

Abstract
The hydroxylation of aromatic substrates catalyzed by coupled binuclear copper enzymes has been observed with side-on-peroxo-dicopper(II) (P) and bis-μ-oxo-dicopper(III) (O) model complexes. The substrate-bound-O intermediate in [Cu(II)2(DBED)2(O)2]2+ (DBED=N,N′-di-tert-butyl-ethylenediamine) was shown to perform aromatic hydroxylation. For the [Cu(II)2(NO2-XYL)(O2)]2+ complex, only a P species was spectroscopically observed. However, it was not clear whether this O-O bond cleaves to proceed through an O-type structure along the reaction coordinate for hydroxylation of the aromatic xylyl linker. Accurate evaluation of these reaction coordinates requires reasonable quantitative descriptions of the electronic structures of the P and O species. We have performed Cu L-edge XAS on two well-characterized P and O species to experimentally quantify the Cu 3d character in their ground state wavefunctions. The lower per-hole Cu character (40±6%) corresponding to higher covalency in the O species compared to the P species (52±4%) reflects a stronger bonding interaction of the bis-μ-oxo core with the Cu(III) centers. DFT calculations show that 10-20% Hartree-Fock (HF) mixing for P and ~38% for O species are required to reproduce the Cu-O bonding; for the P species this HF mixing is also required for an antiferromagnetically coupled description of the two Cu(II) centers. B3LYP (with 20% HF) was, therefore, used to calculate the hydroxylation reaction coordinate of P in [Cu(II)2(NO2-XYL)(O2)]2+. These experimentally calibrated calculations indicate that the electrophilic attack on the aromatic ring does not involve formation of a Cu(III)2(O2−)2 species. Rather, there is direct electron donation from the aromatic ring into the peroxo σ* orbital of the Cu(II)2(O22−) species, leading to concerted C-O bond formation with O-O bond cleavage. Thus, species P is capable of direct hydroxylation of aromatic substrates without the intermediacy of an O-type species.
1 INTRODUCTION
The coupled binuclear copper (CBC) proteins bind molecular oxygen (O2) to form a side-on μ − η2 : η2 peroxo dicopper(II) species (P) (Scheme 1, top).1-6 The CBC proteins include hemocyanin (Hc), catechol oxidase (CO), tyrosinase (Ty) and NspF and are involved in a range of biological functions. Hemocyanin is the dioxygen transport protein in arthropods and mollusks. Tyrosinase and NspF7 catalyze the hydroxylation of phenols to o-diphenols (monooxygenation) along with the subsequent two-electron oxidation to the corresponding o-quinones (oxidoreduction) (Scheme 1, center). In addition, NspF can perform the oxygenation of o-aminophenols to nitrosophenols (hydroxyanilinase activity) (Scheme 1, bottom).7 In contrast, catechol oxidase performs only the oxidoreductase reaction. The side-on peroxo species (P) has been observed in all CBC enzymes. However, protein intermediates beyond P are unknown and the molecular mechanisms for the monooxygenation and oxidoreduction reactions remain unclear.
Scheme 1.

Synthetic models of the CBC active site have shown that Cu(I) can bind O2 in several different modes.4,5,8-12 In particular, side-on peroxo Cu(II)2 (P) and bis-μ-oxo Cu(III)2 (O) species have been observed in different ligand systems (Scheme 2).4,5,8,13 The mode of O2 binding depends on the nature of the ligand, its coordination geometry (including denticity and sterics)14 and the type of donor atom (i.e., aliphatic vs aromatic N-ligand, etc.),15 along with the solvent and counterion.4,8 In general, more sterically demanding, tridentate ligands favor the P species whereas strong σ donating, bidentate ligands that have limited steric interactions tend to stabilize the O isomer.4,5,16 The first X-ray crystal structure of a P species was solved by Kitajima and co-workers with a highly sterically hindered diisopropyl-substituted hydrotris(pyrazolyl)borate (TPB) ligand (Scheme 2, bottom left)17,18 that revealed a core almost identical to that of P found in CBC enzymes.19,20 Tolman and co-workers solved the crystal structure of the first O complex with a tridentate 1,4,7-tribenzyl-1,4,7-triazocyclononane (TACNBn3) ligand (Scheme 2, bottom center)21,22 and Stack and co-workers determined a bis-μ-oxo core in a bidentate N, N′-diethyl-N, N′-dimethyl-cyclohexyldiamine (LMECHD) ligand system (Scheme 2, bottom right).23
Scheme 2.

It has also been found that certain P complexes can be in rapid equilibrium with the O species. Stack and co-workers have demonstrated that the Cu(I) complex of a bidentate ligand, N,N′-di-tert-butyl-ethylenediamine (DBED), reacts with O2 to form a mixture of PDBED (~95%) and ODBED (~5%) intermediates with the latter being the reactive oxidant in o-hydroxylation.24,25 Reaction of PDBED with a phenolate substrate presumably requires axial phenolate binding to one Cu followed by a trigonal bipyramidal rearrangement of the phenolate into the equatorial Cu2O2 plane. This makes the phenolate a strong donor ligand that leads to O-O bond cleavage and formation of a bis-μ-oxo Cu(III)2-phenolate complex (Scheme 3). This is followed by C-O bond formation via an electrophilic aromatic substitution (EAS) mechanism. Thus, in the [Cu(II)2(DBED)2(O2)]2+ model system, the O species is responsible for hydroxylation of the phenolate substrate.25 In contrast to the above model system, Karlin and co-workers have demonstrated that the [Cu(I)2(NO2-XYL)]2+ complex reacts with O2 to exclusively form a PNO2−XYL intermediate that is thought to directly hydroxylate the aromatic ring of the linker (Scheme 4).26-28
Scheme 3.

Scheme 4.

Due to the potential relevance of both the P and O species to the hydroxylation reaction performed by tyrosinase and NspF, it is important to gain quantitative experimental insight into the electronic structures of these two systems. UV-Vis absorption and resonance Raman (rR) spectroscopic studies have demonstrated that the P and O species have distinct spectral features characteristic of their unique Cu2O2 cores (Scheme 2).17,23,29 X-ray absorption spectroscopy (XAS), which is a powerful element specific tool for electronic structure determination, was done at the Cu K-edge to reveal side-on peroxo species to be Cu(II) with a pre-edge transition at ~8979 eV, whereas bis-μ-oxo complexes have a pre-edge at ~8981 eV characteristic of Cu(III).30 Both species, however, are EPR silent with no direct experimental quantification of the copper-dioxygen bonding interactions.
Here, for the first time, we have quantified directly the Cu-O bonding interaction in the P and O species using Cu L-edge XAS, which provides a sensitive probe of the charge on the copper ions and thus probes the covalent donor interactions of the peroxo and oxo ligands with the Cu centers. Two well-characterized systems, PTPB (1) (Scheme 2, left) and OTEED (2) (TEED = N,N,N′,N′-tetraethylethylenediamine, similar to the ligand in Scheme 2, bottom right), are used for the P and O species, respectively. The experimentally determined electronic structures of 1 and 2 are used to calibrate density functional theory (DFT) calculations to find the percent Hartree-Fock (HF) mixing required in a functional to quantitatively reproduce the covalent Cu-O bonding interactions. It is important to emphasize that DFT calculations with different amounts of HF mixing have been found to give quantitatively different descriptions of the bonding,31,32,33 and the determination of a meaningful reaction coordinate requires an accurate description of the electronic structure.
Using the experimentally calibrated DFT functional, we have investigated the hydroxylation mechanism in the [Cu(II)2(NO2-XYL)(O2)]2+ system that is also a potential model for tyrosinase and NspF.26,34,35 This model complex is particularly interesting because it allows the study of the o-hydroxylation in a system where only the P species is present and the substrate does not bind to Cu, in contrast to the phenolate substrate-bonded O species in [Cu2(DBED)2(O2)]2+ . The goal is to determine whether the O-O cleaves along the reaction coordinate forming the O species before hydroxylation or whether the P intermediate is a viable candidate for hydroxylation in tyrosinase and NspF.
2 EXPERIMENTAL SECTION
2.1 Sample Preparation
The [{HB(3,5−iPr2pz)3Cu}2(O2)], 1 (PTPB), and [(LTEEDCu)2(O2)]2+, 2 (OTEED), complexes were prepared as described before.18,36 Both complexes are temperature sensitive and were handled under inert N2 atmosphere and at dry ice temperatures during sample preparation.
2.2 L-edge X-ray Absorption Measurements and Data Analysis
For Cu L-edge XAS data collection, the solid samples were spread thinly over double-sided adhesive conducting graphite tape mounted on an Al sample paddle. The paddles were transferred onto a magnetic manipulator in an antechamber pre-chilled with liquid N2 and then transferred into the main chamber and affixed to an Al block, which was cooled through conduction by a continuous flow of liquid He into a Cu block in an internal cavity. The temperature was monitored using a Lakeshore temperature controller and maintained at ~50 K during the course of data collection.
Cu L-edge X-ray absorption spectra were recorded at SSRL on the 31-pole wiggler beam line 10-1 under ring operating conditions of 50-100 mA and 3 GeV with a spherical grating monochromator with 1000 lines/mm and set at 30 μm entrance and exit slits. Sample measurements were performed using the total electron yield mode, where the sample signal (I1) was collected with a Galileo 4716 channeltron electron multiplier aligned to 45° relative to the copper paddle. The signal was intensity-normalized (I1/I0) using the photocurrent of a gold grid reference monitor (I0). Data for all samples were recorded in a sample chamber maintained below 10−6 Torr, isolated from the ultra-high vacuum beam line by a 1000 Å Al window.
External energy calibration was accomplished by L-edge measurements on CuF2 before and after those of the sample. The L3 and L2 peak maxima were assigned to 930.5 and 950.5 eV, respectively. The variance in this calibration energy measured prior to and after each sample scan was used to shift linearly the experimental spectra between calibration scans. Spectra presented here are 3-5 scan averages, which were processed by fitting a second-order polynomial to the pre-edge region and subtracting it from the entire spectrum as background, resulting in a flat post-edge. The data were normalized to an edge jump of 1.0 at 1000 eV. The rising edges of the data were subtracted as arctangents. The total area under the L3 and L2 peaks was calculated by integrating the intensity between 925 and 955 eV. The integrated area was compared to that for D4h [CuCl4]2− to obtain per-hole covalency numbers. Complex 2 decomposed to a Cu(II) species during data collection. The data for the pure decomposition Cu(II) product of 2 was also collected and subtracted from that of 2 to obtain the clean Cu(III) data. This led to a larger error of ±6% in 2 compared to ±4% in 1 for subsequent d-hole character determination.
2.3 K-edge X-ray Absorption Measurements and Data Analysis
The solid samples were ground in boron nitride at liquid N2 temperature to form a homogeneous mixture that was pressed into a pellet and sealed between Kapton tape windows in a 1 mm aluminum spacer. The samples were maintained below 10 K during data collection using an Oxford Instruments CF 1208 continuous-flow liquid helium cryostat.
Cu K-edge X-ray absorption spectra were measured at SSRL on the unfocused 20-pole, 2.0-T wiggler beam line 7-3 under storage ring parameters of 50-100 mA and 3 GeV. A Rh-coated pre-monochromator, flat, bent mirror was used for harmonic rejection and vertical collimation. A Si(220) double crystal monochromator was used for energy selection. Transmission data were collected with N2-gas-filled ionization detector (I1). Internal energy calibration was accomplished by simultaneous measurement of the absorption of a Cu foil placed between two ionization chambers (I1 and I2) situated after the sample. The first inflection point of the foil spectrum was assigned to 8980.3 eV. The energy-calibrated transmission data (I1/I0) (k = 13.4 Å−1) were processed by fitting a second-order polynomial to the pre-edge region and subtracting this from the entire spectrum as background. A one-region spline of order 2 was used to model the smoothly decaying post-edge region. Normalization of the data was achieved by scaling the spline function and data such that the value of the spline equals 1.0 at 9000 eV. This background subtraction and normalization was done using PySpline.37,38 Since some photodamage was observed over time, data presented in Figure S1 are the first scans of each data set to eliminate any spectral changes from photoreduction.
2.4 Computational Details
Spin-unrestricted broken-symmetry (BS) density functional theory (DFT) calculations were performed using the Gaussian 0939 or ORCA 2.6.3540 packages. Structures of 1 and 2 were calculated using the pure functional BP86 (Becke GGA exchange41 with Perdew 1986 nonlocal correlation42) and the functional BLYP (Becke GGA exchange41 with Lee, Yang, and Parr correlation43,44) modified to include 10, 20 and 38% Hartree-Fock.45 The B3LYP hybrid functional (with 20% HF mixing)46 was used to calculate the hydroxylation reaction coordinate, since it gives more accurate electronic structures than BP86, as determined in Section 3.2.2. The TZVP basis set was used for the Cu, N and O atoms. The SVP basis set was used for B, C and H atoms. For the reaction coordinate analysis TZVP was also used for the ortho-C and ortho-H that participate in the hydroxylation reaction.
Geometry optimizations of 1 and 2 in vacuum were performed starting from crystallographically derived parameters18 where the modified crystal structure of [(LMECHDCu)2O2]2+23 (Scheme 2, bottom right) was used for 2. The crystal structure of [Cu(II)2(H-XYL-O−)(OH−)]2+47 was used to optimize the hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+ with a para-NO2 substituted xylyl. The O⋯O unit (with O⋯O = 2.43 Å, Table S9) in the hydroxylated product was further modified into a peroxo (with O-O = 1.42 Å) and optimized to obtain the structure of the PNO2−XYL intermediate. The reaction coordinate for [Cu(II)2(NO2-XYL)(O22−)]2+ was calculated from this starting structure of PNO2−XYL. The second reaction coordinate studied was for [Cu(II)2(NH3)4(O22−)]2+, where the starting side-on peroxo structure was optimized from the modified the crystal structure of 1.
The self-consistent field (SCF) calculations were set to a tight convergence criterion. All fully optimized structures were verified as minima by frequency calculations that gave no imaginary frequency. The νO−O and νC−O obtained from the frequency calculations reported here were scaled by a factor of 0.965.48,49 Transition state (TS) structures were confirmed to have a single imaginary mode corresponding to the reaction coordinate. Intrinsic reaction coordinate (IRC) calculations showed that the TS structures are along the path between the reactants and products. Single point calculations on PNO2−XYL and the transition state structure were performed using the polarizable continuum model (PCM)50 using the default parameters for CH2Cl2 (EPS = 8.93) to check for any effect on ΔG‡. Dispersion and relativistic effects were included using a VDW51 and DKH52 Hamiltonian where indicated in the text. QMForge53 was used to calculate molecular orbital compositions via Mülliken population analysis54 and for Mayer bond orders.55,56 Wave-functions were visualized and orbital contours were generated in VMD.57 The two-dimensional potential energy surfaces were generated in gnuplot.
In agreement with the experimentally determined spin of P complexes, PNO2−XYL is well described by a broken-symmetry (BS) wave function with antiferromagnetically coupled Cu(II)’s, yielding a MS = 0 ground state electronic structure. The BS energies of PNO2−XYL and structures along the reaction coordinate leading to the TS (with BS spin expectation, <S2>, values of 0.7-1.0) were corrected for spin contamination from the triplet (ST = 1) excited state to obtain the singlet (ST = 0) energies using the spin corrected method by Yamaguchi et al. given by Equation 1:58
| (1) |
For the transition state structure and the ones that follow with BS <S2> values greater than 1.0, due to the formation of additional spins on the xylyl ring and the distal oxygen, the BS energies are contaminated by the triplet (ST = 1) and the quintet (ST = 2) excited states. Equation 1 and 259 are used for spin correction for the ST = 1 and ST = 2 excited states, respectively:60
| (2) |
3 RESULTS AND ANALYSIS
3.1 L-edge X-ray Absorption Spectroscopy
The normalized, edge subtracted Cu L-pre-edge X-ray absorption spectra of [{HB(3,5−iPr2pz)3Cu}2(O2)], 1 (PTPB), and [(LTEEDCu)2(O2)]2+, 2 (OTEED), are presented in Figure 1 (left). The 2nd derivative of the expanded L3 regions of the spectra of 1 and 2 are shown in Figure 1 (right). The Cu L-edge involves an electric dipole-allowed Cu 2p→3d transition. The 2p5 core configuration of the final state undergoes spin-orbit coupling to give two peaks split by ~20 eV, the J = 3/2 L3-edge at ~930 eV and the J = 1/2 L2-edge at ~950 eV with an intensity ratio of ~2:1, respectively.61 The L2-edge is ~1.5 times broader than the L3-edge due to an additional Coster-Kronig Auger decay channel for this excited state.62 The L3- and L2-pre-edge features are followed by weak 2p→4s and 2p→continuum edge transitions at ~10 eV higher energies (the intensity of the Δl = −1 transition is ~30 times lower than that of the Δl = +1 transition). The L3-edge of 1 occurs at 931.0 eV, which is equivalent to the energy position of L3 in D4h [CuCl4]2− and comparable to L3 transitions for many tetragonal Cu(II) complexes.63 The L3-edge of 2 is at 932.9 eV, shifted 1.9 eV to higher energy, consistent with a change in oxidation state to Cu(III).64 An analogous trend in energy is observed for the L2-edge.
Figure 1.

Cu L-edge XAS spectra of 1 (red line) and 2 (blue line). (Left) Normalized Cu L-edge XAS spectra. The weak edge jumps were simulated with arctangent functions and subtracted from the entire spectrum. The intense peaks at ~930 and ~950 eV represent the L3-edge (2p3/2→3d transition) and the L2-edge (2p1/2→3d transition), respectively. (Right) Smoothed second derivative of the Cu L3-edge region.
The Cu 2p→3d L-edge intensity can be used to experimentally obtain the amount of unoccupied metal d character in the ground state wavefunction and thus quantify the copper-dioxygen bonding. Since the Cu 2p orbital is localized on the Cu center and the 2p→3d transition is electric dipole-allowed, the total intensity of the L3- and L2-pre-edges reflects the Cu d character in the . The oxygen and nitrogen ligand valence 2p orbitals undergo bonding/antibonding interactions with the Cu 3d orbitals leading to the mixing of some ligand 2p character (β2) into the ground state wavefunction.65 Thus, increasing ligand character (covalency) in the ground state decreases the metal 3d character and therefore the intensity of the 2p→3d transition by β2 (Equation 3).
| (3) |
Correlating the L-edge intensity of 1 and 2 with that of the well characterized D4h [CuCl4]2− complex (61 ± 4% Cu d character in the ) gives a quantitative estimate of the amount of Cu unoccupied 3d character in their ground state wavefunctions. Table 1 lists the Cu characters obtained from the total integrated intensity of the L-edge spectra of 1 and 2.66 Accounting for two 3d holes per Cu for 2 and one hole for 1 and D4h [CuCl4]2−, the unoccupied Cu character of 1 and 2 are 52 ± 4% and 40 ± 6%, respectively. The decrease in Cu d character in 1 and 2 compared to that in D4h [CuCl4]2− indicates stronger covalent mixing of the Cu centers with the ligand (predominantly oxygen) valence orbitals. Furthermore, the higher covalency of 2 relative to 1, indicates that the Cu centers in the OTEED species have a stronger bonding interaction with the oxygen bridging ligands than in the PTBP species, consistent with shorter Cu(III)-O bonds in the OTEED complex (~1.80 Å versus ~1.90 Å).
Table 1.
Cu L-Edge X-ray Absorption Edge Energies (eV) and Cu Character in Ψ*LUMO of 1 and 2.
| 2p → 3d Energya | Cu Character in Ψ*LUMO | ||
|---|---|---|---|
| L3-edge | L2-edge | (% Hole) | |
| 1 | 931.0 | 951.0 | 52 ± 4 |
| 2 b | 932.9 | 952.8 | 79 ± 6 |
Energy resolution is ~0.1 eV. The energy is determined from the minimum intensity of the second derivative.
Two-hole system (see text for detail).
3.2 Calculations
3.2.1 Geometric Structure
It has been established that the amount of Hartree-Fock (HF) mixing in DFT functionals affects calculated geometric and electronic structures.31,32,67,68 Therefore, we have performed DFT calculations with varying HF mixings to determine the amount that best reproduces geometric and electronic structures of 1 and 2. Specifically, we have used the pure functional BP86 and BLYP modified with 10%, 20% (B3LYP), and 38% HF mixing to calculate the structures of 1 (Figure 2, left) and 2 (Figure 2, right). Selected structural parameters from the DFT-optimized structures of the two complexes are compared to those obtained from crystallography in Tables 2, 3, S1 and S2. The crystal structure of 1 is available,18 whereas no crystal structure of 2 exists; [(LMECHDCu)2O2]2+ (Scheme 2, bottom right) most closely resembles complex 2 and was used for comparison.23
Figure 2.

Schematic representations of (left) 1 and (right) 2. H atoms are omitted for clarity.
Table 2.
Comparison of Select Structural Parameters of 1 from DFT Calculations and Crystallography. All Bond Lengths are in Å.
| Parameter | X-ray18 | BP86 | 10% HF | B3LYP | 38% HF |
|---|---|---|---|---|---|
| ∠Cu1,O3,O4,Cu2a | 180.0 | 174.6 | 179.8 | 180.0 | 179.8 |
| O3-O4 | 1.413 | 1.450 | 1.446 | 1.457 | 1.484 |
| Cu1-Cu2 | 3.556 | 3.705 | 3.728 | 3.718 | 3.681 |
| Ave Cu-O | 1.914 | 1.993 | 1.999 | 1.997 | 1.985 |
| Ave Cu-Neq | 1.997 | 2.007 | 2.022 | 2.035 | 2.058 |
| Ave Cu-Nax | 2.260 | 2.244 | 2.283 | 2.300 | 2.322 |
Dihedral angle of the Cu2O2 core
Table 3.
Comparison of Select Structural Parameters of 2 from DFT Calculations and Crystallography. All Bond Lengths are in Å.
| Parameter | X-ray23 | BP86 | 10% HF | B3LYP | 38% HF |
|---|---|---|---|---|---|
| ∠Cu1,O3,O4,Cu2a | 173.7 | 180.0 | 180.0 | 180.0 | 180.0 |
| O3-O4 | 2.344 | 2.372 | 2.363 | 2.349 | 2.329 |
| Cu1-Cu2b | 2.748 | 2.797 | 2.781 | 2.775 | 2.755 |
| Ave Cu-Ob | 1.808 | 1.834 | 1.825 | 1.818 | 1.804 |
| Ave Cu-Nb | 1.938 | 2.006 | 2.008 | 2.009 | 2.010 |
Dihedral angle of the Cu2O2 core
EXAFS data of 2 (Cu⋯Cu = 2.75 Å, Cu-O = 1.80 Å, Cu-N = 1.92 Å)30
In the case of complex 1, the experimental planar Cu2O2 core is better reproduced with Hartree–Fock mixing. With BP86, the Cu2O2 core is butterflied by 5.4° (Table 2 ∠Cu1,O3,O4,Cu2) while inclusion of 10% HF gives a planar structure. The calculated Cu⋯Cu and O–O distances along the series with increasing HF are characteristic of P complexes but are slightly longer than the crystallographic distances (variations in Cu⋯Cu and O–O along the series are within 0.17 Å and 0.07 Å, respectively, of the X-ray parameters). A similar variation in the DFT optimized bond lengths of P species has been observed before.15 Based only on these structural parameters there is no significant advantage of any functional.
The calculated structures of 2 show a planar Cu2O2 core paralleling the crystal structure of [(LMECHDCu)2O2]2+ . The cores are planar for all O species studied to date (Table S3).21-23,36,69-74 A slight improvement in the bond lengths of the Cu2O2 core (Cu⋯Cu, O⋯O, and Cu–O) is observed with 20-38% Hartree–Fock mixing. The experimental and calculated difference in the Cu⋯Cu and O⋯O distances along the series of differing HF mixing are within 0.05 Å and 0.03 Å, respectively, and smaller than the difference observed for the P complexes. All the other calculated distances of 2 are in reasonable agreement with the crystal structure of [(LMECHDCu)2O2]2+ (within ~0.08 Å). Again, there is no significant dependence of the geometric structure on the choice of functional.
3.2.2 Electronic Structure
For coupled binuclear Cu(II)2 systems, the half-occupied d orbitals on the two Cu(1,2) centers form symmetric (d1 + d2) and antisymmetric (d1 − d2) combinations. In O22−, the degenerate π* orbitals are occupied (HOMOs), while the σ* is unoccupied (LUMO) (Figure 3, right). When O22− binds to Cu in 1, the π* splits into (orbital in the Cu2O2 plane) and (orbital out of the Cu2O2 plane). The undergoes σ bonding and antibonding interactions with the symmetric combination of Cu dx2−y2 orbitals to form the HOMO-2 O22− π*σ + 2Cu2+ (dx2−y2 + dx2−y2)] and the LUMO [2Cu2+ (dx2−y2 + dx2−y2) − O22− π*σ), respectively (Figure 3, center and Figure 4, left). The peroxo σ* similarly forms a bonding and antibonding pair with the antisymmetric combination of filled Cu dx2−y2 orbitals to form the HOMO [2Cu2+ (dx2−y2 − dx2−y2 + O22− σ*] and the LUMO+1 [O22− σ* − 2Cu2+ (dx2−y2 − dx2−y2)], respectively (Figure 3, center and Figure 4, left). The LUMO+1 peroxo σ* is ~3-6 eV higher in energy than the Cu based LUMO. There is a small amount of backbonding from the occupied Cu d orbitals into the σ* orbital that results in some σ* character in the HOMO (Figure 4).
Figure 3.

Schematic molecular orbital diagram for P species.
Figure 4.

The two key unoccupied MOs and their occupied counterparts in P and O. The σ* orbital in P has small backbonding from a occupied Cu dx2−y2 orbital that is not shown here.
As HF mixing is increased, the calculated electronic structure of 1 shows decreasing amounts of σ donation from the filled O22− into the symmetric 2Cu (dx2−y2 + dx2−y2) LUMO (Table S4). The resulting Cu d-hole character for the different functionals is given in Table 4. The closest match to the experimental covalency for 1 (52 ± 4% unoccupied Cu character) is obtained with 10-20% HF (49-57% Cu character). The addition of a HF contribution to the functional also leads to spin polarization of the α and β LUMOs as reflected by the increasing spin expectation value (Table 4) consistent with a Broken Symmetry (MS = 0) singlet ground state. The spin polarized α and β holes reside in the 3dx2−y2 orbitals on each Cu and undergo covalent mixing with the peroxo orbital (Figure 5, left). Spin polarization is not observed with the pure functional (BP86) where both the α and β holes are delocalized over both Cu’s and the amount of d-hole per Cu is underestimated (45% Cu character) indicating that this functional is too covalent (Figure 5, right). Thus, some HF mixing (10-20%) is required to reproduce the experimental covalency of 1, which results in spin polarization of the Cu(II)’s consistent with the antiferromagnetically coupled description of 1.
Table 4.
Per Cu Mülliken Population in the α Plus β Unoccupied Orbitals from Spin-Unrestricted Calculations of 1 and 2. Spin Expectation Values Given in Parenthesis.
| Functional | 1 (LUMO) | 2 (LUMO & LUMO+l)a |
|---|---|---|
| BP86 | 45 (0.00) | 66 (0.00) |
| 10% HF | 49 (0.45) | 68 (0.00) |
| B3LYP | 57 (0.61) | 71 (0.00) |
| 38% HF | 70 (0.90) | 76 (0.00) |
Numbers reported here are for two holes per 3d8 Cu(III).
Figure 5.

Isosurface plots (isovalue 0.04 au) of α and β LUMO of 1 from B3LYP in the BS (MS = 0) state (left) and BP86 (right) spin-unrestricted calculations.
When P isomerizes to O, the O–O bond is cleaved. This lowers the peroxo σ* in energy, oxidizes the Cu(II) to Cu(III) and reduces the O22− to 2O2− leading to complex 2.75 This results in two Cu based LUMO’s that are the symmetric and antisymmetric combinations of the Cu dx2−y2 orbitals and are antibonding with the oxo in-plane 2p orbitals (Figure 4, right). These LUMO and LUMO+1 are separated by <0.2 eV such that both contribute to the single L pre-edge feature in Figure 1. Note that in the P complex, the σ* is ~3-6 eV higher in energy with little Cu character and does not contribute to the pre-edge.
The calculated electronic structure of 2 shows increasing amounts of Cu d character in the LUMO and LUMO+1 as % HF mixing is increased (Table S5). Table 4 summarizes the total d-hole character per Cu for the different functionals using Mülliken populations. The closest match to the experimental covalency for 2 (79 ± 6% Cu hole) is obtained with 38% HF (77% Cu hole). Both the LUMO and LUMO+1 are delocalized and highly covalent with 36.4% and 40.4% Cu character, respectively (Figure 6).
Figure 6.

Isosurface plots (isovalue 0.04 au) of α, β LUMO and LUMO+1 of 2 from 38% HF spin-unrestricted calculations.
Thus, HF mixing is required to reproduce the experimentally observed electronic structures of both P and O species. The closest agreement with the experimental covalency of 1 is with 10-20% HF, whereas ~38% HF best reproduces that of 2. We have, therefore, selected B3LYP (with 20% HF) to study the electrophilic aromatic substitution (EAS) reaction coordinate in the [Cu(II)2(NO2-XYL)(O2)]2+ complex to determine the point of O-O bond cleavage with respect to the transition state. To this end, the reaction coordinate obtained for the PNO2−XYL structure will be compared with that of the ODBED complex studied previously with the B3LYP functional as well.24,25
3.3 Reaction Coordinate
3.3.1 PNO2−XYL Intermediate and Hydroxylated Product
In order to evaluate the EAS reaction coordinate of [Cu(II)2(NO2-XYL)(O2)]2+, we first obtained the geometry optimized structures of the PNO2−XYL reactant and the hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+. The calculated νo−o for PNO2−XYL (Figure 7A) is 879 cm−1 (the experimental νo−o is 747 cm−1)28, 76, 77 and described with a BS singlet (MS = 0) ground state with spin polarized α (2% Cu1 and 48% Cu2 in the LUMO) and β holes (48% Cu1 and 2% Cu2 in the LUMO) in the 3dx2−y2 orbital on each Cu (Table S6). The total Cu character is consistent with Cu L-edge XAS data for 1 (Table 1). Selected structural parameters are given in Table S8 (PNO2−XYL). The Cu2O2 core is butterflied to 138° to accommodate the xylyl bridge and is consistent with the split O22− charge transfer transitions (360 nm and 435 nm) from absorption studies.28,77 The Cu–Cu and O–O distances are 3.57 Å and 1.42 Å, respectively, while the average Cu–O bond length is 2.04 Å. These distances are characteristic of P species.15 The distance between the proximal oxygen (O3) and the nearest ortho-carbon of the arene (C12) is 2.79 Å. Based on this unconstrained structure, the C-O and O-O distances of 2.80 Å and 1.42 Å, respectively, were used as the starting point for a scan of the two-dimensional reaction coordinate presented in Section 3.3.2.
Figure 7.

B3LYP optimized structures of A) the PNO2−XYL complex, and B) the final hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+. Crystallographic parameters are given in parentheses.
The final hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+ formed in the EAS reaction (Figure 7B) (see Section 3.3.3) has the O–O bond cleaved with a calculated distance of 2.43 Å (crystallographic O–O = 2.43 Å47).78 The C–O bond is fully formed at 1.33 Å (crystallographic C–O = 1.33 Å) and the phenolate product bridges the two Cu(II) centers. All other calculated structural parameters compare well with the crystal structure (Table S9). The Cu2O2 core and the resulting phenolate are nearly planar (∠CuOOCu = 177°calc, 178°x−ray; ∠C15C12O3 = 180°calc, 178°x−ray). The experimental νC−O of 1320 cm−1 is in good agreement with the calculated frequency of 1302 cm−1.28 Formation of this hydroxylated product will be discussed in Section 3.3.3.
3.3.2 2-D Reaction Coordinate of O–O Bond Cleavage and C–O Bond Formation
Reaction of the PNO2−XYL intermediate (Figure 7A) to form the hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+ (Figure 7B), requires cleavage of the O3–O4 and C12–H11 bonds and formation of the C12–O3 and O4-H11 bonds. In Section 3.3.3, it is shown that the cleavage of the C12–H11 bond (and the subsequent formation of the O4-H11 bond) occurs after the transition state for C12–O3 formation, consistent with the experimental kinetic isotope effect of ~1.0. In this section, the cleavage of the O3–O4 and formation of the C12–O3 bonds are evaluated. The two-dimensional potential energy surface (PES) of the electronic energy as a function of the O3–O4 and C12–O3 distances is presented in Figure 8. The C12–O3 was decreased in steps of 0.20 Å from 2.80 Å to 1.60 Å and for every point the O3–O4 bond was elongated (1.45, 1.60, 1.80, 2.10, and 2.28 Å). This 2-D PES compares the energetics of the two possible reaction pathways: i) concerted reaction where the C12–O3 forms concurrently with O3–O4 cleavage (pink curve in Figure 8) and ii) step-wise reaction where the C12–O3 forms after complete O3–O4 cleavage (green curve in Figure 8). Figure 8 shows that the concerted reaction is energetically favorable and gives the lowest energy route on the PES (pink curve). The alternate route of first forming ONO2−XYL (followed by C12–O3 bond formation) (green curve) is higher in energy by ~20 kcal/mol. The lowest energy route (pink line) is re-plotted in Figure 9, top, with additional points added near the transition state (TS) and at the end point K (the unconstrained species with C12–H11 still present) with a C12–O3 of 1.41 Å.
Figure 8.

2-D potential energy surface (PES) showing electronic energy changes as a function of O3–O4 and C12–O3 distances for [Cu(II)2(NO2-XYL)(O2)]2+. The pink line indicates the lowest energy route on the PES from A, the 2.80 Å C12–O3 PNO2−XYL starting structure, leading to K, the end species (not shown here). The green line indicates the path taken to first form ONO2−XYL followed by C12–O3 bond formation.
Figure 9.

(Top) Potential energy surface of the formation of phenolate along the C12–O3 coordinate for [Cu(II)2(NO-XYL)(O2)]2+ extracted from the lowest energy path of the 2-D PES (Figure 8, pink line) along O3–O4 and C12–O3 distances with additional points added near the transition state (TS) and at the end point K. All hydrogens except H11 are omitted for clarity. (Bottom) Transition state structure for C12–O3 bond formation. The pink arrows represent the mode of the imaginary frequency of −547 cm−1 with the length of the arrows representing the relative displacement of the atoms.
TS Geometric and Electronic Structure
The transition state along the C12–O3 bond formation reaction coordinate occurs at a C12–O3 distance of 1.88 Å (G) (Figure 9, top). This has one imaginary frequency of −547 cm−1 (with a displacement ratio of 0.63 O3 : −0.25 O4 : −0.48 C12 : 0.44 H11) (Figure 9, bottom, magenta arrow) showing O3–O4 bond cleavage occurs concurrently with C12–O3 bond formation. The calculated energy barriers, ΔH and ΔG, corrected for spin contamination, are ~24 kcal/mol and ~26 kcal/mol, respectively. When dispersion and relativistic effects are also included,79,80 the barrier height is lowered by ~5 kcal/mol and within DFT accuracy of the experimental activation energies (experimental ΔH‡ = 13 ± 2 kcal/mol, ΔG‡ = 15 ± 2 kcal/mol).28,81,82 An intrinsic reaction coordinate (IRC) calculation confirms that the TS is along the EAS reaction coordinate.
In going from point A to the TS structure (G), the O3–O4 distance increases from 1.42 Å to 1.97 Å while the Cu1-Cu2 distance decreases from 3.57 Å to 3.29 Å (Table S8). A decrease in the Cu-O distances is also observed (Cu-Oave is 2.04 Å at point A and 1.94 Å at point G, Table S8). The key angles to note are the ∠Cu1O3O4Cu2 dihedral angle of the Cu2O2 core that becomes more planar (138° at point A and 159° at point G) and the phenolate angle (∠C15C12O3) that becomes more perpendicular (122° at point A and 113° at point G). Another important geometric change observed at the TS is distortion of C12 of the xylyl ring from a planar sp2 to a pseudo tetrahedral sp3 hybridized state (∠C15C12H11 is 177° at point A and 158° at point G). The C12–H11 bond distance decreases consistently from 1.079 Å at point A (ν(C−H) = 3209 cm−1) to 1.075 Å (at point D) before the TS and at the TS it increases slightly to 1.080 Å (ν(C−H) = 3195 cm−1) and continues to increase (Table S8). The kinetic isotope effect (KIE) is calculated to be 1.00, which matches the experimentally observed KIE of ~1.0.83,27
Changes in the electronic structure from point A to G were investigated by monitoring the Mayer bond order (MBO) (Table 5) that show that as the C12–O3 bond is strengthened, the O3–O4 bond is weakened. To obtain a reference for a full C–O bond, structure K was reoptimized with the C12–H11 proton removed (structure L, C12–O3 MBO of 1.180) (Table 5).84 The O-O MBO of structure A (0.733) is used as the reference for an unperturbed side-on peroxo O-O bond. Thus, at the TS, the C12–O3 is found to be 25% formed while 63% of the O–O bond is broken (the TS of the EAS reaction for [(DBED)2Cu(III)2(O2−)2]2+ had 27% of the C–O bond formed while the O⋯O bond was fully broken25).
Table 5.
Distances and Mayer Bond Order (MBO) for C12–O3 and O3–O4 along the C12–O3 Coordinate for [Cu(II)2(NO2-XYL)(O2)]2+.
| Structure | C12–O3 Å |
C12–O3 MBO |
O3–O4 MBO |
CO Formed % |
OO Formed % |
|---|---|---|---|---|---|
| A | 2.800 | <0.01 | 0.733 | 100 | |
| B | 2.600 | <0.01 | 0.723 | 99 | |
| C | 2.400 | <0.01 | 0.709 | 97 | |
| D | 2.200 | <0.01 | 0.688 | 94 | |
| E | 2.000 | <0.01 | 0.635 | 87 | |
| F | 1.900 | 0.030 | 0.572 | 3 | 78 |
| G (TS)a | 1.880 | 0.296 | 0.272 | 25 | 37 |
| H | 1.850 | 0.426 | 0.133 | 36 | 18 |
| I | 1.800 | 0.511 | 0.095 | 43 | 13 |
| J | 1.600 | 0.730 | 0.035 | 62 | 5 |
| K a | 1.409 | 0.893 | <0.01 | 76 | |
| L b | 1.303 | 1.180 | <0.01 | 100 | |
Fully optimized structures with no constraints.
Optimized structure with C12–H11 broken and H+ removed.
Inspection of the Mülliken charge and spin densities on the different fragments (Cu1, Cu2, O3, O4, xylyl-ring (NO2-C6H3), and rest of the ligand) provides insight into the electronic structure changes that occur along the reaction coordinate (Tables 6, 7, S10 and S11). The two Cu centers remain spin polarized and antiferromagnetically coupled from points A to G (+0.52 spin on Cu1 and −0.52 spin on Cu2 at point G, Table 7) and thus are described as Cu(II) centers.85 It is important to note that the xylyl ring becomes noticeably positive with the Mülliken charge increasing from +0.02 at point A to +0.25 at point G (Table 6, combined charge on fragments (NO2-C6H2) and H11). In addition, at point G there is a net β spin of −0.29 on the xylyl ring whereas the net spin at point A is 0.00 (Table 7, combined spin on fragments (NO2-C6H2) and H11). This increase in positive Mülliken charge and β spin on the xylyl ring is due to a 23% α electron donation from the π electron cloud of the xylyl ring (Table 8) (Figure 10, orbital 188α in A) into the O22− σ* orbital (Figure 10, orbital 210α in A)86 forming orbital 197α at point G (Figure 10). Thus, at the TS, the O3–O4 weakens due to α electron donation from the xylyl ring. As the peroxide elongates and starts to cleave, the holes on the O22− σ* orbital are spin polarized leading to a partial β hole on the distal O4 (+0.52 α spin) and a partial α hole on the proximal O3 (−0.21 β spin) (Table 7). The α hole on the O3 is smaller than the β hole on the distal O4 because of the α electron donation from the xylyl ring to O3. Thus, at the TS the C12–O3 bond is strengthened while the O3–O4 bond is weakened in a concerted manner due to the transfer of partial α electron density from the ring into the O3–O4 bond.
Table 6.
Mulliken Charge of Key Species along the C12–O3 Coordinate for [Cu(II)2(NO2-XYL)(O2)]2+.
| 2.80 A |
1.88 G (TS)a |
1.409 Ka |
|
|---|---|---|---|
| Cu1 | 0.38 | 0.41 | 0.40 |
| Cu2 | 0.37 | 0.44 | 0.42 |
| O3 | −0.25 | −0.42 | −0.48 |
| O4 | −0.23 | −0.35 | −0.31 |
| (NO2-C6H2) | −0.05 | 0.13 | 0.06 |
| H11 | 0.07 | 0.12 | 0.12 |
| rest | 1.78 | 1.80 | 1.90 |
Fully optimized structures with no constraints.
Table 7.
Mülliken Spin and Spin Expectation of Key Species along the C12–O3 Coordinate for [Cu(II)2(NO2-XYL)(O2)]2+.
| 2.80 A |
1.88 G (TS)a |
1.409 Ka |
|
|---|---|---|---|
| Cu1 | 0.44 | 0.52 | 0.59 |
| Cu2 | −0.44 | −0.52 | −0.57 |
| O3 | 0.02 | −0.21 | −0.03 |
| O4 | −0.01 | 0.52 | 0.96 |
| (NO2-C6H2) | 0.00 | −0.31 | −0.98 |
| H11 | 0.00 | 0.02 | −0.07 |
| rest | −0.01 | −0.01 | 0.03 |
| <S2> | 0.87 | 1.10 | 1.95 |
Fully optimized TS and K end species with no constraints.
Table 8.
Change in Mülliken Population of the Filled Orbitals along the C12–O3 Coordinate Compared to Structure A. All Numbers are in % of an electron.
| G (1.88) | K (1.409) | |||
|---|---|---|---|---|
| α | β | α | β | |
| Cu1 (d orbitals) | −2 | −9 | 1 | −16 |
| Cu2 (d orbitals) | −9 | −3 | −13 | −3 |
| O3 (p orbitals) | 0 | 24 | 16 | 22 |
| O4 (p orbitals) | 34 | −17 | 50 | −41 |
| (NO2-C6H3) | −23 | 5 | −54 | 38 |
Figure 10.

Key orbitals along the C12–O3 reaction coordinate for [Cu(II)2(NO2-XYL)(O2)]2+. The unoccupied peroxo σ* orbitals (210 α, β in A) are lowered in energy along the coordinate. An α electron is donated from the occupied xylyl HOMO (orbital 188 in A) to the peroxo σ* orbital leaving an α hole on the xylyl (orbital 198 α in K). The β hole ends up on the distal O4 (oxyl pz-like orbital, 197β in K; the z-axis is perpendicular to the Cu2O2 plane).
Species K
From the TS to the end of the reaction before H11 is lost as a proton, the optimized C12–O3 decreases from 1.88 Å at point G to 1.41 Å at point K. The O3-O4 distance increases from 1.97 Å to 2.49 Å from point G to K while the C12–H11 has elongated from 1.08 Å in G to 1.12 Å in K (Table S8). The Cu2O2 core becomes mostly planar with the ∠Cu1O3O4Cu2 dihedral angle increasing from 159° at point G to 174° at point K. In addition, the phenolate angle (∠C15C12O3 in Figure 11) increases from 113° at point G to 145° at point K (Table S8) but is still not planar. This bent phenolate angle has an important role that will be explored below.
Figure 11.

Structure of species K. All hydrogens except H11 are omitted for clarity.
Inspection of the electronic structure at point K reveals that the two Cu centers continue to be spin polarized (+0.59 spin on Cu1 and −0.57 spin on Cu2, Table 7) with no increase in Mülliken charge (Table 6) and thus remain antiferromagnetically coupled Cu(II) centers. The β spin character on the xylyl ring has increased to −0.98 while the α spin character on the distal O4 has increased to +0.96 (Table 7). The increase in β spin density on the xylyl ring is due to an additional 31% α electron donation (Table 8) from the ring to the O3-O4 from point G to K, leaving an α hole in a phenolate-like orbital on the ring (Figure 10 K, orbital 198α and a β spin character of -0.98). The increase of α spin character on the distal O4 is a result of the complete cleavage of the O3-O4 bond that leads to full spin polarization of the O⋯O σ* with the α and β holes localized on O3 and O4, respectively. Another important change in the electronic structure at point K is that the overall Mülliken charge on the xylyl ring has decreased from 0.25 at the TS to 0.18 (Table 6). This decrease in charge on the ring is due to β electron donation from O3 to form the C12–O3 bond with 38% of the electron density residing on the xylyl ring (Table 8). Thus, the α and β electrons from the xylyl ring and O3, respectively, pair for the concerted C12–O3 bond formation with O3–O4 bond cleavage. However, only 76% of the C12–O3 is formed with the O3–O4 completely broken (Table 5) as the C12–H11 bond is still present. The C12–H11 bond cleavage is explored in Section 3.3.3.
1 Versus 2 Electron Donation from Substrate
At the end of the above reaction coordinate at point K, a partial α electron has been transferred from the xylyl ring to the O⋯O forming an antiferromagnetically coupled diradical pair with a β spin on the xylyl ring (substrate) and an α spin on the distal O4. Interestingly, when an unconstrained benzene was used as the substrate for this reaction using a side-on peroxo [Cu(II)2(NH3)4(O22−)]2+ structure as a model (species X in Figure 12, Table S13), the species formed at the end of the reaction and before C-H bond cleavage, had no diradical character on the substrate and distal O4 (species Z with a C-O distance of 1.36 Å in Figure 12, Table S13 and S15). Evaluation of the reaction coordinate for [Cu(II)2(NH3)4(O22−)]2+ reveals that the side-on peroxo species X does react with the substrate to proceed through a transition state, Y, with a small amount of diradical character on the aromatic substrate (spin of +0.23) and the distal O4 (spin of −0.19) (Table S15) and a phenolate angle, ∠CCO, of 119° (Table S13, point Y with a C-O distance of 2.08 Å).87 The diradical character, however, disappears farther along the reaction as the substrate rotates (∠CCO = 163°at point Z, Figure 13, right) to allow the second π electron of the HOMO orbital on the ring to overlap the peroxo based LUMO leading to α, β electron pair donation from the substrate.88 Thus, the only spins observed are the two antiferromagnetically coupled Cu centers (+0.53 on Cu1 and −0.54 on Cu2, Table S15) that remain Cu(II) throughout the reaction coordinate.89 If at point Z (Figure 12) the benzene ring is constrained with respect to the Cu2O2 plane to a ∠CCO of 145°, consistent with the covalently linked xylyl ring of [Cu(II)2(NO2-XYL)(O2)]2+ at point K (Figure 13, left) , the species does contain diradical character. Upon removing the constraint on the ring in this spin polarized complex, the ring rotates by ~20° allowing a second electron to transfer and eliminate the diradical character.90
Figure 12.

Potential energy surface for the EAS reaction of benzene with [Cu(II)2(NH3)4(O22−)]2+.
Figure 13.

Structures of species K versus Z formed at the end of the EAS reaction coordinates for [Cu(II)2(NO2-XYL)(O2)]2+ and [Cu(II)2(NH3)4(O22−)]2+, respectively, but before ortho-H+ cleavage. All hydrogens except ortho-H are omitted for clarity.
3.3.3 C12–H11 Cleavage and Completion of the Reaction
In order to fully form the hydroxylated product [Cu(II)2(NO2-XYL-O−)(OH−)]2+, the C12–H11 proton was removed and the distal O4 protonated. However, due to the large distance between H11 and O4, an exogenous proton acceptor is potentially needed to facilitate this transfer. In these calculations, methylamine is used as a model for this exogenous base.91 Op’t Holt et al. showed that the use of stronger/weaker proton acceptors did not significantly change the reaction coordinate.25 The effect of the C12–H11 bond cleavage was modeled with a 2D PES by adding methylamine to the nine structures from C to K in Figure 9 (C12-Nmethylamine constrained to 2.80 Å) with the lone pair on the N pointing towards the ortho-H11. For each of the nine structures, the N-H11 distance was scanned from 2.06, 1.80, 1.60, 1.40, 1.20 to 1.03 Å, resulting in cleavage of the C12–H11 bond. The 2-D PES of the electronic energy calculated as a function of the C12–O3 and Nbase–H11 bond distances is shown in Figure 14. The electronic energy is scaled relative to that of Ab (C12–O3 = 2.80 Å; C12–H11 = 1.08 Å), which has the same structure as A in Figure 9 but with base present (base is also present in structures Cb, Kb and Lb). The lowest energy path (red line) from Cb (C12–O3 = 2.40 Å; C12–H11 = 1.08 Å) to Kb (C12–O3 = 1.41 Å; C12–H11 = 1.12 Å) has essentially the same geometric and electronic structures as those without base present in Figure 9. Thus, the TS state remains unchanged in the presence of a base.
Figure 14.

2-D potential energy surface (PES) for [Cu(II)2(NO2-XYL)(O2)]2+ showing electronic energy changes as a function of C12–O3 and Nbase–H11 distances with methylamine as the base. The red line indicates the lowest energy route on the PES starting from Ab through Cb to Kb and finally forming the the deprotonated species Lb. Lb is ~41 kcal/mol lower in energy than Ab.
When the C12–O3 bond is constricted sufficiently to 1.41 Å (Kb), it becomes energetically favorable for the proton to be transferred to the nearby base forming structure Lb (C12–O3 = 1.32 Å; C12–H11 = 1.73 Å), which is ~41 kcal/mol lower in energy than Ab. Along the reaction coordinate from Kb to Lb (Tables S16-18), the diradical character on the xylyl ring and distal O4 disappears as the C12–H11 bond is elongated from 1.12 Å to 1.25 Å and the ∠CCO becomes more planar (Table S18). This reflects the additional transfer of the second electron from the xylyl ring (Figure 10 K, orbital 196β) to the hole on the distal O4 (Figure 10 K, orbital 197β). Further C12–H11 bond elongation to 1.73 Å in Lb completes C12-O3 bond formation (MBO of 1.18).
Finally, transfer of a proton from the base to the distal O3, gives another ~40 kcal/mol energy stabilization. Thus, formation of the hydroxylated species [Cu(II)2(NO2-XYL-O−)(OH−)]2+ via a base is energetically favorable. The hydroxylated species formed has a structure that is very similar to that of Lb. Protonation of the distal O, therefore, has no substantial effect on the Cu2O2 core.
4 DISCUSSION
L-edge XAS experimentally quantified the amount of Cu character in the ground state wavefunction (Cu 3d character in the unoccupied orbitals) to be 52 ± 4% in 1 (PTPB) and 79 ± 6% in 2 (OTEED). The 52% Cu character in 1 corresponds to one hole per Cu(II) center, whereas, the 79% Cu character in 2 corresponds to two holes per Cu(III) center. The lower per-hole Cu character and thus higher covalency in 2 compared to 1, indicates that the bis-μ-oxo core has a stronger bonding interaction with the Cu centers. The results from L-edge XAS have been correlated to DFT calculations to determine that a HF mixed hybrid functional is required to reproduce the quantitative electronic structures of 1 and 2. In addition to increasing the Cu 3d-hole character, inclusion of HF mixing leads to spin polarization in 1 with each hole localized on a different Cu. Thus, HF mixing is required to produce the singlet ground state with an antiferromagnetically coupled pair of Cu(II) centers (Figure 5).
B3LYP (with 20% HF), which gives a reasonable comparison to the L-edge XAS data for 1 and 2 was then employed to evaluate the EAS reaction coordinate in [Cu(II)2(NO2-XYL)(O2)]2+ that has an experimentally defined P intermediate. Stack and co-workers have previously shown that the O isomer in [(DBED)2Cu2(O2)]2+, another potential tyrosinase model, is capable of performing aromatic hydroxylation.24,25 The goal here was to determine whether the PNO2−XYL in Cu(II)2(NO2-XYL)(O2)]2+ converts to an ONO2−XYL intermediate along the reaction coordinate for EAS. This is an ideal system to study because the substrate does not bind to the Cu, in contrast with the [(DBED)2Cu(III)2(O)2]2+ system, where the phenolic substrate binds to one of the Cu centers and then undergoes EAS. Two key differences are observed in comparing the reaction coordinate of [Cu(II)2(NO2-XYL)(O2)]2+ to the previously developed model of [(DBED)2Cu(III)2(O)2]2+. First, the calculations show that for [Cu(II)2(NO2-XYL)(O2)]2+, the Cu centers remain antiferro-magnetically coupled Cu(II) throughout the reaction and are not oxidized to Cu(III). The aromatic ring of the crosslinker transfers π electron density directly to the peroxo σ* orbital resulting in O-O bond cleavage with concerted C-O bond formation (Scheme 5). Thus, the P intermediate is best interpreted to be the reactive species in [Cu(II)2(NO2-XYL)(O2)]2+ that performs the EAS reaction.
Scheme 5.
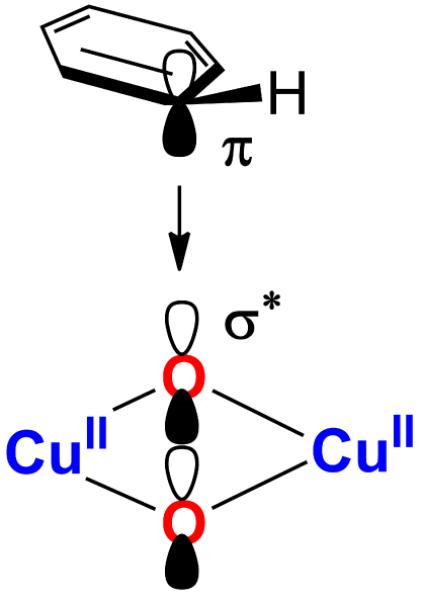
Second, instead of transferring a pair of electrons from the aromatic ring in [Cu(II)2(NO2-XYL)(O2)]2+ to form the C-O bond, α and β electrons are transferred from the xylyl ring and the peroxo π*, respectively, due to the limited flexibility of the xylyl chelate (Figure 15). The α counterpart of the peroxo π* ends up on the distal oxygen (Od) resulting in a radical on the ring and a radical on Od that are antiferromagnetically aligned. This diradical character is eliminated as the o-C-H cleaves and a second electron is transferred from the ring to Od via the Op-Cu-Od bonding network. This one versus two electron attack is due to the ∠CCO phenolate angle (Figure 13). In [Cu(II)2(NO2-XYL)(O2)]2+, where the rotation of the aromatic ring is constrained by covalent linkage to be less than 145°, only one electron transfer to peroxide is observed. As the ∠CCO becomes more planar with weakening of the o-C-H bond, the second electron is transferred. In contrast, in the EAS reaction coordinate where the aromatic ring is allowed to freely rotate (Figure 13), the ∠CCO becomes >163° and a two electron attack is observed as found for O in [(DBED)2Cu2(O2)]2+.25
Figure 15.

Schematic mechanism of aromatic hydroxylation performed by PNO2−XYL in [Cu(II)2(NO2-XYL)(O2)]2+. The red and black arrows represent transfer of α and β electrons, respectively. In going through the transition state (TS) to K, a pair of α and β electrons are transferred from the xylyl ring and peroxo π* orbital, respectively, to concertedly form the C-O bond and cleave the O-O bond. This produces an α spin on Od that is paired with the β radical on the ring. In proceeding from K to L and subsequently to the hydroxylated product, the o-H+ is transferred to the Od via a base. As the o-C-H bond is cleaved, its α and β electrons are transferred to the phenolate and Od, respectively, leading to the full formation of the phenolate C-O bond.
Thus, both the P and O species are capable of performing aromatic hydroxylation via their separate mechanisms. The question now in tyrosinase and NspF is whether P is the reactive species or converts to O upon phenolic substrate binding. The strength of the substrate-Cu interaction in the enzyme will be a key determining factor. While no substrate-bound oxy-CBC intermediate has been characterized spectroscopically, inhibitor binding studies have shown that substrate analogs bind to the Cu(II) in tyrosinase.92,93 These studies also show that the interaction of the substrate with the protein pocket influences this binding. Thus, the combined influence of the Cu and protein pocket interactions will determine the nature of the Cu2O2 species; the stronger and more equatorial the substrate binding, the more favorable the conversion of P to O. These combined interactions will also determine the orientation of the substrate and whether the transfer of the electron pair is concerted during this electrophilic reaction.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the National Institutes of Health (NIH) (E.I.S., DK31450; K.O.H., P41 RR001209 and GM103393; K.D.K., GM28962) and the Japan Society for the Promotion of Science (JSPS) (K.F., 25109505) for research support. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (NIGMS) and the National Center for Research Resources (NCRR).
Footnotes
SUPPORTING INFORMATION
Cu K-edge XAS spectra of 1 and 2; details of the DFT-optimized geometric and electronic structures of 1 and 2; key geometries and electronic structures along the reaction coordinates for [Cu(II)2(NO2-XYL)(O2)]2+ (structures A-L) and [Cu(II)2(NH3)4(O2)]2+ (structures X-Z); full Gaussian 09 reference.
Notes and References
- (1).Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- (2).Solomon EI, Baldwin MJ, Lowery MD. Chem. Rev. 1992;92:521–542. [Google Scholar]
- (3).Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- (4).Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- (5).Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- (6).Fukuzumi S, Karlin KD. Coord. Chem. Rev. 2013;257:187–195. doi: 10.1016/j.ccr.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ginsbach JW, Kieber-Emmons MT, Nomoto R, Noguchi A, Ohnishi Y, Solomon EI. Proc. Natl. Acad. Sci. U.S.A. 2012;109:10793–10797. doi: 10.1073/pnas.1208718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hatcher LQ, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- (9).Jacobson RR, Tyeklár Z, Farooq A, Karlin KD, Liu S, Zubieta J. J. Am. Chem. Soc. 1988;110:3690–3692. [Google Scholar]
- (10).Baldwin MJ, Ross PK, Pate JE, Tyeklár Z, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1991;113:8671–8679. [Google Scholar]
- (11).Schatz M, Becker M, Thaler F, Hampel F, Schindler S, Jacobson RR, Tyeklár Z, Murthy NN, Ghosh P, Chen Q, Zubieta J, Karlin KD. Inorg. Chem. 2001;40:2312–2322. doi: 10.1021/ic000924n. [DOI] [PubMed] [Google Scholar]
- (12).Zhang CX, Kaderli S, Costas M, Kim E-I, Neuhold Y-M, Karlin KD, Zuberbühler AD. Inorg. Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- (13).A third end-on peroxo Cu(II)2 species has also been observed.
- (14).Comba P. Coord. Chem. Rev. 2000;200-202:217–245. [Google Scholar]
- (15).Park GY, Qayyum MF, Woertink J, Hodgson KO, Hedman B, Narducci Sarjeant AA, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2012;134:8513–8524. doi: 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Que L, Tolman WB. Angew. Chem. Int. Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- (17).Kitajima N, Fujisawa K, Moro-oka Y, Toriumi K. J. Am. Chem. Soc. 1989;111:8975–8976. [Google Scholar]
- (18).Kitajima N, Fujisawa K, Fujimoto C, Moro-oka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J. Am. Chem. Soc. 1992;114:1277–1291. [Google Scholar]
- (19).Magnus KA, Hazes B, Ton-That H, Bonaventura C, Bonaventura J, Hol WG. Proteins. 1994;19:302–309. doi: 10.1002/prot.340190405. [DOI] [PubMed] [Google Scholar]
- (20).Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. J. Biol. Chem. 2006;281:8981–8990. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- (21).Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbühler AD, Tolman WB. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- (22).Mahapatra S, Halfen JA, Wilkinson EC, Pan G, Wang X, Young VG, Cramer CJ, Que LJ, Tolman WB. J. Am. Chem. Soc. 1996;118:11555–11574. [Google Scholar]
- (23).Mahadevan V, Hou Z, Cole AP, Root DE, Lal TK, Solomon EI, Stack T. J. Am. Chem. Soc. 1997;119:11996–11997. [Google Scholar]
- (24).Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- (25).Op’t Holt BT, Vance MA, Mirica LM, Heppner DE, Stack TDP, Solomon EI. J. Am. Chem. Soc. 2009;131:6421–6438. doi: 10.1021/ja807898h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nasir MS, Cohen BI, Karlin KD. J. Am. Chem. Soc. 1992;114:2482–2494. [Google Scholar]
- (27).Karlin KD, Nasir MS, Cohen BI, Cruse RW, Kaderli S, Zuberbühler AD. J. Am. Chem. Soc. 1994;116:1324–1336. [Google Scholar]
- (28).Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1998;120:7841–7847. [Google Scholar]
- (29).Lam BM, Halfen JA, Young VG, Hagadorn JR, Holland PL, Lledós A, Cucurull-Sánchez L, Novoa JJ, Alvarez S, Tolman WB. Inorg. Chem. 2000;39:4059–4072. doi: 10.1021/ic000248p. [DOI] [PubMed] [Google Scholar]
- (30).DuBois JL, Mukherjee P, Collier AM, Mayer JM, Solomon EI, Hedman B, Stack TDP, Hodgson KO. J. Am. Chem. Soc. 1997;119:8578–8579. [Google Scholar]
- (31).Szilagyi RK, Metz M, Solomon EI. J. Phys. Chem. A. 2002;106:2994–3007. [Google Scholar]
- (32).Atanasov M, Comba P, Martin B, Müller V, Rajaraman G, Rohwer H, Wunderlich S. J. Comput. Chem. 2006;27:1263–1277. doi: 10.1002/jcc.20412. [DOI] [PubMed] [Google Scholar]
- (33).This discussion of HF mixing needed in DFT calculations will be discussed further in another report.
- (34).Cruse RW, Kaderli S, Karlin KD. J. Am. Chem. Soc. 1988;110:6882–6883. [Google Scholar]
- (35).Blackburn NJ, Strange RW, Farooq A, Haka MS, Karlin KD. J. Am. Chem. Soc. 1988;110:4263–4272. [Google Scholar]
- (36).Cole AP, Mahadevan V, Mirica LM, Ottenwaelder X, Stack T. Inorg. Chem. 2005;44:7345–7364. doi: 10.1021/ic050331i. [DOI] [PubMed] [Google Scholar]
- (37).Tenderholt AL, Hedman B, Hodgson KO. PySpline. Stanford Synchrotron Radiation Laboratory; Stanford CA: 2006. [Google Scholar]
- (38).Tenderholt A, Hedman B, Hodgson KO. AIP Conf. Proc. 2006;882:105–107. [Google Scholar]
- (39).Frisch MJ, et al. Gaussian 09 Revision A.1. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- (40).Neese F. ORCA: An ab initio, DFT, and semiempirical electronic structure package, Version 2.6 revision 35. Universität Bonn; Bonn–Germany: 2008. [Google Scholar]
- (41).Becke AD. Phys. Rev. A. 1988;83:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- (42).Perdew J. Phys. Rev. B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- (43).Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- (44).Miehlich B, Savin A, Stoll H, Preuss H. Chem. Phys. Lett. 1989;157:200–206. [Google Scholar]
- (45).BLYP gives very similar results as BP86.
- (46).Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- (47).Karlin KD, Hayes JC, Gultneh Y, Cruse RW, McKown JW, Hutchinson JP, Zubieta J. J. Am. Chem. Soc. 1984;106:2121–2128. [Google Scholar]
- (48).Merrick JP, Moran D, Radom L. J. Phys. Chem. A. 2007;111:11683–11700. doi: 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
- (49). http://cccbdb.nist.gov/vibscalejust.asp.
- (50).Tomasi J, Mennucci B, Cammi R. Chem. Rev. 2005;105:2999–3094. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- (51).Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132:154104–154119. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- (52).Jansen G, Hess BA. Phys. Rev. A. 2011;39:6016–6017. doi: 10.1103/physreva.39.6016. [DOI] [PubMed] [Google Scholar]
- (53).Tenderholt AL. QMForge, v. 2.1. Stanford University; Stanford CA: 2007. http://qmforge.sourceforge.net. [Google Scholar]
- (54).Mulliken R. J. Chem. Phys. 1955;23:1833–1840. [Google Scholar]
- (55).Mayer I. Chem. Phys. Lett. 1983;97:270–274. [Google Scholar]
- (56).Bridgeman AJ, Cavigliasso G, Ireland LR, Rothery J. J. Chem. Soc., Dalton Trans. 2001:2095–2108. [Google Scholar]
- (57).Humphrey W, Dalke A, Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- (58).Yamaguchi K, Jensen F, Dorigo A, Houk KN. Chem. Phys. Lett. 1988;149:537–542. [Google Scholar]
- (59).Kitagawa Y, Saito T, Ito M, Nakanishi Y, Shoji M, Koizumi K, Yamanaka S, Kawakami T, Okumura M, Yamaguchi K. Int. J. Quantum Chem. 2007;107:3094–3102. [Google Scholar]
- (60).For example, the BS spin expectation value for species A (C-O = 2.80 Å) is 0.87 (Table 7) with contamination from only the ST = 1 excited state. Thus, equation 1 is used for energy correction. At the TS, the BS spin expectation value is 1.10 (Table 7) with spin contamination from the ST = 1 and ST = 2 excited states. An average spin expectation of 0.83 (average of the spin expectation values from species A to F, Table S11) was used as the spin contamination from the ST = 1 state at the TS with the rest from the ST = 2 state.
- (61).de Groot FMF. Physica B. 1995;208-209:15–18. [Google Scholar]
- (62).Fuggle JC, Alvarado SF. Phys. Rev. A. 1980;22:1615–1624. [Google Scholar]
- (63).George SJ, Lowery MD, Solomon EI, Cramer SP. J. Am. Chem. Soc. 1993;115:2968–2969. [Google Scholar]
- (64).Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).DeBeer George S, Metz M, Szilagyi RK, Wang H, Cramer SP, Lu Y, Tolman WB, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2001;123:5757–5767. doi: 10.1021/ja004109i. [DOI] [PubMed] [Google Scholar]
- (66).High covalency can lead to shake-up satellite features on the L-edge which redistribute the intensity (vide in f ra), and therefore, total integrated intensities for 1 and 2 were used for comparison to square planar [CuCl4]2−.
- (67).Solomon EI, Szilagyi RK, DeBeer George S, Basumallick L. Chem. Rev. 2004;104:419–458. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- (68).Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J. Am. Chem. Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- (69).Mizuno M, Hayashi H, Fujinami S, Furutachi H, Nagatomo S, Otake S, Uozumi K, Suzuki M, Kitagawa T. Inorg. Chem. 2003;42:8534–8544. doi: 10.1021/ic0345166. [DOI] [PubMed] [Google Scholar]
- (70).Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. J. Am. Chem. Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- (71).Hayashi H, Fujinami S, Nagatomo S, Ogo S, Suzuki M, Uehara A, Watanabe Y, Kitagawa T. J. Am. Chem. Soc. 2000;122:2124–2125. [Google Scholar]
- (72).Mahapatra S, Young VG, Kaderli S, Zuberbühler AD, Tolman WB. Angew. Chem. Int. Ed. 1997;36:130–133. [Google Scholar]
- (73).Straub BF, Rominger F, Hofmann P. Chem. Commun. 2000:1611–1612. doi: 10.1021/ic991173w. [DOI] [PubMed] [Google Scholar]
- (74).Zhou L, Powell D, Nicholas KM. Inorg. Chem. 2006;45:3840–3842. doi: 10.1021/ic052121b. [DOI] [PubMed] [Google Scholar]
- (75).Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J. Am. Chem. Soc. 1999;121:10332–10345. [Google Scholar]
- (76).DFT calculations with B3LYP and the all-electron TZVP/SVP basis set generally overestimate the O-O stretching frequency in side-on peroxo Cu(II)2 species by ~100 cm−1. In the PNO2−XYL complex, the frequency is overestimated by 132 cm−1. This likely reflects an overestimation of the bend of the Cu2O2 dihedral angle, which has previously been shown in ref 77 to increase the O-O stretching frequency.
- (77).Pidcock E, Obias HV, Abe M, Liang H-C, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1999;121:1299–1308. [Google Scholar]
- (78).Based on the available crystal structure of [Cu(II)2(H-XYL-O−)(OH−)]2+.
- (79).Siegbahn PEM, Blomberg MRA, Chen S-L. J. Chem. Theory Comput. 2010;6:2040–2044. doi: 10.1021/ct100213e. [DOI] [PubMed] [Google Scholar]
- (80).The reaction coordinates obtained with and without dispersion and relativistic effects are similar. The electronic structures of 1 and 2 also do not change significantly when dispersion and relativistic effects are added.
- (81).Karlin KD, Kaderli S, Zuberbühler AD. Acc. Chem. Res. 1997;30:139–147. [Google Scholar]
- (82).The calculated ΔE‡ with 10 percent HF added, instead of 20 percent HF, is 23 kcal/mol, which is reduced to 13 kcal/mol when dispersion and relativistic effects are included.
- (83).NIH shift experimentally observed with a methyl substituted at the C12 position (Figure 7) is calculated to take place after the TS and therefore does not affect the TS.
- (84).The C-O MBO in a fully optimized molecule of NO2-phenolate and NO2-phenol (without any Cu) are 1.807 and 1.233, respectively.
- (85).There is a slight increase in charge and spin on the Cu centers that is due to approximately 11 percent α and β electron donation (2 percent α, 9 percent β from Cu1 and 9 percent α, 3 percent β from Cu2) from the coppers to the peroxo moiety (Table 8).
- (86).At point G the peroxo π* and σ* orbitals are hybridized (Figure S2, orbitals 197 and 198). The peroxo σ* (orbital 210 in Figure S2 A) is lowerd in energy relative to the peroxo π*σ (LUMO, 197) at point G. For a planar Cu2O2 core, the peroxo π*σ HOMO is in the plane and σ antibonding to Cu, whereas the π*v is out of the Cu2O2 plane and non-bonding to Cu. In the xylyl system, the peroxo π*σ and π*v orbitals hybridize because the Cu2O2 core is butterflied.
- (87).The diradical formation is due to the peroxo π* and σ* orbitals hybridizing to maximize overlap with the π cloud of the substrate HOMO and resulting in greater overlap with the peroxo β LUMO compared to the peroxo α LUMO (Figure S3, point Y).
- (88).Similar results are obtained with a five coordinate Cu system, [Cu(II)2(NH3)6(O22−)]2+, instead of a four coordinate Cu species and with NO2 substituted at the para position of the benzene.
- (89).At the TS (Table S15, point Y), the Mülliken spins on the Cu centers decrease. However, the Mülliken charges (Table S14) also decrease showing that the Cu centers are not oxidized to Cu(III)’s.
- (90).Synthetic modifications designed to experimentally probe for or induce radical intermediates, along with computational studies, are planned for future investigations.
- (91).Similar results are obtained with water as the exogenous base.
- (92).Winkler ME, Lerch K, Solomon EI. J. Am. Chem. Soc. 1981;103:7001–7003. [Google Scholar]
- (93).Wilcox DE, Porras AG, Hwang YT, Lerch K, Winkler ME, Solomon EI. J. Am. Chem. Soc. 1985;107:4015–4027. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.