

Abstract
Objectives
Determine the bidirectional transfer of pravastatin across the dually perfused term human placental lobule and its distribution between the tissue, maternal and fetal circuits.
Study design
The transfer of pravastatin was determined in the Maternal-to-Fetal (n=11) and Fetal-to-Maternal (n=10) directions. Pravastatin was co-perfused with its [3H]-isotope and the marker compound antipyrine (20 μg/mL) and its [14C]-isotope. The concentration of pravastatin in the perfused tissue, the maternal and fetal circuits was determined using liquid scintillation spectrometry. Inside-out vesicles prepared from placental brush border membranes were utilized to investigate the role of efflux transporters in transplacental transfer of pravastatin.
Results
Pravastatin was transferred from the maternal to the fetal circuit and vise versa. In the Maternal-to-Fetal direction, the distribution of pravastatin at the end of experiment was as follows: 14 ± 5% of the drug was retained by the tissue, 68 ± 5% remained in the maternal circuit, and 18±4% was transferred to the fetal circuit. The normalized transfer of pravastatin (Clearance index) to antipyrine in the Fetal-to-Maternal direction (0.48 ± 0.07) was higher than its transfer in the Maternal-to-Fetal direction (0.36 ± 0.07, p<0.01). Furthermore, pravastatin inhibited the ATP-dependent uptake of the paclitaxel and estrone sulfate.
Conclusions
The transfer of pravastatin across the dually perfused placental lobule suggests that fetal exposure to pravastatin is plausible. The higher transfer of pravastatin in the Fetal-to-Maternal direction than the reverse as well as its inhibition of the ATP-dependent uptake of [3H]-paclitaxel and [3H]-estrone sulfate strongly suggest the involvement of efflux transporters in decreasing its transfer across the placenta, and support pravastatin favorable pharmacokinetic profile in pregnancy.
Keywords: Pravastatin, Preeclampsia, Pregnancy, Transplacental Transfer
INTRODUCTION
Preeclampsia complicates up to 5% of pregnancies and is a major cause of maternal and neonatal morbidity and mortality. It also shares pathogenic similarities and many risk factors with adult cardiovascular diseases. To date, prevention of preeclampsia using various supplements and classes of medications had limited success. On the other hand, prevention of cardiovascular mortality and other cardiovascular events in non-pregnant patients using 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, e.g. statins, has been widely accepted.1,2 The beneficial effects of the cholesterol-lowering drug pravastain in reversing several pathophysiologic pathways in cardiovascular disease which are also associated with preeclampsia such as angiogenic imbalance, endothelial injury, inflammation, and oxidative stress provided biological plausibility for its use in prevention preeclampsia.3
The use of pravastatin is contraindicated in pregnant patients ( Pregnancy Category “X”) and current recommendations suggest discontinuing the medication before conception or immediately if the patient becomes pregnant.4 Pravastatin is one of the HMG-CoA reducatese inhibitors thus it decreases the synthesis of cholesterol and its derivatives that are important for normal fetal growth and development. To date, data obtained from experimental animals5,6 and limited data from humans7,8 suggest that pravastatin does not increase the risk of major congenital anomalies.9 However, the use of other HMG-CoA reductase inhibitors (simvastatin, lovastatin, atorvastatin, cerivastatin) may be associated with an increase in the incidence of skeletal malformations10 and was attributed to the difference in physicochemical properties between the hydrophilic pravastatin versus the other lipophilic HMG-CoA reductase inhibitors.
The molecular weight of pravastatin is 446.5 and its protein binding is between 43 and 54%.11 The low permeability of pravastatin across biological membranes including human placenta is to be expected because of its hydrophilic properties.12 On the other hand, it appears that both uptake and efflux transporters contribute to pravastatin distribution across biological membranes. Thus, it was demonstrated that permeability of pravastatin through intestinal membranes was higher than that expected from its hydrophilicity suggesting involvement of uptake transporters.11 Furthermore, the renal clearance of pravastatin exceeds the glomerular filtration rate,11,13 suggesting that renal tubular secretion contributes to the elimination of this compound. Since intestinal and kidney transporters are important components of absorption and tubular secretion, the interaction of pravastatin with transporters in other tissues, including placenta is possible. The placenta contains multiple drug transporters, some of which are efflux transporters that appear to be specifically dedicated to removal of exogenous and endogenous toxic compounds from the fetal back to the maternal circulation, and thus protect the developing fetus. They include the ATP-biding cassette (ABC) transporters such as P-glycoprotein (P-gp), several multidrug resistant associated proteins (MRPs), and breast cancer resistant protein (BCRP). They are usually located on the apical brush border membrane of the syncytiotrophoblasts. Therefore, the intrauterine exposure of the fetus to pravastatin should be determined.
Therefore, the aim of this investigation was to determine the bidirectional transfer of pravastatin across term human placenta, the involvement of efflux transporters, and distribution of pravastatin between placental tissue, maternal and fetal circuits. The aims were achieved by using the ex vivo technique of dual perfusion of placental lobule and in vitro preparations of inside-out vesicles from placental apical membranes.
MATERIALS AND METHODS
Chemicals
[3H]-pravastatin (specific activity, 5 Ci/mmol), [3H]-paclitaxel (specific activity, 45.1 Ci/mmol) and [3H]-estrone sulfate (specific activity, 45.6 Ci/mmol) were purchased from American Radiolabeled Chemicals (St. Louis, MO), Moravek Biochemicals, Inc (Brea, CA), and PerkinElmer (Boston, MA), respectively.
The non-radioactive pravastatin and all other chemicals including radioactive [14C]-antipyrine (specific activity, 6.5 mCi/mmol) were purchased from Sigma-Aldrich (St. Louis, Mo).
Clinical Material
Placentas from uncomplicated term (37 – 42 weeks) singleton pregnancies (n = 21) were obtained immediately after vaginal or abdominal deliveries from the Labor and Delivery Ward of the John Sealy hospital, the teaching hospital of University of Texas Medical Branch, Galveston, Texas, according to a protocol approved by the Institutional Review Board. Exclusion criteria included maternal infections, systemic diseases, multifetal pregnancies, and drug or alcohol abuse during pregnancy.
Transplacental Transfer and Distribution of Pravastatin
To determine the bidirectional transfer of pravastatin from the Maternal to Fetal and Fetal to Maternal circulations and the distribution of pravastatin in each compartment, we used the technique of dual perfusion of placental lobule as previously described by Miller et al. and earlier reports from our laboratory.14,15
Following the control period, the maternal and fetal perfusates were replaced with fresh medium. Human serum albumin, at a concentration of 3 mg/mL, was added to the media of both the maternal and fetal circuits. The non-ionizable, lipophilic marker compound antipyrine 20 μg/mL and its [14C]-isotope (1.5 μCi) were co-transfused with pravastatin to account for interplacental variations and to normalize the transfer of pravastatin to that of antipyrine.
Antipyrine and pravastatin were added either to the maternal or fetal reservoirs according to the transfer direction investigated i.e., from the maternal to fetal (M→F) or fetal to maternal (F→M), respectively. The initial concentration of pravastatin in the donor circuit was 50 ng/mL. This concentration corresponds to the reported serum levels of pravastatin following the administration of 40 mg dose daily.11
The perfusion system was utilized in its closed-closed configuration (re-circulation of the media). The concentrations of compounds were determined in 0.5 mL aliquots taken from the maternal and fetal arteries and veins at 0, 5, 10, 15, 30, 40, 50, 60, 90, 120, 150, 180, 210 and 240 minutes. The amount of radioactivity in both the maternal and fetal perfusate were determined by liquid scintillation spectrometry in the [3H] and [14C]-channels simultaneously (1900TR; Packard Instruments, Inc, Shelton, CT).
At the end of each experiment, the perfused area was dissected from the adjoining placental tissue, weighed, and homogenized in a volume of saline equal to four times its weight. 1 mL of 1M NaOH was added to 1 mL of the homogenate and the samples were incubated for 12 hours at 60 ºC in the dark to allow for luminescence decay. Scintillation cocktail was added to each sample, counted and the concentration of pravastatin calculated.
Effect of pravastatin on the uptake of [3H]-paclitaxel and [3H]-estrone sulfate by the placental vesicles
To assess the involvement of efflux transporters and the functional kinetics of transport mediated by transporters such as P-gp, MRP and BCRP, we evaluated the effect of pravastatin on the ATP-dependent uptake of [3H]-paclitaxel and [3H]-estrone sulfate which are known prototypic substrates of these efflux transporters. The method for preparing inside out vesicles (IOV) from placental brush border membranes is that originally described by Ushigome16 and modified in our laboratory as previously reported.17 The effect of a range of pravastatin concentrations (0–100 μM) on the uptake of 40 nM [3H]-paclitaxel and 100 nM [3H]-estrone sulfate was determined in 140 μL of buffer (46 mM MOPs-TRIS, 65 mM KCl, 8 mM MgCl2, pH 7.4) using 40 μg or 80 μg of membrane protein, respectively. The reactions were initiated by the addition of either 4 mM ATP, or 4 mM AMP for control. The reactions were terminated after 1 or 3 min, respectively by the addition of 3 mL ice cold buffer followed by rapid filtration (Brandel Cell Harvester) to isolate the vesicles. The amount of [3H]-substrates retained on the filter was determined by liquid scintillation analysis. Active uptake was calculated as the difference in the amount of [3H]-substrate in the presence of ATP and AMP. The IC50 was determined from plots of the percent of the [3H]-substrate uptake versus the log of pravastatin concentration.
Data and Statistical Analysis
The maternal to fetal (M→F) and fetal to maternal (F→M) transfer rates (%) were calculated as follows: (Crecipient/Cdonor) x 100, where Crecipient is the final concentration of drug in recipient circuit and Cdonor is the initial concentrations of drug in donor circuit. The clearance index (Clindex) is the ratio between transfer rate of the drug to that of the marker compound antipyrine.
Statistical analyses were performed using GraphPad Prism, version 5.01 (Graph-Pad Software Inc, La Jolla, CA). All reported values are expressed as mean ± SD. Statistical significance of the differences observed between groups were calculated by the Mann-Whitney U-test and considered significant when P <0.05.
RESULTS
Placental Transfer of Pravastatin in the Maternal to Fetal Direction and its Distribution
In this investigation, transplacental transfer of antipyrine was approximately 50%, and is consistent with previous reports from our laboratory.15 Pravastatin rapidly crossed the placenta and appeared in the fetal circuit within the initial five minutes of its perfusion. The concentration of Pravastatin in the fetal circuit at the end of the 4-hour experimental period was 8.8 ± 1.8 ng/mL, and the fetal/maternal concentration ratio was 0.26 ± 0.06. The normalized transfer of Pravastatin (Clearance index) to antipyrine in the Maternal-to-Fetal direction was 0.36 ± 0.07 (Figure 1).
Figure 1.
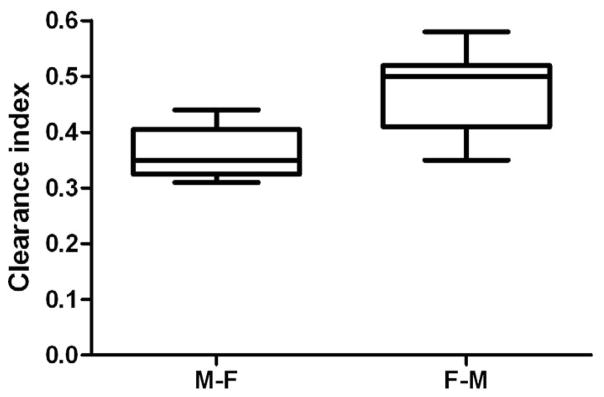
Normalized Maternal to Fetal and Fetal to Maternal transfer of Pravastatin. In order to compare M-F and F-M rate of Pravastatin transfer the initial concentration of the Pravastatin in the donor circuits was similar. Normalized (to antipyrine) transplacental transfer of Pravastatin in the Fetal to Maternal (n=11) direction was statistically higher than its transfer in the opposite Maternal to Fetal (n=10) direction.
The distribution of Pravastatin, at the end of 4-hour perfusion was as follows: 14 ± 5% of the drug was retained by the tissue, 68 ± 5% remained in the maternal circuit, and 18 ± 4% was transferred to the fetal circuit.
Placental Transfer of Pravastatin in the Fetal to Maternal Direction and its Distribution
Following the addition of Pravastatin to the fetal (donor) reservoir, the drug rapidly crossed the placenta and appeared in the maternal (recipient) circuit in less than 5 minutes. At the end of the 4 hours of perfusion period, the concentration of Pravastatin in the maternal circuit reached 6.4 ± 1.0 ng/mL which represents 13 ± 2% of its initial concentration in the donor reservoir. The maternal/fetal concentration ratio of Pravastatin was 0.19 ± 0.03, and the normalized transfer of Pravastatin to antipyrine in the Fetal-Maternal direction was 0.48 ± 0.07 which was significantly higher (p < 0.01) from its transfer from the Maternal to Fetal direction (Figure 1).
Furthermore, the retention of pravastatin by the perfused lobule at the end of the 4-hour experimental period reached 20 ± 4% of its initial concentration in the donor circuit.
Effect of pravastatin on the uptake of [3H]-paclitaxel and [3H]-estrone sulfate by inside-out vesicles prepared from placental apical membranes
Pravastatin inhibited the ATP-dependent uptake of [3H]-paclitaxel and of [3H]-estrone sulfate (Figure 2 A&B) with IC50 values of 11 μM and 30 μM, respectively.
Figure 2.
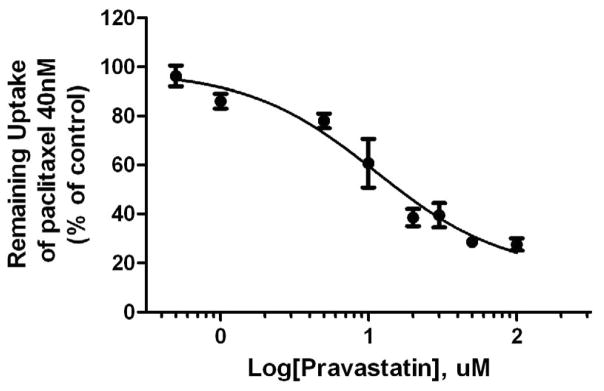
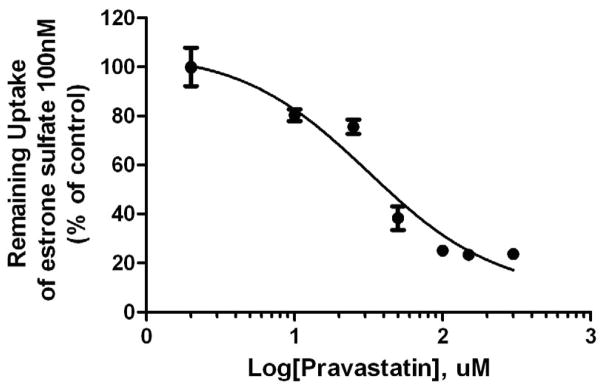
Figure 2A. Inhibition of ATP-dependent transport of [3H]-paclitaxel in placental IOV by pravastatin.
Pravastatin inhibited ATP-dependent uptake of paclitaxel (40 nM) with the IC50 values of 11 μM. Each point represents the mean ± S.D. from 3 experiments in duplicate.
Figure 2B. Inhibition of ATP-dependent transport of [3H]-estrone sulfate in placental IOV by pravastatin.
Pravastatin inhibited ATP-dependent uptake of estrone sulfate (100 nM) with the IC50 values of 30 μM. Each point represents the mean ± S.D. from 3 experiments in duplicate.
COMMENT
The successful use of pravastatin for treatment of cardiovascular disease which shares several pathophysiologic pathways associated with preeclampsia provided biological plausibility for its use to prevent preeclampsia. However, today the data on human use of pravastatin during pregnancy is limited.
The aim of this investigation was to determine the bidirectional transfer of pravastatin across term human placenta, the involvement of efflux transporters, and distribution of pravastatin between placental tissue, maternal and fetal circuits.
The data obtained in this investigation revealed that pravastatin crosses the human placenta in both the Maternal to Fetal (18%) and Fetal to Maternal (13%) directions. The transfer of pravastatin in both directions suggests that intrauterine fetal exposure to the drug is possible and that the main route for drug elimination from the fetus is its transfer back to the maternal circulation.
The observed difference in the extent of pravastatin transfer in Maternal to Fetal versus Fetal to Maternal directions is attributed to the differences in the experimental conditions as previously discussed18 and was normalized to the transfer of the marker compound antipyrine because its transfer rate is proportionately affected by the same experimental variables. The normalized transfer of pravastatin in the Fetal to Maternal direction 0.48 ± 0.07 was significantly higher than its transfer in the opposite direction 0.36 ± 0.07, p < 0.01 (Figure 1) suggesting the involvement of placental efflux transporters localized on the apical membranes. The data obtained from perfusion experiments were further substantiated by in vitro experiments which revealed that pravastatin inhibited the ATP-dependent uptake of [3H]-paclitaxel (Figure 2A) and [3H]-estrone sulfate (Figure 2B) the known prototypic substrates of efflux transporters.17,19 These results are in agreement with previous reports indicating that pravastatin is a predominant substrate of the multidrug resistance-associated protein 2 (MRP2)20 and breast cancer resistant protein (BCRP).21,22 These transporters restrict drug transfer from the maternal circulation to the fetus and thus may protect the fetus from exogenous or endogenous toxic substrates. Other placental transporters23,24 that could also contribute to efflux of pravastatin are P-glycoprotein (P-gp)25 and a bile salt export pump (BSEP).26 The expression of these transportes was not studied in this paper, and should be further investigated.
The low lipophilicity of pravastatin12 and its extrusion by placental efflux transporters back into the maternal circuit suggests that its transfer across the placenta to the fetal circuit should be limited. However, the higher than anticipated transfer rate of pravastain in the Maternal to Fetal direction suggests that placental uptake transporters could be important regulators of pravastatin transfer across the placenta. The method of dual perfusion of placental lobule has been validated over the years and proven to mimic drug transfer in vivo during last few weeks of gestation, but it also has its limitations. The limitations include; the absence of maternal and fetal tissue that could affect drug distribution; the absence of maternal and fetal hormones and enzymes that might influence placental disposition of the medication; data obtained cannot be extrapolated to earlier gestational age placentas. The contribution of placental uptake transporters to the bio-distribution of pravastatin in feto-placental compartment should be further investigated.
The main reason why pravastatin (in particular) and statins in general were classified as FDA category X was due to lack of indications for their use in pregnancy, and concerns about their safety. Recently, there has been a lot of interest in the use of pravastatin to prevent preeclampsia in high risk women. In fact the Eunice Kennedy Shriver National Institute of Child Health and Human Development Obstetric-Fetal Pharmacology Research Units Network started a double blinded placebo controlled pilot trial to collect preliminary maternal-fetal safety data for pravastatin during pregnancy, and evaluate its pharmacokinetics when used as a prophylactic daily treatment in high-risk pregnant women (ClinicalTrials.gov Identifier NCT01717586). Pravastatin has not been shown to be teratogenic in animal and human cohort studies. In addition, data from the long term cardiovascular trials in non-pregnant women and men suggest a favorable maternal safety and pharmacokinetic profiles. We believe that the data presented in this manuscript will add strength to what is already available and support the push for reclassification of pravastatin.
Acknowledgments
Source of financial support: Obstetric-Fetal Pharmacology Research Units (OPRU) network (NICHD U10-HD 047891).
The authors sincerely appreciate the support of the physicians and nurses of the Labor & Delivery Ward of the John Sealy Hospital, the teaching hospital at UTMB, Galveston, Texas, the Perinatal Research Division, and the Publication, Grant, & Media Support division of the Department of Obstetrics & Gynecology.
Footnotes
Disclosure: None of the authors have a conflict of interest.
Presentation information: This work was presented at Society for Maternal-Fetal Medicine, 33rd Annual Meeting, San Francisco, CA, February 11–16, 2013
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shepherd J, Cobbe SM, Ford I, et al. West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia 1995. Atheroscler Suppl. 2004;5(3):91–7. doi: 10.1016/j.atherosclerosissup.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 2.The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;5;339(19):1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 3.Costantine MM, Cleary K. Eunice Kennedy Shriver National Institute of Child Health and Human Development Obstetric--Fetal Pharmacology Research Units Network. Pravastatin for the prevention of preeclampsia in high-risk pregnant women. Obstet Gynecol. 2013;121(2 Pt 1):349–53. doi: 10.1097/aog.0b013e31827d8ad5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drugs.com [Internet] Pravastatin Pregnancy and Breastfeeding Warnings. Drug Information Online. Copyright 1996–2012 Cerner Multum, Inc. Version: 12.02. [Revision Date: 2012-03-21, 9:32:12 AM]. Available from: http://www.drugs.com/pregnancy/pravastatin.html.
- 5.Tanase TM, Hirose K. Reproduction studies of pravastatin sodium administered during the period of fetal organogenesis in rats. Yakuri to Chiryo. 1987;15(12):4883–94. [Google Scholar]
- 6.Tanase TM, Asai M, Hirose K. Reproduction studies of pravastatin sodium administered during the period of fetal organogenesis in rabbits. Yakuri to Chiryo. 1987;15(12):5005–11. [Google Scholar]
- 7.Teelucksingh S, El-Youssef J, Sohan K, Ramsewak S. Prolonged inadvertent pravastatin use in pregnancy. Reprod Toxicol. 2004;18(2):299–300. doi: 10.1016/j.reprotox.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Taguchi N, Rubin ET, Hosokawa A, et al. Prenatal exposure to HMG-CoA reductase inhibitors: effects on fetal and neonatal outcomes. Reprod Toxicol. 2008;26(2):175–7. doi: 10.1016/j.reprotox.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Hosokawa A, Bar-Oz B, Ito S. Use of lipid-lowering agents (statins) during pregnancy. Can Fam Physician. 2003;49:747–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Edison RJ, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outscomes following gestational exposure to statins. Am J Med Genet. 2004;131(3):287–981. doi: 10.1002/ajmg.a.30386. [DOI] [PubMed] [Google Scholar]
- 11.Hatanaka T. Clinical pharmacokinetics of pravastatin: mechanisms of pharmacokinetic events. Review Clin Pharmacokinet. 2000;39(6):397–412. doi: 10.2165/00003088-200039060-00002. [DOI] [PubMed] [Google Scholar]
- 12.Hamelin BA, Turgeon J. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Review. Trends Pharmacol Sci. 1998 Jan;19(1):26–37. doi: 10.1016/s0165-6147(97)01147-4. [DOI] [PubMed] [Google Scholar]
- 13.Singhvi SM, Pan HY, Morrison RA, Willard DA. Disposition of pravastatin sodium, a tissue-selective HMG-CoA reductase inhibitor, in healthy subjects. Br J Clin Pharmacol. 1990;29(2):239–43. doi: 10.1111/j.1365-2125.1990.tb03626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller RK, Wier PJ, Maulik D, di Sant’Agnese PA. Human placenta in vitro: characterization during 12 h of dual perfusion. Contrib Gynecol Obstet. 1985;13:77–84. [PubMed] [Google Scholar]
- 15.Nanovskaya T, Deshmukh S, Brooks M, Ahmed MS. Transplacental transfer and metabolism of buprenorphine. J Pharmacol Exp Ther. 2002;300(1):26–33. doi: 10.1124/jpet.300.1.26. [DOI] [PubMed] [Google Scholar]
- 16.Ushigome F, Koyabu N, Satoh S, et al. Kinetic analysis of P-glycoprotein-mediated transport by using normal human placental brush-border membrane vesicles. Pharm Res. 2003;20(1):38–44. doi: 10.1023/a:1022290523347. [DOI] [PubMed] [Google Scholar]
- 17.Hemauer SJ, Patrikeeva SL, Nanovskaya TN, Hankins GD, Ahmed MS. Opiates inhibit paclitaxel uptake by P-glycoprotein in preparations of human placental inside-out vesicles. Biochem Pharmacol. 2009 Nov 1;78(9):1272–8. doi: 10.1016/j.bcp.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanovskaya TN, Patrikeeva S, Zhan Y, Hankins GD, Ahmed MS. Transplacental transfer of oseltamivir carboxylate. J Matern Fetal Neonatal Med. 2012;25(11):2312–5. doi: 10.3109/14767058.2012.693993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson JE, Heddle C, Rozkov A, et al. High-activity p-glycoprotein, multidrug resistance protein 2, and breast cancer resistance protein membrane vesicles prepared from transiently transfected human embryonic kidney 293-epstein-barr virus nuclear antigen cells. Drug Metab Dispos. 2010;38(4):705–14. doi: 10.1124/dmd.109.028886. [DOI] [PubMed] [Google Scholar]
- 20.Yamazaki M, Kobayashi K, Sugiyama Y. Primary active transport of pravastatin across the liver canalicular membrane in normal and mutant Eisai hyperbilirubinaemic rats. Biopharm Drug Dispos. 1996;17(8):645–59. doi: 10.1002/(SICI)1099-081X(199611)17:8<645::AID-BDD986>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 21.Matsushima S, Maeda K, Kondo C, et al. Identification of the hepatic efflux transporters of organic anions using double-transfected Madin-Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistance-associated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast cancer resistance protein. J Pharmacol Exp Ther. 2005;314(3):1059–67. doi: 10.1124/jpet.105.085589. [DOI] [PubMed] [Google Scholar]
- 22.Elsby R, Smith V, Fox L, et al. Validation of membrane vesicle-based breast cancer resistance protein and multidrug resistance protein 2 assays to assess drug transport and the potential for drug-drug interaction to support regulatory submissions. Xenobiotica. 2011 Sep;41(9):764–83. doi: 10.3109/00498254.2011.578761. [DOI] [PubMed] [Google Scholar]
- 23.Vähäkangas K, Myllynen P. Drug transporters in the human blood-placental barrier. Br J Pharmacol. 2009 Oct;158(3):665–78. doi: 10.1111/j.1476-5381.2009.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano MA, Macias RI, Briz O, et al. Expression in human trophoblast and choriocarcinoma cell lines, BeWo, Jeg-3 and JAr of genes involved in the hepatobiliary-like excretory function of the placenta. Placenta. 2007 Feb-Mar;28(2–3):107–17. doi: 10.1016/j.placenta.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Chen C, Mireles RJ, Campbell SD, et al. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005 Apr;33(4):537–46. doi: 10.1124/dmd.104.002477. [DOI] [PubMed] [Google Scholar]
- 26.Hirano M, Maeda K, Hayashi H, Kusuhara H, Sugiyama Y. Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J Pharmacol Exp Ther. 2005;314(2):876–82. doi: 10.1124/jpet.105.084830. [DOI] [PubMed] [Google Scholar]