

Abstract
Induction of experimental autoimmune encephalomyelitis (EAE) in susceptible animals requires reactivation of encephalitogenic CD4+ T cells by antigen presenting cells (APCs) in the central nervous system (CNS). However, it has remained unresolved from where APCs in the CNS acquire myelin antigen (Ag) for T cell activation and under which conditions, i.e. whether only during EAE or also in the naïve CNS. Here, we investigated the kinetics of myelin Ag uptake by CNS APCs during EAE and in the naïve CNS. Our results show that during EAE CX3CR1+CD11b+ microglia were the first APCs in the CNS to contain myelin Ag upon induction of disease, albeit in very small numbers. Dendritic cells (DCs) arrived in the CNS in sizable numbers significantly later (day 5 post-immunization (p.i.)), without detectable myelin Ag, but acquired it by day 7 p.i. Furthermore, a sharp increase in neuroantigen-containing DCs coincided with the onset of EAE symptoms. Importantly, in naïve mice a low but consistent number of microglia contained myelin Ag, suggesting release by oligodendrocytes under steady state conditions. Although microglia isolated from naïve brain and spinal cord did not elicit a strong CD4+ T cell response in vitro, myelin Ag-containing microglia may still play a local role in modulating encephalitogenic CD4+ T cell responses in early EAE prior to the arrival of other professional APCs such as DCs. Finally, newly arriving DCs in the CNS not yet loaded with myelin Ag before the onset of EAE may be a potential therapeutic target.
Keywords: EAE, T cells, antigen presenting cells, myelin basic protein, microglia localization
INTRODUCTION
Multiple Sclerosis (MS) is the most common autoimmune disease of the central nervous system (CNS), affecting over 400,000 people in the United States alone (1). Hallmarks of the disease are focal demyelinated lesions and transection of axons which are believed to be mediated by infiltrating inflammatory cells including CD4+ and CD8+ T cells, B cells, and antigen presenting cells (APCs) such as dendritic cells (DC) and macrophages (2–5).
Autoreactive CD4+ T cells are thought to be important contributors to the disease process (6). To mediate disease, myelin-reactive CD4+ T cells have to migrate to the CNS where they encounter their cognate myelin antigen (Ag) presented by MHC class II molecules on APCs and become activated and exert their effector functions (7, 8). Along these lines, it has been shown that impaired expression of MHC class II molecules in the CNS abrogates EAE, as does depletion of APCs (9–11). Potential APCs found in the CNS during EAE and MS are tissue-resident microglia as well as infiltrating DCs, macrophages, B cells, and neutrophils (12–14). Parenchymal microglia rapidly upregulate MHC Class II molecules in response to a variety of inflammatory stimuli, suggesting that they may play a role in Ag presentation in the CNS (15–17); however many current molecular markers used to identify microglia are also present on peripheral macrophages which makes it difficult to ascertain their origin. CNS-resident astrocytes have been shown in vitro to upregulate MHC Class II molecules and present Ag to T cells (18), however this has not been demonstrated in vivo. Additionally, they do not upregulate the costimulatory molecules CD80 or CD86 (19), and therefore may be unable to activate T cells. It has been shown that infiltrating DC are critical for Ag presentation to encephalitogenic T cells in EAE (11, 17, 20). Recently it has been suggested that during the early stages of EAE, myelin Ag presentation takes place in the subarachnoid space and leptomeninges (21).
Presumably, this would require the translocation of myelin Ag from oligodendrocytes/myelinated axonal membranes to the perivascular spaces for Ag presentation to occur.
Oligodendrocytes represent the most likely source of myelin Ag presented by APCs to CD4+ T cells in MS and its animal model experimental autoimmune encephalomyelitis (EAE) as they are the CNS-resident cells that are responsible for myelination by extending their own membranes around neuronal axons (22). However, oligodendrocytes themselves do not express MHC Class II molecules and thus cannot directly present myelin Ag to CD4+ T cells (23–26). Therefore, an important unanswered question is from where and under which conditions myelin Ag is translocated to APCs in the CNS for Ag presentation and the activation of pathogenic CD4+ T cells. Furthermore, little is known about the kinetics and cellular distribution of myelin Ag uptake by APCs in the naïve CNS or during disease conditions.
Here we show that myelin Ag was constitutively present in a small number of CNS-resident microglia in naïve mice. Upon induction of EAE, CD4+ T cells, DCs, neutrophils and macrophages infiltrated the CNS in increasing numbers. Myelin Ag was not detected in DCs infiltrating the CNS before day 5 after induction of EAE, whereas as a small percentage of microglia contained myelin Ag already by day one after immunization. Onset of EAE coincided with a sudden spike in the number of infiltrating DCs and macrophages in the CNS, the majority of which contained myelin Ag. In contrast, disease remission was paralleled by a strong decline in CNS APCs associated with myelin Ag. Our data suggest that myelin uptake by CNS APCs is a dynamic process shaped by inflammatory cells during the course of disease.
MATERIALS AND METHODS
Animals, immunization, and EAE scoring
Six to eight week old female C57BL/6 and SJL mice were obtained from The Jackson Laboratory. 2D2 transgenic (tg) mice, DRB1*15:01 tg mice, CX3CR1+/GFP mice, and HLA-DR2 tg shiverer (Mbp−/−) mice were bred in-house and all animals were maintained in pathogen free conditions under the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas at San Antonio. Mice were fed and watered ad libitum. To induce EAE, six to eight week old mice were immunized subcutaneously (s.c.) with 300 μg MOG35-55 peptide emulsified 1:1 in Complete Freund’s Adjuvant (CFA) containing 5 mg/ml Mycobacterium tuberculosis. Pertussis toxin (PTX) (400 ng/ml) was given intraperitoneally (i.p.) on day 0 and day 2 relative to immunization. Clinical scores were monitored daily and assigned a score based on the following symptoms: 0, no clinical disease; 1, flaccid tail; 2, partial hind limb paralysis; 3 total hind limb paralysis; 4, front and hind limb paralysis; 5, moribund or dead. For adoptive transfer, donor SJL mice were immunized s.c. with 200 μg PLP139-151 peptide emulsified in CFA. On day 10, inguinal and popliteal lymph nodes and spleens were obtained and single cell suspensions were prepared. Cells were adjusted to a concentration of 1×107 cells/ml in Dulbecco’s Modified Essential Media (DMEM) containing 10% FCS and 1% L-glutamine and placed into a 24 well culture plate with 10 μg/ml PLP139-151 Ag and 20 ng/ml IL-23 and placed into a 37°C incubator with 5% CO2. On day 4, cells were removed from culture, washed two times and resuspended in serum free media, and 2×107 cells were then injected i.p. into each recipient SJL mouse.
Immunofluorescence staining, antibodies and quantification
Antibodies to myelin basic protein (MBP, 7H11), myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein (PLP) were purchased from Neuromics. Fluorochrome conjugated antibodies to CD4 (GK1.5), CD11b (M1/70), CD11c (N418), CD19 (1D3), DEC205 (205yekta), I-A/I-E (MHC class II, NIMR-4, M5/114/15/2 or 10–3.6 as haplotype-appropriate) were purchased from eBioscience, Inc. Ly-6G (1A8) was purchased from BD Biosciences. 4′,6-diamidino-2-phenylindole (DAPI) was purchased from Sigma Aldrich. Immunofluorescent staining was performed as follows: Murine brains and spinal cords were obtained at designated time points, placed in OCT freezing compound and frozen at −80°C. Tissue was cut into 4–20 μm thick transverse sections and staggered onto glass slides. Alternatively, tissue was cut into 20–60 μm-thick transverse sections and placed in fixative solution in 24-well non-tissue culture treated plates (Fisher) and placed on slides following staining procedure. Sections were fixed (fixation buffer, eBioscience) on ice, then permeabilized (permeabilization buffer, eBioscience) and blocked with appropriate serum prior to staining. Purified primary antibodies were allowed to incubate on sections overnight at 4°C in a closed, humid chamber. Secondary antibodies and directly labeled primary antibodies were then added for an hour at room temperature (RT) in a closed, humid chamber. Slides were rinsed in between incubations with either phosphate buffered saline (PBS), PBS+0.05% Tween-20 (PBST) or PBS+0.01% Triton-X (PBT) depending on antigen of interest. Coverslips were mounted with ProLong Gold Antifade Reagent (Invitrogen) and slides were allowed to dry and cure overnight at RT in the dark. Images were acquired on a Zeiss LSM510 confocal laser scanning microscope using Zen 2009 (Zeiss) acquisition software. Image analysis was performed using Imaris 3D/4D software version 7.2 (Bitplane) as described below.
In vitro titration and detection of myelin antigen by confocal microscopy
For in vitro titration of myelin antigens, naïve splenocytes were obtained from six to eight week old C57BL/6 mice and processed into single cell suspensions. CD11c+ dendritic cells were positively selected for using magnetic separation on AutoMACS (Miltenyi, 94.1% purity). Additionally, single cell splenocyte suspensions from 2D2 tg mice were enriched for MOG35-55 peptide-reactive CD4+ T cells using negative selection on AutoMACS (Miltenyi, 95.3% purity). CD11c+ and CD4+ cells were cultured overnight with increasing concentrations of recombinant rat MOG (rMOG) protein alone or in combination with 100 ng/ml of lipopolysaccharide (LPS). Cells were spread onto positively charged “plus” slides (Fisher) at a concentration of 1×107 cells per ml and stained according to the protocol described in immunofluorescence staining section. Slides were imaged with a Zeiss LSM510 confocal laser scanning microscope and analyzed using Imaris software.
Reconstructing Neuroantigen Loading in Three-Dimensions
The three-dimensional structure of APCs in the CNS was reconstructed from confocal laser scanning microscopy z-stack images taken 0.2–0.3 μm apart from 1.2 to 59.6 μm in depth using Imaris software depending on thickness of CNS tissue being imaged. Quantification of the absolute number of APCs was performed for each image by taking advantage of the fact that Imaris can recognize the center of mass in each nucleus. The number of nuclei present in each 3D stacked image which could be radially expanded to co-localize with APC cell surface markers within 5–10 μm in >75% of all possible directions were counted as APCs using Imaris 7.2 software. To distinguish myelin Ag that was associated with an APC, including myelin Ag colocalizing to the cell surface, from Ag in the extracellular microenvironment, APCs were digitally enclosed using the reconstructed 3D surface (Supplemental Fig. S2).
Cytokine ELISPOT
Cytokine ELISPOT assays were performed as described previously (27). Briefly, ELISPOT plates (Multiscreen IP; Millipore) were coated with 1μg/ml IFN-γ-specific (AN-18; eBioscience) or IL-17-specific capture mAb (17F3; Bio X Cell) diluted in PBS. The plates were blocked with 1% BSA in PBS for 1 h at room temperature and then washed four times with PBS. Cells were added with or without Ag and incubated for 24 h at 37°C. The plates were washed three times with PBS and four times with PBST, and IFN-γ-specific biotinylated detection mAb (R4-6A2, eBioscience) or IL-17-specific biotinylated detection mAb (TC11-81-14; BioLegend) was added and allowed to incubate overnight. The plate was washed four times with PBST and incubated with streptavidin-alkaline phosphatase (Invitrogen). Cytokine spots were visualized by 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium phosphatase substrate (Kirkegaard & Perry Laboratories). Image analysis of ELISPOT assays was performed on a Series 2 ImmunoSpot analyzer and software (Cellular Technology) as described previously (28). In brief, digitized images of individual wells of the ELISPOT plates were analyzed for cytokine spots, based on the comparison of experimental (containing T cells and APC with Ag) and control wells (T cells and APC, no Ag). After separation of spots that were touching or partially overdeveloped, nonspecific noise was gated out by applying spot size and circularity analysis as additional criteria. Spots that fell within the accepted criteria were highlighted and counted.
Flow Cytometry and Cell Sorting
Spleen, lymph nodes, brain and/or spinal cord tissues were removed from naïve mice following cardiac perfusion or EAE mice without perfusion as previously described (29). Single cell suspensions were obtained from tissue by mechanical isolation. Extracellular myelin removal was performed for all brain and spinal cord suspensions according to manufacturer’s instructions. (Miltenyi Biotec). CD11b+ microglia were sorted on a BD FACSAria II (BD Bioscience) (96.5% purity) from brain and spinal cord of naïve C57BL/6 mice following myelin removal on AutoMACS (Miltenyi). For myelin-reactive CD4+ T cells, single cell suspensions were prepared from draining lymph nodes of MOG35-55 immunized C57BL/6 mice, and then enriched for CD4+ T cells using negative selection with magnetic beads on AutoMACS (Miltenyi) followed by positive selection using FACS on BD FACSAria II (BD Bioscience) at a final purity of 99.7%. For surface staining, cell suspensions were blocked with 2% mouse serum or Fc block (eBioscience) for 20 min on ice, then stained with directly labeled antibodies for 45 min. Following staining, cells were washed with 10X volume of PBS and fixed using fixative agent (eBioscience). For intracellular staining, fixed cells were permeabilized in 1X permeabilization buffer (eBioscience) for 15 min., then incubated with directly labeled antibodies for 45 min. Cells were then washed with 10X volume of 1X permeabilization buffer and analyzed or sorted on a BD FACSAria II (BD Bioscience). Data were analyzed with FACSDiva software (BD Bioscience).
Statistical Analysis
Statistical evaluation was performed using JMP SAS 8 software. Comparison across groups of infiltrating and MBP-associated cells was evaluated using analysis of covariance (ANOVA). T cell activation was evaluated using a 2-tailed Student’s t test. p values ≤ 0.05 were considered significant.
RESULTS
Distribution of potential APCs and MHC class II in the CNS during EAE and in naïve mice
It has remained unresolved which APCs are the most critical for presentation of neuroantigen to CD4+ T cells for induction of EAE, and whether myelin Ag is presented in the naïve CNS and by which APCs. To begin to address these questions, we investigated the presence of cells with the potential to present myelin Ag based on myeloid lineage and expression of MHC class II molecules in the CNS of naïve mice and animals with EAE.
EAE was induced in C57BL/6 mice with MOG35-55 peptide and animals were evaluated for clinical disease as described (2). In parallel, a group of age and gender matched C57BL/6 mice was maintained unimmunized. The distribution of potential APCs and other inflammatory cells was examined in brain tissue slices of these mice by H&E (Fig. 1A, H) and immunofluorescence (Fig. 1B–G; 1I–N) staining at peak EAE (usually around day 21 p.i). As expected, inflammatory infiltrates containing large numbers of CD4+ T cells were detected in the CNS of C57BL/6 mice with acute EAE, in contrast to the few meningeal-restricted cells found in naïve animals (Fig. 1B versus 1I). CD11c+ DCs were abundantly present and clustered in CNS lesions in mice with EAE, whereas they were exceedingly rare in the CNS of naïve mice and only found contained within the meninges (Fig. 1C, 1J). Large clusters of CD11b+ cells which included microglia and infiltrating macrophages were found in the highest numbers during EAE (Fig. 1D). They were distributed evenly and occurred in lower numbers mostly throughout the parenchymal white matter in CNS of naïve mice and represent the resident microglial population (Fig. 1K, Fig. 6). Similarly, neutrophils (Ly-6G+) were virtually absent from the CNS of naive mice, while they were detected in low numbers in mice with EAE (Fig. 1E, 1L). CD19+ B cells were not detected in notable numbers in the CNS of mice with EAE in this study (Fig. 1F, 1M). Finally, expression of MHC class II molecules was upregulated in inflammatory lesions in the CNS of mice with EAE, whereas it was rarely detected in the CNS of naïve mice (Fig. 1G, 1N), and only on CD11b+ microglia.
FIGURE 1.
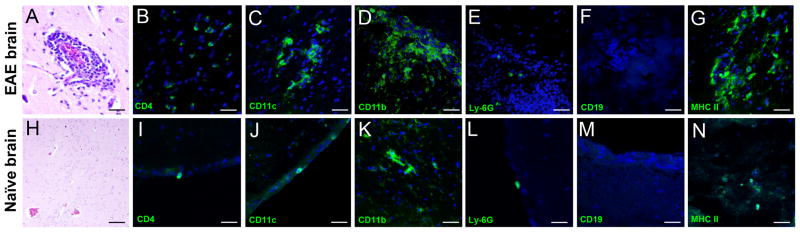
Detection of inflammatory cells and MHC class II expression in the CNS. Brains were obtained from EAE mice at the peak of clinical EAE (day 21 pi, top row), and naïve mice (bottom row). (A and H) Hematoxylin and eosin (H&E) staining of an inflammatory perivascular focus in CNS. Confocal images of IF staining (green) for (B and I) CD4+ T cells; (C and J) CD11c+ dendritic cells; (D and K) CD11b+ microglia/infiltrating macrophages; (E and L) Ly-6G+ neutrophils; (F and M) CD19+ B cells; and (G and N) MHC II+ cells. Cell nuclei are stained with DAPI. As expected, only microglia were present in significant numbers in the naïve parenchyma (K), whereas other cell types were found only in meninges prior to immunization (I, J and L). (F and M) CD19+ B cells were not detected in the CNS of naïve or EAE mice. (G and N) IF staining for MHC class II molecule shows increased expression during EAE while expression is low in the naïve brain.
FIGURE 6.
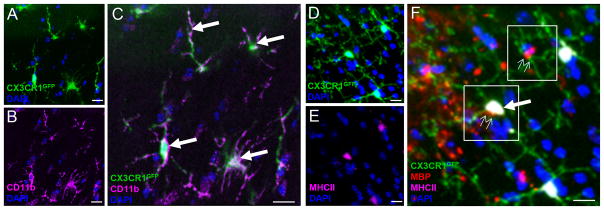
Detection of myelin Ag in CNS-resident microglia in naïve brain. (A–C) Expression of markers CX3CR1 and CD11b in brain tissue sections from naïve CX3CR1+/GFP mice (A) CX3CR1 (green); (B) CD11b (pink); (C) merge A+B. CX3CR1+CD11b+ cells are indicated by arrows. (D–F) CX3CR1+CD11b+ microglia containing MBP (F, small arrows, cells positive for red signal); some of these are also MHCII+ (F, large arrow). Scale bars = 10 μm.
Thus, activated microglia/macrophages and DCs were the predominant potential APCs in CNS lesions in the MOG35-55 peptide-induced EAE model in our studies. In contrast, microglia were the only potential APC identified in the CNS parenchyma of naïve mice.
Visualizing uptake of neuroantigen by myeloid cells in the CNS during EAE
Next, we asked which myeloid cells contained myelin Ag during EAE and whether it was presented on MHC II molecules for T cell activation. To address this issue, EAE was induced in C57BL/6 mice with MOG35-55 peptide or via adoptive transfer of PLP139-151-reactive T cells in SJL mice as previously described (30). CNS tissue was obtained at the peak of disease and analyzed by confocal microscopy for colocalization of MBP, MOG, or PLP with myeloid cells.
Shown in Fig. 2A–C are representative confocal images of MBP, MOG, or PLP expression in the CNS of naïve mice. Specificity of staining was confirmed using CNS tissue from MBP-deficient shiverer mice which, as expected, did not show MBP staining (Fig. 2D). Importantly, all three myelin Ag colocalized with DCs and microglia/macrophages in EAE mice (Fig. 2E–H, arrows), and were detected within DCs and macrophages/microglia (Fig. 2I). A similar kinetic of colocalization of myelin antigens (e.g. MBP, MOG, PLP) with DCs and microglia was observed when EAE was induced by adoptive transfer of PLP139-151-specific T cells into SJL mice (Supplemental Fig. S1). Consistent with our earlier results, infiltrating CD11b+ inflammatory macrophages only acquired myelin Ag (e.g. PLP) upon entering the parenchymal white matter at the onset of EAE (day 7; Supplemental Fig. S1A). Shown in Fig. 2I, MBP was clearly detectable within APCs at high magnification (100X; MBP+) and could easily be distinguished from APCs that did not contain MBP in internal slices of z-stacks made from each lesion area (Fig. 2I, MBP−; Supplemental Fig. S3). Only a small percentage of neutrophils in the CNS showed staining for MBP (Fig. 4E) or MOG (not shown), arguing against a major role of these phagocytic cells for the presentation of myelin Ag during EAE (Fig. 4).
FIGURE 2.

Myelin antigen localization to myeloid cells in naïve and EAE CNS. (A) MBP (B) MOG and (C) PLP staining in naïve mice cerebellum showing characteristic staining pattern for white matter myelin tracts. (D) MBP staining in naïve MBP−/− shiverer mice confirms specificity of IF staining. (E–H) Myelin Ag and APCs colocalize at lesions during peak of disease. (I) APCs containing MBP (MBP+) can be distinguished from those without (MBP−) in internal z-stack slices. (J) MK16 Ab colocalizes with MBP at APCs. (K) CD4+ T cells in close proximity to MBP+ APCs in and around lesions. Scale bars = 50 μm (A–D, K), 20 μm (E–H) and 5 μm (I and J).
FIGURE 4.

Myelin Ag is first detected in microglia in the CNS during EAE. C57BL/6 mice were immunized with MOG35-55 peptide for induction of EAE as previously described. Animals were monitored and disease scored daily. (A) Representative mice were scored for EAE and sacrificed as indicated over the course of EAE (n = 3 per day) and brain tissue frozen and sectioned. Tissue sections were stained as indicated and evaluated by confocal microscopy analysis for (B) CD4+ T cells, (C) CD11c+, (D) CD11b+, or (E) Ly-6G+ cells. Cells in (C–E) were also stained with anti-MBP mAb. Numbers shown represent mean ± S.D., ANOVA (p≤0.002).
To determine if myelin Ag detected in CNS APCs was processed and loaded onto MHC II molecules for Ag presentation to T cells, we took advantage of the MK16 antibody, which specifically recognizes the MBP85-99 peptide bound to HLA-DR2 molecules (31). EAE was induced in HLA-DR2b (DRB1*15:01) tg mice with MOG35-55 peptide and CNS tissue was obtained at the peak of disease for confocal imaging. Staining of brain tissue from HLA-DR2b tg mice with EAE with anti-MBP and anti-CD11c and -CD11b mAb confirmed that MBP colocalized with APCs in a pattern similar to that observed in C57BL/6 mice (Fig. 2J). Importantly, MK16 staining colocalized to APCs (Fig. 2J, large arrow) and overlapped with the MBP+ staining (Fig. 2J, small arrow, box). In contrast, no MK16 staining above background levels was detected in HLA-DR2b tg shiverer mice (MBP−/−) or HLA-DR4 tg mice with EAE (not shown), confirming the specificity of MK16 staining. Finally, CD4+ T cells (small arrow) were found adjacent to CNS APCs containing MBP, consistent with Ag presenting function by these cells (Fig. 2K).
Taken together, the results show that myelin Ag is primarily taken up and presented by CD11b+ microglia/infiltrating macrophages and CD11c+ DCs during EAE. The data suggest that the contribution of other potential APCs to neuroantigen presentation in the CNS is minor during the clinical disease course.
Detection of myelin antigen in APCs by confocal microscopy corresponds to T cell activation threshold for cytokine production
We showed that myelin Ag, e.g. MBP and MOG, were detected by confocal microscopy in several APC subsets in the CNS over the course of EAE. However, it remained unresolved how sensitive this technique was; for example, was myelin Ag detected by confocal microscopy in CNS APCs at levels that could be considered sufficient for the activation of encephalitogenic T cells?
To begin to address this issue we developed a semi-quantitative in vitro assay that replicated confocal imaging of myelin Ag in CNS tissue. Splenocytes were pulsed with recombinant rat MOG1-125 protein, and cell spreads were prepared for confocal imaging in order to determine the concentration of MOG that could be detected in APCs. In parallel, MOG-specific 2D2 T cells were incubated with MOG1-125 protein-pulsed splenocytes to determine by cytokine ELISPOT assay for IFN-γ, IL-17, and GM-CSF at what Ag concentrations T cell responses were induced.
The results show that MOG protein could be detected internalized within cells by internal slices of z-stacks taken by confocal microscopy starting at 0.01 μg/ml of Ag (Fig. 3A, arrows). Importantly, as determined by cytokine ELISPOT assay, this concentration of MOG protein was suboptimal for the activation of MOG35-55-specific T cells for the production of IFN-γ, IL-17, and GM-CSF (Fig. 3B). At 1 μg/ml of Ag, similarly strong results were detected by confocal microscopy as well as cytokine ELISPOT assay (Fig. 3A, 3B).
FIGURE 3.

MOG detection by confocal microscopy corresponds to activation threshold of pathogenic T cells. CD11c+ cells from Wt B6 mice and CD4+ cells from 2D2 TCR-tg mice were sorted by FACS and co-cultured in a 1:1 ratio with increasing concentrations of rMOG1-125 protein. (A) Detection of increasing levels of rMOG in CD11c+ DCs by confocal microscopy; shown is internal slice from z-stack depicting inside of cells; scale bars = 20 μm (B) Detection of MOG-specific T cell responses by cytokine ELISPOT assay upon incubation with DCs pulsed with increasing concentrations of rMOG protein. Shown is mean ± S.D. of number of cytokine spots over three separate experiments with three replicates each.
Taken together, the results suggested that confocal microscopy was a highly sensitive technique for detection of myelin Ag internalization by APCs. Since myelin Ag was detected in vitro by confocal microscopy in APCs at concentrations below optimal T cell activation threshold the data suggests that this technique could similarly detect myelin Ag in CNS APCs at low concentrations, conceivably at concentrations that were not sufficient to induce optimal activation of encephalitogenic T cells in situ.
Myelin antigen is first detected in microglia in the CNS during EAE
Next, we asked whether APCs found in the CNS during EAE constitutively contained myelin Ag for presentation to T cells, and if not, at what time point after induction of disease was it acquired.
To address this question, EAE was induced in C57BL/6 mice with MOG35-55 peptide as described (30). Representative animals were sacrificed daily starting one day after immunization and CNS tissue sections were obtained for confocal microscopy analysis and quantification of CD4+ T cells and various APC populations present. Quantification was performed for the total number of CD4+ T cells, as well as APCs containing MBP as a surrogate marker for myelin Ag uptake, as described in the Methods section by Imaris image analysis software and Supplemental Fig. S2.
The results show that CD4+ T cells infiltrated the CNS in substantial numbers as early as day 5 following immunization and a rapid rise in CD4+ T cells coincided with the onset of EAE around day 11 (Fig. 4A, 4B). The number of CD4+ T cells further increased until the peak of EAE, occurring around day 21, followed by a dramatic decrease in numbers upon remission by day 23 (Fig. 4A, 4B). A similar kinetic was observed for CD11c+ DCs (Fig. 4C). Consistent with previous reports, DCs appeared first in the meningeal areas and several days later became detectable in the perivascular spaces and surrounding tissues (data not shown) (21).
Interestingly, DCs detected in the CNS until day 5 did not contain MBP (Fig. 4C), MOG or PLP (data not shown) indicating that these cells had not acquired myelin Ag until after their arrival in the CNS. Similar results were observed upon induction of passive EAE via adoptive transfer (Supplemental Fig. S1). However, by day 7 approximately 70–80% of DCs contained MBP and this percentage remained relatively stable throughout the course of disease.
The number of CD11b+ microglia/infiltrating macrophages substantially increased around day 13 following the onset of EAE, and, similarly to CD4+ T cells and DCs, cell numbers peaked with clinical disease, at approximately day 21 (Fig. 4D).
Parallel to the decline in clinical disease symptoms beginning around day 23, the number of DCs decreased substantially, falling to levels nearly three times less than at disease peak. Although the absolute number of DCs and microglia/macrophages decreased during the remission of EAE, the proportion of myelin-associated DCs and microglia/macrophages remained consistently high. By day 35 the number of DCs present in the CNS of EAE mice was negligible (Fig. 4C).
As shown in Fig. 4E, Ly-6G+ neutrophils were also detected in the CNS approximately 5 days following induction of EAE. Similar to DCs and microglia/macrophages, numbers of neutrophils also peaked at the height of clinical EAE. However, although the number of neutrophils increased, overall they were dramatically lower in numbers as compared with DCs or CD11b+ microglia. Of note, ≤ 25% of neutrophils contained myelin Ag over the course of EAE (Fig. 4E), in strong contrast to DCs and microglia/infiltrating macrophages (Fig. 4C, 4D). Of note, astrocyte numbers also increased, and colocalized to lesions at peak EAE and later timepoints, however GFAP+ cells were not found to contain myelin Ag (not shown).
CD11b+ cells in the CNS include CNS-resident microglia and infiltrating macrophages from the immune periphery. Thus, in order to specifically dissect the timepoint at which peripheral macrophages infiltrated into the CNS we used the well characterized CX3CR1+/GFP mice to visualize CNS-resident microglial cells as previously reported (32–34). CD11b+GFP+ (CX3CR1+) microglia were observed throughout early EAE (Fig. 5A, 5B), some of which were also MBP+ (Fig. 5C, small arrows), whereas CD11b+GFP− (CX3CR1−) infiltrating macrophages were not detected until day 4 p.i. (Fig. 5F). Myelin Ag could not be detected in infiltrating macrophages for several days after entering the CNS similar to DCs. Of note, CNS-resident microglia and infiltrating macrophages were morphologically distinct with CX3CR1+CD11b+ cells having long, ramified processes (Fig. 5A), and CX3CR1−CD11b+ cells being more rounded with larger cell bodies (Fig. 5F) until the onset of clinical symptoms, when they became indistinguishable by morphology alone.
FIGURE 5.

Peripheral macrophages infiltrate into the CNS by day 4 of EAE. CX3CR1+/GFP mice were immunized with MOG35-55 peptide for induction of EAE and scored for disease as previously described. Confocal microscopy analysis for expression of CX3CR1, CD11b and MBP shows (A) CX3CR1 (green); (B) CD11b (pink); (C) merge A+B. CX3CR1+CD11b+ cells are indicated by large arrows, of which a small percentage are also MBP+ (small arrows), and are found primarily in the corpus callosum (green box), whereas (D–F) CX3CR1−CD11b+ cells (F, large arrow) are only detectable by day 4 p.i. in swollen meninges (red box) and do not contain MBP Ag. Scale bars = 10 μm.
Taken together, the data showed that the number of CNS-resident microglia, as well as infiltrating macrophages and DCs containing myelin Ag strongly increased after the onset of EAE. The observation that a small percentage of microglia contained myelin Ag already by day one after immunization, whereas DCs appeared to first migrate to the CNS and subsequently acquire myelin Ag, may have important implications for EAE pathogenesis.
Only rare microglia in the CNS of naïve mice contain myelin antigens
Our results showed that myelin Ag was detected in microglia as early as day 1 after immunization for EAE (Fig. 4D). The presence of myelin Ag this early after induction of EAE in the absence of other inflammatory cells such as T cells or DCs raised the question whether microglia and/or other APCs in the CNS of naive mice constitutively take up and present myelin Ag. To begin to address this question, brain tissue was obtained from naive CX3CR1+/GFP mice and examined by immunofluorescence staining and confocal microscopy for the presence of myelin Ag in microglia. First, we confirmed that CNS-resident microglia in naive mice were positive for both CX3CR1 and CD11b (Fig. 6A–C. Furthermore, we noted that a small percentage of CX3CR1+ cells were also positive for MBP and MHC II (Fig. 6D–F, small arrows), suggesting that some microglia had the potential to activate CD4+ T cells via Ag presentation. Of note, CX3CR1−CD11b+ infiltrating macrophages were very rare in the parenchymal CNS (not shown).
The results showed that a small proportion (3%) of CX3CR1+CD11b+ microglia were positive for MBP and some also expressed MHCII (Fig. 6E, 6F), suggesting that these CNS-resident cells were potentially capable of presenting myelin Ag in naïve mice. Finally, MBP+ microglia in the naïve CNS frequently also contained MOG, and/or PLP (not shown), allowing MBP to be used as a surrogate marker for myelin Ag. Myelin Ag-containing microglia were mostly restricted to the corpus callosum and nearby subependymal area of the brain similar to those found in very early EAE (Fig. 5A).
In contrast, Ly-6G+ neutrophils, CX3CR1−CD11b+ infiltrating macrophages and CD11c+ DCs were only very rarely detected in the CNS of naïve mice and were only found associated with the meninges (Fig. 1). Importantly, neither submeningeal DCs nor neutrophils detected in the naive brain stained positive for MBP, MOG or PLP (not shown).
Thus, a small fraction of microglia contained myelin Ag in the CNS of naive mice, which could be reflective of the scavenging of myelin debris in the naïve CNS. Furthermore, it is conceivable that myelin Ag-containing microglia in the naïve CNS may play a role in the activation or regulation of encephalitogenic T cells.
Microglia isolated from the naïve CNS do not activate myelin-reactive T cells in the absence of exogenous antigen
Since a small number of microglia in the CNS of naïve mice contained myelin Ag the question arose whether these cells could activate myelin-specific T cells. Alternatively, and not mutually exclusive, these cells could function in a regulatory role during development of EAE. To begin to address this question, microglia were isolated from the CNS of naïve mice by FACS (> 96% purity) and tested with or without exogenous myelin Ag for their potential to activate neuroantigen-reactive T cells.
The results show that microglia isolated from the brains of naïve C57BL/6 mice only induced significant production of IFN-γ or IL-17 by MOG35-55-peptide-reactive CD4+ T cells when exogenous MOG peptide was added (Fig. 7). Microglia-induced T cell responses (Fig. 7, middle and right sets of bars) were substantially lower as compared with responses induced by spleen APCs (Fig. 7, left set of bars). Pre-activation of microglia with LPS enhanced T cell responses (Fig. 7, microglia, right versus middle set of bars) but did not result in significant T cell cytokine production in the absence of MOG35-55 peptide (not shown).
FIGURE 7.

T cell activation by naïve CD11b+ microglia. CD11b+ microglia from naïve CNS (brain and spinal cord) and CD4+ from draining lymph nodes (DLNs) of d10 EAE mice were sorted by FACS and co-cultured at a 1:1 ratio in the presence of MOG35-55 (Ag) and/or LPS. ELISPOT assay was performed to determine the number of IFN-γ or IL-17-producing cells. Splenocytes with and without exogenous Ag shown for comparison. Shown is mean ± S.D. of number of cytokine spots with appropriate medium control subtracted. *significant increase in spots over background control, (n ≥ 10, Student’s t test p ≤ 0.05).
Overall, the data indicate that microglia in naive CNS are inefficient at activating neuroantigen-reactive T cells.
DISCUSSION
In this study we investigate the kinetics of myelin Ag uptake by potential CNS APCs over the course of EAE and in the naïve CNS. Our results show that CNS-resident microglia were the first APCs to contain myelin Ag after the induction of EAE, as early as one day after immunization. In contrast, peripheral macrophages and DCs appeared in substantial numbers later during the course of EAE. Of note, DCs or infiltrating macrophages arriving in the CNS did not contain myelin Ag and it took several days before Ag became detectable. Other potential APCs, such as astrocytes or neutrophils were not significantly loaded with myelin Ag throughout the course of EAE.
In the naïve brain, as expected, and early after induction of EAE, a substantial number of microglia were present. In contrast, DCs were only rarely observed in the CNS of naïve mice, and when present found predominantly in the meninges, whereas in EAE they arrived in the parenchyma approximately by day 5 relative to immunization. While the critical role of DCs for the induction of EAE has been appreciated (9,10), it has not been resolved whether DCs or other APCs are critical for providing presentation of myelin Ag to encephalitogenic T cells for the initiation and/or propagation of EAE. Along these lines, it is noteworthy that approximately 8% of microglia in the brain contained MBP as early as one day after induction of EAE. In strong contrast, infiltrating DCs and neutrophils did not appear to contain myelin Ag during the first days after induction of EAE. CD11b+CX3CR1− infiltrating macrophages were detected in the EAE CNS as early as day 4 p.i. and similar to other infiltrating cells did not contain myelin Ag initially.
GFAP+ astrocytes were present in the naïve CNS, but did not colocalize to any of the myelin Ag tested either before or after induction of EAE (not shown). However, we observed many astrocytes surrounding lesions at peak and later timepoints, as has been previously reported (35), consistent with an important role in the disease process.
Neutrophils represent a relatively small percentage of the cells infiltrating the CNS during EAE. It has been reported that these cells can take up and present Ag via MHC class II molecules under certain conditions (36) and could conceivably play a role in promoting T cell responses. Therefore, we determined the number of myelin containing neutrophils in our model. Interestingly, neutrophils infiltrated into the CNS very early during EAE; however, they acquired myelin Ag only several days later and the number of Ag-containing neutrophils remained constantly low over the course of EAE, arguing against a significant role for the cells in promoting T cell responses via Ag presentation.
At the onset of clinical EAE symptoms (day 11), the number of DCs and microglia/infiltrating macrophages in the CNS increased dramatically. At this time point, the majority of DCs and microglia contained myelin Ag (70–80%). In striking contrast, while the number of Ly-6G+ neutrophils in the CNS also increased until the onset of EAE, myelin Ag was only detectable in about 20% of the cells.
The numbers of microglia/infiltrating macrophages and DCs continued to increase until the peak of EAE (around day 21), and the cells continued to exhibit high loading with myelin Ag. Upon recovery of the animals, usually by day 23, the numbers of DCs and microglia decreased dramatically but most of the remaining cells continued to contain myelin Ag. This is particularly noteworthy for microglia since they continued to be present in the CNS in considerable numbers after disease remission, whereas DCs had almost disappeared by day 39 p.i.
Detection of myelin Ag in APCs in the CNS does not necessarily reflect Ag presentation to T cells and requires evidence of processing and loading of myelin peptides onto MHC II. Therefore, to address this issue we used a bi-specific antibody (MK16) that recognizes MBP85-99 peptide bound to HLA-DR2 molecules (31). Using this tool, we confirmed that detection of MBP in HLA-DR2+ DCs and microglia overlapped with staining for the MK16 antibody, thus suggesting that detection of myelin Ag in APCs corresponded with presentation of myelin peptides by MHC II molecules. Furthermore, we observed colocalization of CD4+ T cells with myelin Ag loaded APCs, consistent with Ag presentation by these cells to encephalitogenic T cells.
Myelin Ag was detected in a low number of microglia in the naïve CNS. However, upon induction of EAE the number of microglia containing myelin Ag increased substantially and for the first several days they were the only cell population with a significant number of cells containing myelin Ag. Thus, it is possible that microglia play a role in the early phases of myelin Ag presentation to encephalitogenic T cells. Conceivably, microglia may activate the first wave of encephalitogenic T cells arriving in the CNS resulting in further recruitment of T cells and DCs, the latter of which subsequently promote extensive T cell activation resulting in CNS pathology and demyelination. It has been shown previously that CNS-resident microglia are not only capable of presenting Ag (35, 37–42) but are potent APCs in vitro that activate myelin-reactive T cells (37, 43–46), however these findings have been challenged by others (47). More recently, it has been suggested that microglia are inefficient as compared to peripheral dendritic cells unless they are specifically activated towards a DC phenotype, such as stimulation with GM-CSF (48, 49) or genetic CX3CR1 deficiency (50), however it remains possible that this type of microglial activation of T cells occurs very early in EAE, prior to DC appearance in the CNS as has been suggested previously (40).
After this point the role of microglia in promoting EAE may become secondary to that of DCs and possibly shift to other functions such as removal of myelin debris to limit CNS damage.
The role of astrocytes in EAE has remained controversial. Some studies have suggested a role for astrocytes in promoting EAE, whereas work by Zipp and others has suggested a regulatory role for this cell population (51–53). Our studies did not detect myelin Ag in astrocytes. Therefore, the results argue against a critical role of astrocytes for Ag presentation to encephalitogenic T cells. We have not fully addressed the possibility that myelin peptides were bound externally to MHC II expressed by astrocytes, but it must be noted that the expression of MHC II by astrocytes in vivo in the CNS is controversial (54, 55).
Novel information provided by our studies concerns the distribution of APCs and loading of these cells with myelin Ag in the brain of naïve mice. In the naïve brain we observed that the level of MHC Class II expression was relatively low and colocalized with microglia. DCs and neutrophils were only rarely detected, and if, then only in the meninges of the naïve brain and without containing myelin Ag. Similarly, in naïve mice, astrocytes did not constitutively express MHC class II and we observed no colocalization of astrocytes with myelin Ag.
Importantly, 3 – 4% of microglia in the brains of naïve mice contained myelin Ag, and in a very small number of mice the percentage of myelin-containing microglia was up to 20% of the cells. This contrasted with the lack of myelin Ag detected in rarely observed DCs. These observations raise several questions: 1) what is the mechanism whereby myelin Ag moves from myelin sheaths to microglia in the naïve CNS? Presumably, this mechanism is not dependent on inflammatory mediators in the absence of EAE. 2) Does myelin Ag presented by microglia in the naïve CNS play a role for the activation of encephalitogenic T cells arriving in the brain after being activated in the immune periphery? Or, does uptake of myelin Ag by resting microglia in the naïve CNS represent phagocytosis and removal of myelin debris as housekeeping function and serve to prevent undesired autoimmune pathology? In this view, myelin uptake by microglia in the absence of inflammatory stimuli may not be conducive to T cell activation and rather serve to maintain T cell tolerance. Answering these questions will require future mechanistic studies; however, insights gained could potentially lead to novel approaches to prevent Ag presentation to autoreactive T cells in MS patients.
Our findings suggest that upon induction of EAE, myelin Ag containing microglia may be the APCs initially encountered by encephalitogenic T cells in the CNS. Conceivably, this could result in the activation of these T cells and promote subsequent recruitment of DCs and additional autoreactive T cells into the CNS. Once a certain threshold of DCs and myelin-reactive T cells has accumulated in the CNS, overt clinical disease may ensue. At this point, DCs may be requisite and sufficient for promotion of disease. Should DCs not arrive in sufficient numbers or be functionally compromised in the CNS to take over further activation from microglia, as shown by Becher and colleagues, the disease process may not develop.
Lastly, we propose that uptake of myelin antigen within the CNS and outside the CNS (e.g. cervical draining lymph nodes) are not mutually exclusive concepts, but may go together in a sequential fashion. In the EAE model we favor a view where myelin antigen is first taken up by CNS APCs, presented to myelin-specific T cells resulting in reactivation, cytokine production and enhanced demyelination and subsequent drainage of myelin to the periphery, e.g. cervical draining lymph nodes. However, in certain situations myelin antigen may be released in the CNS (e.g. trauma) and drain to the periphery, where it may under conditions of a “perfect storm”, e.g. a simultaneously occurring upper respiratory infection, result in the activation and priming of encephalitogenic T cells. A better understanding of the cellular players and antigen presentation mechanisms may lead to novel therapeutic approaches for MS.
Supplementary Material
Acknowledgments
This work was supported by grants SC1GM095426 (A.E.C.), NS-52177 and G12MD007591 (T.G.F.) from the National Institute of Health, and grants RG3499 and RG3701 from the National Multiple Sclerosis Society (T.G.F.), and support from the Jesse H. & Mary Gibbs Jones Endowed Chair (T.G.F.).
We would like to thank Dr. Neal Guentzel and Colleen Witt for helpful discussions and the RCMI imaging core under the direction of Dr. Colleen Witt for assistance with confocal imaging and analysis.
Abbreviations
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- PLP
proteolipid protein
- EAE
experimental autoimmune encephalomyelitis
- DC
dendritic cells
References
- 1.Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron. 2006;52:61–76. doi: 10.1016/j.neuron.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 3.Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, McFarlin DE, McFarland HF. Fine Specificity and Hla Restriction of Myelin Basic Protein-Specific Cytotoxic T-Cell Lines from Multiple-Sclerosis Patients and Healthy-Individuals. Journal of Immunology. 1990;145:540–548. [PubMed] [Google Scholar]
- 4.Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. Journal of Experimental Medicine. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun DM, Whitaker JN, Huang ZG, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8(+) T cells are encephalitogenic and produce severe disease in C57BL/6 mice. Journal of Immunology. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 6.Compston A, Coles A. Multiple sclerosis. The Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 7.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986:9. [Google Scholar]
- 8.Owens T, Renno T, Taupin T, Krakowski M. Inflammatory cytokines in the brain: does the CNS shape immune responses? Immunol Today. 1994:15. doi: 10.1016/0167-5699(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 9.Stuve O, Youssef S, Slavin AJ, King CL, Patarroyo JC, Hirschberg DL, Brickey WJ, Soos JM, Piskurich JF, Chapman HA, Zamvil SS. The role of the MHC class II transactivator in class II expression and antigen presentation by astrocytes and in susceptibility to central nervous system autoimmune disease. Journal of Immunology. 2002;169:6720–6732. doi: 10.4049/jimmunol.169.12.6720. [DOI] [PubMed] [Google Scholar]
- 10.Huitinga I, van Rooijen N, de Groot CJA, Uitdehaag BMJ, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990:172. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 12.McMahon EJ. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 13.McMahon EJ, Bailey SL, Miller SD. CNS dendritic cells: Critical participants in CNS inflammation? Neurochemistry International. 2006;49:195–203. doi: 10.1016/j.neuint.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, Danilenko DM, Eastham-Anderson J, Slavin AJ, Linington C, Bernard CCA, Martin F, Zamvil SS. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Annals of Neurology. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in Degenerative Neurological Disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 16.Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 17.McLaurin J, Williams K, Ulvestad E, Antel JP. Activation of Microglia Following Myelin Phagocytosis. Journal of Neurochemistry. 1994;62:S71–S71. [Google Scholar]
- 18.Fontana A, Fierz W, Wekerle H. Astrocytes Present Myelin Basic-Protein to Encephalitogenic T-Cell Lines. Nature. 1984;307:273–276. doi: 10.1038/307273a0. [DOI] [PubMed] [Google Scholar]
- 19.Falsig J, Pörzgen P, Lund S, Schrattenholz A, Leist M. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. Journal of Neurochemistry. 2006;96:893–907. doi: 10.1111/j.1471-4159.2005.03622.x. [DOI] [PubMed] [Google Scholar]
- 20.Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84:532–543. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 21.Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, Khoury SJ. Localizing central nervous system immune surveillance: Meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Annals of Neurology. 2009;65:457–469. doi: 10.1002/ana.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010;468:8. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- 23.Suzumura A, Silberberg DH, Lisak RP. The Expression of Mhc Antigens on Oligodendrocytes - Induction of Polymorphic H-2 Expression by Lymphokines. Journal of Neuroimmunology. 1986;11:179–190. doi: 10.1016/0165-5728(86)90002-0. [DOI] [PubMed] [Google Scholar]
- 24.Lee SC, Raine CS. Multiple-Sclerosis - Oligodendrocytes in Active Lesions do Not Express Class-Ii Major Histocompatibility Complex-Molecules. Journal of Neuroimmunology. 1989;25:261–266. doi: 10.1016/0165-5728(89)90145-8. [DOI] [PubMed] [Google Scholar]
- 25.Wucherpfennig KW. Autoimmunity in the Central-Nervous-System - Mechanisms of Antigen Presentation and Recognition. Clinical Immunology and Immunopathology. 1994;72:293–306. doi: 10.1006/clin.1994.1145. [DOI] [PubMed] [Google Scholar]
- 26.Wong GH, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature. 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]
- 27.Shive CL, Hofstetter H, Arredondo L, Shaw C, Forsthuber TG. The enhanced antigen-specific production of cytokines induced by pertussis toxin is due to clonal expansion of T cells and not to altered effector functions of long-term memory cells. European Journal of Immunology. 2000;30:2422–2431. doi: 10.1002/1521-4141(2000)30:8<2422::AID-IMMU2422>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Karulin AY, Hesse MD, Tary-Lehmann M, Lehmann PV. Single-cytokine-producing CD4 memory cells predominate in type 1 and type 2 immunity. Journal of Immunology. 2000;164:1862–1872. doi: 10.4049/jimmunol.164.4.1862. [DOI] [PubMed] [Google Scholar]
- 29.Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat Protocols. 2006;1:1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- 30.Ji N, Rao N, Guentzel NM, Arulanandam BP, Forsthuber TG. Anaphylaxis and Mortality Induced by Treatment of Mice with Anti–VLA-4 Antibody and Pertussis Toxin. The Journal of Immunology. 2011;186:2750–2756. doi: 10.4049/jimmunol.1000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krogsgaard M, Wucherpfennig KW, Canella B, Hansen BE, Svejgaard A, Pyrdol J, Ditzel H, Raine C, Engberg J, Fugger L. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2-MBP 85–99 complex. Journal of Experimental Medicine. 2000;191:1395–1412. doi: 10.1084/jem.191.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang DR, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 33.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 34.Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The Fractalkine Receptor but Not CCR2 Is Present on Microglia from Embryonic Development throughout Adulthood. The Journal of Immunology. 2012;188:29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto Y, Ohmori K, Fujiwara M. Microglial and astroglial reactions to inflammatory lesions of experimental autoimmune encephalomyelitis in the rat central nervous system. Journal of Neuroimmunology. 1992;37:23–33. doi: 10.1016/0165-5728(92)90152-b. [DOI] [PubMed] [Google Scholar]
- 36.Gosselin EJ, Wardwell K, Rigby WFC, Guyre PM. Induction of MHC class II on Human Polymorphonuclear neutrophils by granulocyte-macrophage colony-stimulating factor, IFN-gamma, and IL-3. Journal of Immunology. 1993;151:1482–1490. [PubMed] [Google Scholar]
- 37.Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 38.Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 39.Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 40.Ponomarev ED, Shriver LP, Maresz K, Dittell BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of Neuroscience Research. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 41.Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. Journal of Neuroimmunology. 1998;90:113–121. doi: 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- 42.Gehrmann J, Gold R, Linington C, Lannes-Vieira J, Wekerle H, Kreutzberg GW. Microglial involvement in experimental autoimmune inflammation of the central and peripheral nervous system. Glia. 1993;7:50–59. doi: 10.1002/glia.440070110. [DOI] [PubMed] [Google Scholar]
- 43.Ulvestad E. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- 44.Matyszak MK. Microglia induce myelin basic protein-specific T cell anergy or T cell activation, according to their state of activation. Eur J Immunol. 1999;29:3063–3076. doi: 10.1002/(SICI)1521-4141(199910)29:10<3063::AID-IMMU3063>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 45.Zhang GX, Li JF, Ventura E, Rostami A. Parenchymal microglia of naive adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Experimental and Molecular Pathology. 2002;73:35–45. doi: 10.1006/exmp.2002.2441. [DOI] [PubMed] [Google Scholar]
- 46.Mack CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. J Neuroimmunol. 2003;144:68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 47.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 48.Almolda B, González B, Castellano B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. Journal of Neuroimmunology. 2010;223:39–54. doi: 10.1016/j.jneuroim.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 49.Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF Production by Autoreactive T Cells Is Required for the Activation of Microglial Cells and the Onset of Experimental Autoimmune Encephalomyelitis. The Journal of Immunology. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- 50.Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, Forsthuber TG, Cardona AE. Regulation of Adaptive Immunity by the Fractalkine Receptor during Autoimmune Inflammation. The Journal of Immunology. 2013 doi: 10.4049/jimmunol.1300040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aktas O, Waiczies S, Zipp F. Neurodegeneration in autoimmune demyelination: Recent mechanistic insights reveal novel therapeutic targets. Journal of Neuroimmunology. 2007;184:17–26. doi: 10.1016/j.jneuroim.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 52.Xiao BG, Diab A, Zhu J, van der Meide P, Link H. Astrocytes induce hyporesponses of myelin basic protein-reactive T and B cell function. Journal of Neuroimmunology. 1998;89:113–121. doi: 10.1016/s0165-5728(98)00123-4. [DOI] [PubMed] [Google Scholar]
- 53.Hindinger C, Bergmann CC, Hinton DR, Phares TW, Parra GI, Hussain S, Savarin C, Atkinson RD, Stohlman SA. IFN-γ Signaling to Astrocytes Protects from Autoimmune Mediated Neurological Disability. Plos One. 2012;7:e42088. doi: 10.1371/journal.pone.0042088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Keyser J, Zeinstra E, Frohman E. Are astrocytes central players in the pathophysiology of multiple sclerosis? Archives of Neurology. 2003;60:132–136. doi: 10.1001/archneur.60.1.132. [DOI] [PubMed] [Google Scholar]
- 55.Cornet A, Bettelli E, Oukka M, Cambouris C, vellana-Adalid V, Kosmatopoulos K, Liblau RS. Role of astrocytes in antigen presentation and naive T-cell activation. Journal of Neuroimmunology. 2000;106:69–77. doi: 10.1016/s0165-5728(99)00215-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.