effective efferocytosis, and active autophagy are fundamental to lesional stability (27); lesions with large necrotic cores are most vulnerable.
A macrophage is not simply “a big eater”, however, and it does not exist in isolation. Lesional macrophages are polyfunctional and interact with other leukocytes and non-leukocyte client cells (Fig. 1). Macrophage polyfunctionality in the aorta represents either dedicated behavior of individually specialized cells (distinct macrophage subsets) or adaptive behavior elicited by the tissue environment (macrophage plasticity). Regardless of how division of labor is maintained, macrophages – as a cell population that accumulates lipids – secrete cytokines that attract other leukocytes, produce proteinases that digest the extracellular matrix, disturb smooth muscle cell function, and can influence endothelial-dependent vasodilatation (5). Macrophages can also resolve inflammation and promote granulation tissue formation (28). Aside from, or perhaps in addition to, controlling LDL levels, a therapeutic goal for atherosclerosis may be to coax macrophage responses away from inflammation and proteolysis and toward robust cholesterol transport, intelligent cellular flux, and sustained efferocytosis.
Figure 1. Leukocytes in experimental atherosclerosis.

The cartoon depicts leukocyte flux and function in experimental atherosclerosis. Monocytes are made in bone marrow (medullary hematopoiesis) and spleen (extramedullary hematopoiesis). Ly-6Chigh monocytes accumulate preferentially in the lesion and differentiate to macrophages, which ingest lipids or cholesterol crystals, and become lipid-rich “foam cells”. Ly-6Clow monocytes patrol the vasculature and enter lesions less frequently. Concurrently, other leukocytes and platelets participate in inflammation and lesion growth in various consequential ways (see main text). The scanning electron and transmission electron microscope images show, from left to right, monocytes (M) adhering to the endothelium (E), monocytes extending into lumen (Lu) trapped in junctions of the endothelium, endothelium overlying foam cells (FC) in intima, and ruptured endothelium revealing numerous foam cells. Images are from a classic study using Yorkshire pigs (6, 7).
Adaptive immunity in atherosclerosis
Is adaptive immunity – the immune system’s arm that relies on somatic hypermutation, recombination, and clonal cell proliferation to generate potent defense and lasting memory against specific pathogens – important in atherosclerosis? Dendritic cells, which capture antigen and present it to naive T cells, reside in the aorta and affect progression of atherosclerosis (29–31), possibly by interacting with T cells (32) previously activated in lymph nodes and tertiary lymphoid organs (33). T cells are relatively rare in lesion. Even so, specific perturbations, mostly of T cell-associated cytokines, suggest that T helper (TH)-1 and TH17 cells are atherogenic, whereas TH2 and TREG cells are protective (34, 35). TH1-generated interferon (IFN)-γ, for example, activates macrophages and propagates inflammation, whereas TREG-generated interleukin (IL)-10 and transforming growth factor (TGF)-β dampen inflammation. B cells, which are rare in lesions but more prevalent in the adventitia, can be both atheroprotective and atherogenic (36, 37). The innate-like B1 B cells are atheroprotective, possibly because they produce natural IgM antibodies that mark lipids for Fc receptor-mediated removal. The adaptive, B2 B cells, on the other hand, contribute to disease, presumably by interacting with other leukocytes and/or secreting inflammatory cytokines (36). Because cellular and molecular constituents implicated in adaptive immunity regulate atherosclerosis, they can be harnessed, at least experimentally, to alter disease progression.
The generation of antigen-specificity through somatic hypermutation defines adaptive immunity. In atherosclerosis, antibodies and CD4+ T cells that react to lipid moities such as oxLDL have been identified (38, 39). However, lymphocytes can secrete cytokines and proliferate in response to germline-encoded receptor signaling. Innate lymphoid cells, NKT cells, tissue-resident γδT cells, and innate-like B cells such as innate response activator (IRA) B cells, share phenotypic features with their adaptive counterparts but differ in how they perceive and respond to molecular patterns. The relative importance of germline-encoded and somatically-hypermutated receptors in recognizing relevant atherosclerotic antigens and influencing disease progression remains unknown.
Inflammatory professionals
Neutrophils, mast cells, and platelets are detectable in lesions and may influence atherosclerosis and its complications in important ways. Neutrophils, the inflammatory granulocytes, circulate in large numbers and accumulate acutely at sites of injury or infection. In experimental atherosclerosis, neutrophils, whose numbers rise in the blood, help monocyte adhesion or transmigration by releasing alarmins and other preformed granular proteins (40). Neutrophils also contain large quantities of myeloperoxidase, NADPH oxidase, and lipoxygenases, which contribute to oxidative stress, a major determinant of endothelial cell dysfunction, lesion growth, and instability. Mast cells, best known for their role in allergy and anaphylaxis, promote atherosclerosis by releasing the contents of their protease-cytokine-autacoid-rich granules (41). Platelets – the megakaryocyte-derived thrombocytes – play an essential, dual role. During atherosclerosis, they adhere to the endothelium and help monocytes enter lesions (42). During plaque rupture, they form the thrombus that causes ischemia of downstream tissue (5). Thus, neutrophils, mast cells and platelets all promote atherosclerosis by intensifying inflammation.
LEUKOCYTES IN MYOCARDIAL INFARCTION AND HEART FAILURE
Leukocytes as friends and foes of the heart
Small atherosclerotic lesions that do not hinder blood flow are found in many people. Subclinical atherosclerosis manifests itself when lesions reduce tissue oxygen supply. Adluminal lesion growth and inward arterial remodeling gradually narrow the vessel diameter, thereby reducing blood flow. If coronary arterial stenosis reaches more than 80%, downstream heart muscle becomes ischemic, especially when a high cardiac work load increases oxygen demand. Typically, myocardial ischemia leads to chest pain, which occurs during exertion (stable angina pectoris). Plaque rupture, which is precipitated by leukocyte action, suddenly occludes the artery, and causes unstable angina and myocardial infarction (MI). In contrast to atherosclerosis, where chronic low-grade stimulation by native and oxidized lipoproteins mobilizes leukocytes to the vessel wall, in MI, acutely dying myocytes elicit different triggers that nevertheless recruit similar classes of leukocytes. In response to the MI, the leukocyte profile in the myocardium changes drastically. The population of macrophages (43) and dendritic cells (30) that reside in the healthy heart is quickly overwhelmed by inflammatory leukocytes.
Shortly after onset of ischemia, endothelial cells upregulate adhesion molecules, that, along with released chemokines, trigger neutrophil extravasation. Rapid accumulation leads to an early neutrophil peak after injury (28). In skin wounds, neutrophils protect against infection through phagocytosis of bacteria and release of reactive oxygen species. In the ischemic heart, neutrophils phagocytose dead tissue and release inflammatory mediators – an immunological misfire aimed at myocytes that survived the ischemic injury. It is believed that neutrophils are deleterious in myocardial infarction, but they eventually undergo apoptosis and are removed by macrophages.
Aside from neutrophils, inflammatory Ly-6Chigh monocytes are among the earliest responders. Distressed tissue expresses CCL2 (44) that attracts Ly-6Chigh monocytes during the first several days, after which the wounded heart switches to CX3CL1-mediated recruitment of Ly-6Clow monocytes (28). The cause of the switch is unknown, but sequentially accumulating monocyte subsets do pursue differential functions in the infarct (Fig. 2). Although both are phagocytic, the early inflammatory subset is particularly rich in pro-inflammatory mediators. These include proteases, which are tightly regulated to balance wound debridement with tissue destabilization, because unchecked proteolysis may cause infarct expansion or even rupture. During resolution of inflammation, reparative Ly-6Clow monocytes support angiogenesis and extracellular matrix synthesis by providing vascular endothelial growth factor (VEGF) and TGFβ (28). Whether Ly-6Chigh and Ly-6Clow monocytes give rise to distinct macrophage subsets in the infarcted myocardium is unknown, but more intriguing is the possibility that monocytes, as monocytes, impose a lasting effect on the tissue environment before differentiating (or dying, or exiting). Decades of research have argued that macrophages are highly susceptible to microenvironmental cues. Perhaps recruited monocytes influence a tissue in a manner that long-seeded, sessile macrophages, no longer can.
Figure 2. Biphasic monocyte response during early myocardial remodeling.
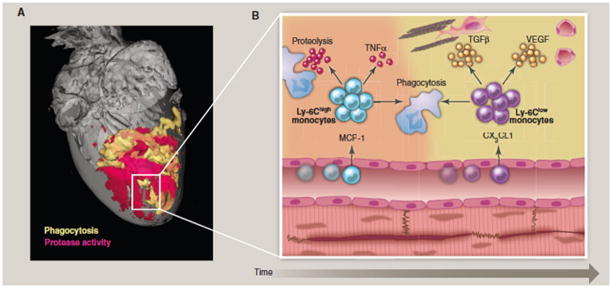
(A) Optical projection tomography of a murine heart with myocardial infarction after injection of molecular imaging reporters for major function of myeloid cells, including protease activity (red) and phagocytic activity (yellow). Unpublished, image courtesy of Dr. Claudio Vinegoni. (B) Magnetic resonance images of murine hearts. The upper panel shows a normal short axis view, while the lower panel depicts a severely dilated and remodeled heart after coronary ligation. Unpublished own data. (C) Cartoon of biphasic monocyte subset activity in the infarct as a function of time. In a first phase, inflammatory Ly-6Chigh monocytes are recruited via MCP-1 and remove necrotic debris. In a second phase, Ly-6Clow monocytes accumulate via CX3CL1 and pursue repair functions that result in a stable scar.
With a short time delay and at lower numbers when compared to the infarct, monocytes also invade the non-ischemic remote myocardium (45). Here, myeloid cells may contribute to various processes: they may cause heart failure by facilitating ventricular dilatation via proteolysis of the collagen matrix that lends mechanical stability to the heart; they may activate fibroblasts and promote interstitial myocardial fibrosis; they may harm myocytes through secretion of pro-apoptotic factors. But myeloid cells could also protect the non-ischemic remote myocardium, for instance though secretion of angiogenesis-promoting cytokines. The function of myeloid leukocytes in this region is yet to be determined.
Lymphocytes are present in low numbers in the infarct and proliferate in draining lymph nodes shortly after ischemic injury (46). CD4+ T cell deficiency delays the transition from Ly-6Chigh to Ly-6Clow monocyte presence and impairs healing of the heart. Likewise, depletion of dendritic cells disturbs resolution of inflammation (47). These observations suggest a link to adaptive immunity, but it is unclear, given that myocardial infarction is a form of sterile injury, whether any processed peptide antigens are recognized and presented. Myocardial infarction may be mobilizing elements of adaptive immunity without necessarily relying on its essential mechanisms.
The emerging picture positions leukocytes as both protective and harmful in myocardial infarction. Ly-6Chigh monocytes, for example, are required during the initial response to ischemia, but can also be destructive if they persist in the infarct too long. Ly-6Clow monocytes, on the other hand, are probably essential to wound repair (28, 48). Optimal healing requires balance: sequential and coordinated cell recruitment that removes dead tissue and strengthens the wound so that the heart performs its vital function is a manifestation of that balance (28); co-morbidities can disrupt it (49). Therapeutic strategies should therefore aim to recalibrate the leukocyte response toward optimal healing. In vivo RNA interference is one strategy that can selectively silence CCR2 at defined time-points, thus limiting the accumulation of Ly-6Chigh monocytes in atherosclerotic plaques and the infarcted myocardium (50). As our understanding of how various leukocytes regulate atherosclerosis and its complications enlarges, so will the potential repertoire of targeted therapeutics.
Leukocytes anchor system-wide mayhem after MI
Acute MI is a severe traumatic event that mobilizes multiple organ systems (Fig. 3). A substantial number of the initially-recruited monocytes derives from a reservoir in the splenic red pulp, where increased angiotensin-2 signaling triggers their mobilization within hours after injury (51). For the first few days after infarction, leukocyte recruitment remains at astonishing levels (48). To meet the demand, cell production increases in the bone marrow and the spleen. How do the myelopoietic sites learn about the need for cells in the heart? Pain and anxiety during acute MI alert the sympathetic nervous system and activate neuro-immune synapses (52). In the bone marrow, sympathetic nerves release noradrenaline, which binds to β3 adrenergic receptors expressed by mesenchymal stem cells (MSC). These cells are part of a housekeeping team that regulates leukocyte progenitor cell activity in the bone marrow niche. In response to noradrenaline, MSC withdraw the HSPC retention factor CXCL12, and liberate HSPC into circulation. These cells then seed the spleen and amplify extramedullary myelopoiesis, enabling the organ to contribute monocytes beyond its baseline reservoir function. Local environmental changes in the spleen, including increased levels of IL-1β and stem cell factor (SCF/kit ligand), orchestrate the emergency cell production after MI (48, 52). Other still unknown soluble factors may act as alarmins to trigger emergency hematopoiesis after MI.
Figure 3. Organ networks that lead to acceleration of atherosclerotic disease after myocardial infarction.
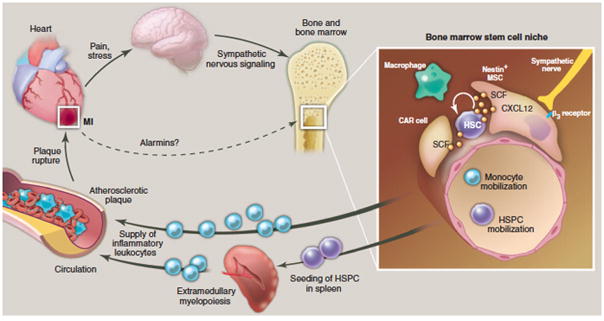
The cartoon depicts events observed in mice acutely after MI that lead to accelerated disease progression in atherosclerotic plaque. The enlarged inset depicts processes in the bone marrow microenvironment. Here, niche cells provide signals that regulate HSC activity, retention and leukocyte production. After MI, increased sympathetic nervous signaling releases noradrenalin in the bone marrow niche, which binds to beta-3 adrenoreceptors on niche cells. These withdraw the soluble factor CXCL12 which results in increased HSC activity and emigration to extramedullary sites. Increased production of leukocytes then feeds an expanded pool of circulating monocytes which are recruited to atherosclerotic plaque in higher numbers, accelerating plaque growth and inflammation. This feedback loop may cause the high re-infarction rates observed clinically. The dotted arrow refers to currently unknown cross-talk between the cardiac wound and the hematopoietic system via alarmins (danger associated molecular patterns, DAMPs), which may also activate the bone marrow after MI. HSC: hematopoietic stem cell. HSPC: hematopoietic stem and progenitor cells. CAR: CXCL12 abundant reticular cell, MΦ: macrophage.
The next turn in a vicious cycle?
Regardless of how many leukocytes are needed in the infarcted myocardium, the activated endothelium that lines (unruptured) atherosclerotic plaques can recruit leukocytes that have become available in increased numbers as a result of the MI. This may partly explain why atherosclerotic lesions grow faster and develop a more advanced phenotype shortly after ischemic injury (53). Because the inflammatory burst after MI generates proteolytic leukocytes, their ‘off-target’ accumulation in atherosclerotic lesions may disrupt the fibrous cap, increasing the likelihood of re-infarction. Leukocyte activity post-MI should therefore be considered as a therapeutic target for secondary prevention.
Derailed infarct healing contributes to heart failure
Myocardial infarction is frequently studied in young mice subjected to coronary ligation. Young and healthy humans, however, rarely suffer from MI. Atherosclerosis, risk factors, and co-morbidities such as diabetes and obesity – many of which have an inflammatory component (see Review by Odegaard and Chawla) – typically precede ischemic injury. The smoldering systemic inflammation impedes infarct healing by interfering with the resolution of local inflammation and delaying the reparative phase (49). Consequently, the infarct expands and the left ventricle dilates, ultimately leading to heart failure (Fig. 2). This frequent syndrome, defined by the inability to pump sufficient quantities of oxygenated blood into peripheral tissues, manifests with shortness of breath and fluid retention, and carries a mortality as high as 50%. Data on leukocyte activity in the chronically failing myocardium are unavailable; however, inflammatory biomarkers, such as C-reactive protein, and inflammatory mediators, such as the cytokines TNF-α and IL-6, increase systemically in heart failure, and leukocytosis associates with disease progression. Leukocytes are present in the myocardium of mice with chronic heart failure induced by pressure overload (54). Given leukocytes’ role in many other disease settings, their function in patients with chronic heart failure should be examined.
Human data suggest that insights obtained in rodents can be translated to the clinic. Leukocytosis and inflammatory monocyte levels correlate with post-MI heart failure in patients (55, 56). After acute MI, the human bone marrow increases activity (57) and releases HSPC into circulation (58), possibly resulting in splenic proliferation (52). Two monocyte subset peaks occur in the blood of patients with acute MI (56), suggesting that the infarcted human myocardium likewise mobilizes monocyte subsets in distinct phases. The similarities between human and rodent data are encouraging, but must be viewed with caution given the many dissimilarities between human biology and rodent models of human disease. That being said, there is immense value in what animal models can reveal. The integration of knowledge from divergent disciplines – human genetics that searches for susceptibility loci using genome-wide association studies; molecular biology that describes and forges links between intracellular metabolic and immune pathways; and cellular immunology that monitors leukocyte communication in murine organ systems – ought to be a focus of a concerted effort in cardiovascular disease research. Among the tools, animal models of atherosclerosis and myocardial infarction, though imperfect, remain uniquely indispensable at identifying a biological process, testing its in vivo function and importance, and rationalizing its targeting in human disease.
HALLMARKS OF LEUKOCYTE FUNCTION IN CARDIOVASCULAR DISEASE
The leukocyte system’s role in cardiovascular disease has several prevailing features. First, the system appears to be maladaptive. The danger signal during atherosclerosis or after myocardial infarction is unlikely to be a microbe, yet leukocytes mobilize powerful anti-microbial and inflammatory molecules that cannot adequately handle either lipids or dying myocytes. Second, the system is heterarchical. Macrophage abundance in atherosclerosis does not imply hierarchy and, indeed, other leukocytes such as dendritic cells may be acting in parallel. Third, the system is collaborative: leukocytes communicate through processes that elicit activation, differentiation, degranulation, and transmigration. Fourth, the system is competitive in that selective ablation of various same-class leukocyte subsets often reveals opposing effects on atherosclerosis. Fifth, the system is pervasive. Leukocytes accumulate in vascular lesions or the myocardium, but they are produced in other tissue and circulate in the blood. Sixth, the system is integrated. Leukocytes do not operate in isolation but belong to a network that connects organ systems (Fig. 3).
CONCLUDING REMARKS
Therapeutic approaches that target leukocytes to treat cardiovascular disease have not yet been realized, partly because the fundamental biology remains somewhat enigmatic (see Review by Tabas and Glass). Important lingering questions include: What key triggers lead to leukocyte participation and activation? What are the initializing danger signals, modes of information transfer, and essential disease amplifiers? What defines the nature of the system’s response (attack versus resolution)? What are the connecting points between organ systems, including lymphatic, endocrine and nervous? Can we identify therapeutic targets that are efficacious yet specific enough to avoid collateral damage? Is it possible to target specific auto-antigens such as LDL while preserving host defense? Answering these questions may be integral to steering leukocyte function away from self-destruction and towards physiological stability, in order to durably prevent and suppress the devastating consequences of cardiovascular disease.
Acknowledgments
We gratefully acknowledge Drs. R. Weissleder and P. Libby for fruitful discussion and critical reading of the manuscript. This work was supported in part by NHLBI grants R01HL095612, R01HL095629, R01HL096576.
References and Notes
- 1.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 2.Bloom DE, et al. The Global Economic Burden of Noncommunicable Diseases. Geneva: World Economic Forum; 2011. [Google Scholar]
- 3.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 4.Stary HC. Atlas of Atherosclerosis: Progression and Regression. The Parthenon Publishing Group Inc; New York: 1999. p. 131. [Google Scholar]
- 5.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 6.Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181. [PMC free article] [PubMed] [Google Scholar]
- 7.Gerrity RG. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191. [PMC free article] [PubMed] [Google Scholar]
- 8.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajamaki K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning-Tobin JJ, et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29:19. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menu P, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spann NJ, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138. doi: 10.1016/j.cell.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swirski FK, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacke F, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robbins CS, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy AJ, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westerterp M, et al. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilgendorf I, Swirski FK. Making a difference: monocyte heterogeneity in cardiovascular disease. Curr Atheroscler Rep. 2012;14:450. doi: 10.1007/s11883-012-0274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Combadiere C, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 23.Saederup N, Chan L, Lira SA, Charo IF. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2−/− mice: evidence for independent chemokine functions in atherogenesis. Circulation. 2008;117:1642. doi: 10.1161/CIRCULATIONAHA.107.743872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Gils JM, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feig JE, et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Potteaux S, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao X, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahrendorf M, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206:2141. doi: 10.1084/jem.20090866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi JH, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497. doi: 10.1084/jem.20082129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi JH, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. 2011;35:819. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 32.Koltsova EK, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012 doi: 10.1172/JCI61758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabner R, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J Exp Med. 2009;206:233. doi: 10.1084/jem.20080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134:33. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Lahoute C, Herbin O, Mallat Z, Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol. 2011;8:348. doi: 10.1038/nrcardio.2011.62. [DOI] [PubMed] [Google Scholar]
- 36.Kyaw T, Tipping P, Toh BH, Bobik A. Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis. Curr Opin Lipidol. 2011;22:373. doi: 10.1097/MOL.0b013e32834adaf3. [DOI] [PubMed] [Google Scholar]
- 37.Kyaw T, Tipping P, Bobik A, Toh BH. Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc Med. 2012;22:48. doi: 10.1016/j.tcm.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Stemme S, et al. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1995;92:3893. doi: 10.1073/pnas.92.9.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yla-Herttuala S, et al. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14:32. doi: 10.1161/01.atv.14.1.32. [DOI] [PubMed] [Google Scholar]
- 40.Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- 41.Sun J, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 42.Huo Y, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 43.Pinto AR, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One. 2012;7:e36814. doi: 10.1371/journal.pone.0036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dewald O, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 45.Lee WW, et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59:153. doi: 10.1016/j.jacc.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann U, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 47.Anzai A, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 48.Leuschner F, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panizzi P, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leuschner F, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dutta P, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright AP, et al. Atherosclerosis and leukocyte-endothelial adhesive interactions are increased following acute myocardial infarction in apolipoprotein E deficient mice. Atherosclerosis. 2010;212:414. doi: 10.1016/j.atherosclerosis.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Engstrom G, Melander O, Hedblad B. Leukocyte count and incidence of hospitalizations due to heart failure. Circ Heart Fail. 2009;2:217. doi: 10.1161/CIRCHEARTFAILURE.108.827071. [DOI] [PubMed] [Google Scholar]
- 56.Tsujioka H, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 57.Assmus B, et al. Acute myocardial infarction activates progenitor cells and increases Wnt signalling in the bone marrow. Eur Heart J. 2012;33:1911. doi: 10.1093/eurheartj/ehr388. [DOI] [PubMed] [Google Scholar]
- 58.Massa M, et al. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105:199. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]