

Abstract
Astrocytes, the most important energy regulator in the brain, support brain energy needs. In the meantime, numerous studies have demonstrated that impaired brain glucose metabolism is closely linked to abnormal astrocytic metabolism in AD. Indeed, the interaction between amyloid plaques and perturbed astrocytic homeostasis contributes to AD pathogenesis and astrocytic metabolic dysfunction is thought to be a trigger for Aβ pathology through oxidative stress and neuroinflammation Moreover, astrocytic metabolic dysfunction may regulate Aβ generation via modulating proteolytic processing of amyloid precursor protein (APP) by β-secretase, γ-secretase, and α-secretase, and may also modulate APP post-translational modifications such as glycosylation, phosphorylation, and tyrosine sulfation. While it is known that metabolic dysfunction of astrocytes contributes to the failure of Aβ clearance, it has also been reported that such dysfunction has neuroprotective property and exhibits no detrimental outcomes. Therefore, the exact role of astrocytic metabolic dysfunction in Aβ pathology remains to be further investigated.
Keywords: Alzheimer's disease, astrocytes, beta-amyloid protein, metabolic dysfunction
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative diorder leading to cognition decline, behavioral symptoms, and eventual loss of social or occupational function. Beta-amyloid (Aβ) plaques (extracellular Aβ deposition) and neurofibrillary tangles (NFT, intracellular deposits of hyper-phosphorylated tau protein) have been identified as two classical pathological hallmarks of AD [1-3]. Accordingly, numerous studies have focused on Aβ generation and deposition as well as on NFT formation as the triggering factors for AD occurrence [4, 5].
By maintaining brain homeostasis, astrocytes are the most important energy regulator in the brain [6, 7]. Indeed, astrocytes play a role of metabolic activation in the brain and support brain energy needs [7, 8]. Over the past decade, numerous studies on astrocytes attempting to elucidate AD etiology have evidenced that impaired brain glucose metabolism is closely associated with abnormal astrocytic function in AD [9-11]. Firstly, the interaction between amyloid plaques and perturbed astrocytic homeostasis has been found in AD. Secondly, it is via oxidative stress and neuroinflammation that metabolic dysfunction of astrocyte contributes to Aβ pathology [12]. In addition, it is known that metabolic dysfunction of astrocyte regulates proteolytic processing of APP by β-secretase, γ-secretase, and α-secretase [13]. Moreover, metabolic dysfunction of astrocytes may also be a regulator of APP post-translational modifications such as glycosylation [14], phosphorylation, and tyrosine sulfation [15-18], and may lead to failure in Aβ clearance [19, 20].
This review focuses on the role of metabolic dysfunction of astrocytes in Aβ pathology. An overview is provided for the proposed general pathogenic mechanisms of metabolic dysfunction of astrocyte in Aβ pathology that are mainly via oxidative stress and neuroinflammation, regulation of proteolytic processing of APP and APP post-translational modification, and failure in Aβ clearance. Finally, the possibility of improving astrocytic function as a potential target for AD prevention and treatment is also discussed.
2. Astrocytes
Astrocytes, large and star-shaped neuroglial cells with many branches, are specialized glial cells that out-number neurons by more than five-fold [21]. They play multifunctional roles including physical support, nutritional supply, and biochemical support to the blood-brain barrier (BBB) and neurons [22-24] as well as to the repair and scarring of injury [25, 26]. Astrocytes are critical mediators of brain homeostasis, such as maintaining the balance of ions and the integrity of BBB in the central nervous system, communicating with neurons and other important structures, releasing growth factors, and regulating neurotransmitter levels [24, 27, 28]. Numerous studies have indicated that astrocytes are involved in all types of brain pathologies from acute lesions such as trauma or stroke to chronic neurodegenerative processes such as AD, Parkinson's disease, multiple sclerosis [29-33], and psychiatric diseases. Many studies also reported a role of astrocytic degeneration and atrophy in various neurodegenerative disorders [34-36].
3. Abnormal glucose metabolic dysfunction in astrocytes: a direct link to AD
Glucose, an essential energy source for the brain, is oxidized through sequential metabolic pathways including glycolysis, the tricarboxylic cycle, and oxidative phosphorylation. Glucose can also be stored as polysaccharide glycogen in the brain. Medical scientists confirm that low glucose metabolism is the early warning signs of AD [37, 38], and glucose metabolic failure is an early event in neurodegenerative disease represented by altered expression of nutrient transporters, metabolic enzymes and molecular components of cellular respiration [39, 40]. Therefore, it is well established that brain glucose metabolism is impaired in AD [41, 42] and alterations in cerebral blood flow and oxygen consumption would decrease most severely with age and neurodegenerative process accompanied by low glucose metabolism [43, 44]. It has been reported that in AD glucose hypometabolism, mostly due to glycolytic breakdown and pyruvate oxidation [45, 46], promotes Aβ pathology, facilitates abnormal hyperphosphorylation of tau [47], damages synaptic transmission function, and leads to the occurrence of cognitive impairment [48, 49].
Astrocytes are thought to play a role in metabolic activation in the brain by promoting glycolysis, glycogenolysis activities, and production of lactate, all of which supports brain energy needs. Astrocytic glycogen breaks down to lactate against hypoglycemic neural injury. Studies have shown that the activities of key glycolytic enzymes in the brains of patients with AD are changed, including a significant increase of pyruvate kinase and lactate dehydrogenase in frontal and temporal cortex, and a significant decrease of glucose 6-phosphate dehydrogenase activity in hippocampus [50, 51]. Furthermore, some glycolytic enzyme activities are correlated with contents of lactate dehydrogenase and glial fibrillary acidic protein (GFAP) in astrocytes, implicating that the increased activity of some glycolytic enzymes may be the result of astrocytic dysfunction developed in the course of AD [50]. AD is also linked to aerobic glycolysis whereby glucose does not go into oxidative phosphorylation in astrocytes. Indeed, it is found that the spatial distribution of aerobic glycolysis correlates spatially with Aβ deposition [52].
4. Metabolic dysfunction from astrocytes: a trigger for Aβ pathology
Although the brain is about 2% of the whole body weight, it consumes about 15% of the cardiac output, 20% of the oxygen, and 25% of the glucose to maintain cerebral functions. Therefore, the brain is such an organ with the highest energy requirements in the human body. It is well-known that astrocyte, as the most important energy regulator in the brain, maintains brain homeostasis and restores ion gradients such as post-synaptic and action potentials, as well as uptake and recycling of neurotransmitters, which all count toward energy consumption in the brain.
The generation and deposition of Aβ in the brain is a conventional molecular trend in the pathogenesis of AD. So far the amyloid hypothesis of AD has been recognized as the main pathological features. Therefore, not surprisingly, compelling studies have supported the notion that metabolic dysfunction of astrocyte is a trigger for Aβ pathology [53, 54] given the importance of astrocytes in the brain.
4.1 Brain homeostasis disorders induced by astrocytic dysfunction: a contributor to Aβ pathology
Increasing evidence has indicated that astrocytes, the most abundant cells in the brain, plays a pivotal role in maintenance of brain extracellular homeostasis and functional recovery from injuries through a variety of means, including regulating intracellular ion homeostasis [55], providing a metabolic support for brain energy [55], regulating metabolism of neurotransmitters, maintaining the structure and function of BBB, clearing the abnormal aggregates in the brain, and regulating the brain immune response and neurodevelopmental processes [56-58]. Astrocytic dysfunction induces and facilitates neurodegeneration, which leads to cognitive impairment found in neurodegenrative diseases, such as AD, Parkinson's disease [59] and Huntington disease [60, 61]. Therefore, it is thought that brain homeostasis disorders induced by astrocytic dysfunction are closely associated with AD.
Brain homeostasis failure leads to an inability to maintain brain physiological balance that causes severe maladjustment [62, 63]. During aging, the capabilities of brain homeostasis are increasingly vulnerable as evidenced by age-related Alzheimer's disease [64, 65]. Therefore, numerous studies have demonstrated that failure in brain homeostasis enhances Aβ deposition and a decline in Aβ clearance. The past several decades have witnessed many important discoveries that provide novel insights into associations between amyloid plaques and perturbed astrocytic homeostasis [10, 55, 65]. Impaired cellular ion homeostasis and energy metabolism not only are triggers for Aβ deposition and a decline in Aβ clearance, but also render neurons vulnerable to Aβ excitotoxicity [66]. It is also known that presenilin mutations perturb calcium homeostasis in astrocytic endoplasmic reticulum, indicating that aberrant calcium homeostasis is linked to altered APP processing [67].
4.2 Oxidative stress and neuroinflammation induced by metabolic dysfunction from astrocytes: a contributor to Aβ pathology
Increasing evidence demonstrates that neuroinflammation and oxidative stress occur throughout the pathological process of AD [68-70]. An important pathological feature of AD is that oxidative stress triggers an active and self-perpetuating cycle of chronic neuroinflammation, which further promotes oxidative stress that eventually leads to irreversible neuronal dysfunction and cell death [69, 71, 72]. Evidence has also indicated that not only oxidative stress and neuroinflammation have a close association with Aβ pathology, but also the interaction between oxidative stress and neuroinflammation enlarges Aβ generation [71, 73].
It is known that senescent astrocytes may link advanced age with increased risk for sporadic AD [65, 74]. It is also known that astrocytes, as a site for the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), are the initiator of oxidative stress that occurs through the whole process of AD pathogenesis [65]. Compelling evidence has also demonstrated that the inflammatory reaction and oxidative stress are generated by activated astrocytes and/or senescent astrocytes that produce a number of inflammatory cytokines including interleukin-6 (IL-6) and IL-1 [65, 74, 75]. Astrocytic metabolic phenotype modified by proinflammatory cytokines is observed in AD while long-term treatment with IL-1β or TNF-α alone enhances glucose utilization in astrocytes [11]. Given that energy failure, increased oxidative stress, and neuroinflammation are critical for Aβ pathology [76, 77], astrocytic metabolism dysfunction may dictate the occurrence of Aβ-related events through mechanisms that are yet to be elucidated.
4.3 Proteolytic processing of APP regulated by metabolic dysfunction from astrocytes
Proteolytic processing of APP
Amyloid precursor protein (APP), an integral membrane protein, is best known as the Aβ precursor molecule. Proteolytic processing of APP commonly includes an amyloidogenic and a non-amyloidogenic pathway. In the amyloidogenic pathway Aβ is generated by the sequential proteolysis of two enzymes: β-secretase and γ-secretase [78]. The non-amyloidogenic pathway is a process whereby α-secretase cleaves APP in the extracellular domain and releases soluble APPα. Following this cleavage, the C-terminal fragment (α-CTF) is cleaved again by γ-secretase to yield 3kDa fragment known as P3.
α-secretase
APP can be cleaved by α-secretase within the APP domain between Lys687 and Leu688, producing a soluble α-APP domain and the C-terminal fragments containing C83. C83 can then be cleaved by γ-secretase at residue 711 or 713 to release a P3 fragment. This process does not yield Aβ peptide [79]. Hence, the α-secretase pathway has a beneficial effect in lowering Aβ peptide levels. Considerable evidence has demonstrated that abnormal astrocytes play a crucial role in Aβ generation and deposition. The production and aggregation of Aβ is regulated by abnormal astrocytes via interfering with APP cleavage. A recent study has discovered that a group of high-energy compounds (HECs), including ATP, GTP, CTP, phosphocreatine, phosphoenol pyruvate, S-adenosylmethionine and acetyl coenzyme A, promotes APP α-secretase-processing with varying potencies whereas their cognate counterparts do not have the same effects that could be eliminated by energy inhibitors [80]. Yao et al [81] have also shown that 2-deoxy-D-glucose (2-DG) increases α-secretase and decreases γ-secretase expression via inducing ketogenesis and sustaining mitochondrial function, and reduces pathology in female mouse model of AD. These findings implicate that improving astrocytic energy supply may be useful in slowing down the progression of AD via enhancing α-secretase-processing.
β-secretase
A large body of evidence supports that β-secretase (beta-site amyloid precursor protein-cleaving enzyme 1, BACE1) is the rate-limiting enzyme for the production of Aβ [82, 83]. In APP transgenic (Tg2576) mice, energy inhibition by several pharmacological agents (insulin, kainic acid, 2-deoxyglucose and 3-nitropropionic acid) caused approximately a 150% increase in cerebral BACE1 levels and a 200% increase in cerebral Aβ40 levels, respectively, when compared with controls, implicating that impaired energy production in the brain may enhance Aβ pathology by elevating BACE1 levels and activities [84]. In addition, bioenergy impairment could also contribute to neuronal apoptosis and elevated β-secretase, thereby promoting AD pathogenesis [85]. It is obvious that bioenergy impairment of astrocytes would be a decisive factor in Aβ generation owing to elevated β-secretase levels and activities.
γ-secretase
γ-secretase is a multi-subunit protease complex that consists of four individual proteins: presenilin (PS1 or PS2), nicastrin, APH-1 (anterior pharynx-defective 1, APH-1α or APH-1β), and PEN-2 (presenilin enhancer 2). As described above, improving astrocytic energy supply by 2-DG lowers γ-secretase level and limits Aβ pathogenesis in vivo [81]. On the contrary, activated astrocytes have an increased expression of the γ-secretase components presenilin-1 and nicastrin [86]. Hence, a hypothesis has been postulated that astrocytic bioenergy impairment triggers γ-secretase overexpression and contributes to Aβ pathogenesis.
4.4 Post-translational modification of APP by metabolic dysfunction from astrocytes ?
Besides many types of proteolytic processing, APP undergoes extensive post-translational modifications including glycosylation, phosphorylation, and tyrosine sulfation [87]. Protein glycosylation, common in Eukaryotic proteins, is the most important and complex form of post-translational modifications. APP contains both N-linked and O-linked sugars [88, 89]. N-linked glycosylations are covalently attached to Asn residues within a consensus sequence (Asn-Xaa-Ser/Thr), enabling prediction of the modification sites and sharing of a common pentasaccharide core (GlcNAc2Man3) recognized by N-glycanase enzymes. Mucin-type, the most prevalent O-linked glycosylation is characterized by an N-acetylgalactosamine (GalNAc) residue linked to the hydroxyl group of Ser or Thr. GalNAc residue is installed by a family of 24 N-acetyl-galactosaminyltransferases, and further elaborated by a series of glycosyltransferases to generate higher O-linked structures. It has been identified that the structures of the core type O-linked glycans are attached at the residues Thr291, Thr292 and Thr576 of the full-length APP695 with the use of electron transfer dissociation and collision [90]. Griffith et al. [91] first presented that APP is modified with O-linked N-acetylglucosamine, namely O-linked to cytoplasmic serine or threonine residues (O-GlcNAc). Numerous results support that the post-translational modification of APP by glycosylation is a key event in determining the processing of the protein [92-94].
Phosphorylation of APP is known to occur on several phosphorylatable amino acid residues in its cytoplasmic and luminal region [95, 96]. It has been found that phosphorylation of APP at Thr668 induces neurodegeneration via regulating the nuclear translocation of the APP intracellular domain [97] while Thr668 phosphorylation may facilitate the BACE1 cleavage of APP to increase Aβ generation [98].
It is also well known that APP is a type I transmembrane protein with a large ectodomain. It is found that APP can undergo sulfation on tyrosine residues within its ectodomain [87]. Furthermore, sulfation is shown to be critical for the effect of heparin on APP processing and Aβ production [17].
Additionally, a number of post-translational modifications to APP have been found in the brains of AD patients and transgenic animal models of AD. As metabolic disorder is an important determinant for post-translational modifications [99-103], a reasonable conclusion is that metabolic disorder will influence AD-associated APP post-translational modifications. However, whether an astrocytic metabolism disorder promotes AD-associated APP post-translational modifications remain unknown. Hence, identification of APP post-translational modifications promoted by astrocytic dysfunction may provide novel therapeutic targets for AD treatment.
4.5 Astrocytes, metabolic disorder and failure of Aβ clearance
Emerging evidence suggests that failure of Aβ clearance in the brain is an important factor in the progression of AD [104]. Multiple cellular and molecular mechanisms have been elucidated that astrocytes are involved in Aβ clearance. Within the progress of Aβ deposition and pathological severity, the ability of astrocytes to clear Aβ is gradually compromised under a cytokine cycle of molecular and cellular cascades induced by astrocytic activation, microglial activation, and Aβ deposition, which all confer risks for AD. It has been recognized that astrocytes can lose energy-generating reactions to clear aggregated proteins when astrocytes show persistent response to the chronic inflammation and oxidative stress induced by microglial activation and/or Aβ deposition [105-107]. Thus, the cytokine cycle of molecular and cellular cascades from astrocytic activation, microglial activation and Aβ deposition may be one of the most important factors in Aβ clearance [108-110].
Astrocytes play many pathophysiological roles, such as biochemical support of endothelial cells that form the BBB, nutrient supply for the nervous tissue, maintenance of ion homeostasis, as well as clearance and degradation of aggregated proteins. Various studies implicate that the BBB plays a role in the deposition of Aβ and faulty clearance of Aβ from the brain [111-113]. Astrocytic BBB disruption is a critical contributor to the failure of Aβ clearance whereas the astrocyte end-feet encircling endothelial cells are in the maintenance of the structural and functional integrity for BBB [114-116]. As the main function of BBB regulates the passage of molecules and leukocytes in and out of the brain, BBB injury by astrocytic disorder would be a major underlying cause of neurodegenerative disorders since astrocytic-endothelial tight junctions disrupt the integrity of the BBB [117-119]. Therefore, the main pathogenesis of AD involves disruption of BBB that subsequently not only delays the Aβ clearance from the brain but also facilitates an increase in Aβ influx from the cerebrospinal fluid (CSF). Metabolic dysfunction of astrocytes may perturb the rapid clearance of Aβ across the BBB via increasing levels of receptor for advanced glycation end products (RAGE) [120-122], downregulating low density lipoprotein receptor-related protein 1(LRP-1) [123, 124], and dysregulating enzymatic degradation (such as matrix metalloproteinases, MMPs) [125]. Furthermore, oxidative stress and neuroinflammation may be critical mediators between Aβ clearance and astrocytic metabolic dysfunction (Figure 1).
Figure 1. Schematic diagram of failure of Aβ clearance by metabolic dysfunction from astrocyte.
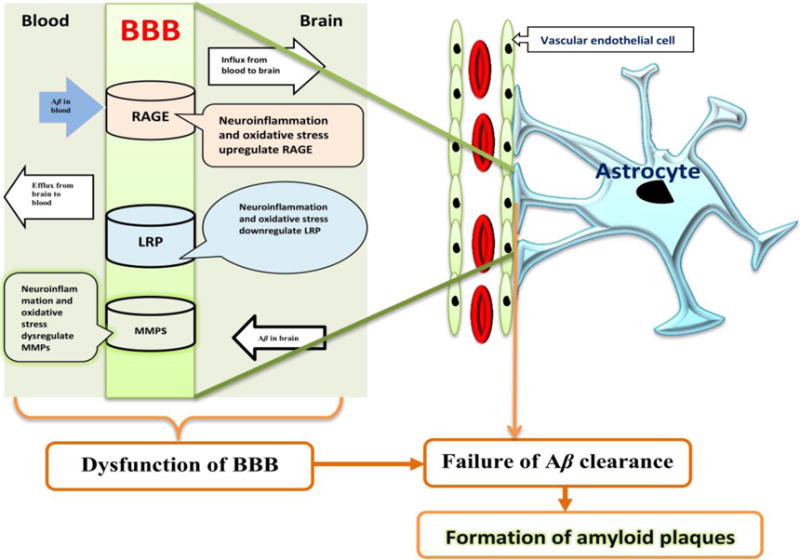
Metabolic dysfunction from astrocyte leads to failure of Aβ clearance by disrupting BBB transporter. Metabolic dysfunction from astrocyte may injury its rapid clearance across the BBB via increasing RAGE level, downregulating LRP-1, and dysregulating enzymatic degradation (such as MMPs). RAGE is a member of multi-ligand immunoglobulin superfamily and cell surface receptor. RAGE promotes influx of circulating Aβ across the BBB from blood to brain, which is antagonized by LRP-1-mediated efflux of Aβ [126, 127]. LRP-1, a multifunctional scavenger and signaling receptor belonging to the low-density lipoprotein receptor family, plays a role in the cellular transport of cholesterol, endocytosis of diverse ligands, transcytosis of ligands across the BBB [124, 128]. LRP-1, abundantly expressed in capillary endothelial cells, is a major clearance receptor responsible for efflux of Aβ from brain to blood [123, 124]. MMPs are involved in BBB damage related to Aβ clearance [125] and formation of Aβ plaques [129]. Neuro-inflammation and oxidative stress may a critical mediator between Aβ clearance and metabolic dysfunction from astrocyte.
5. Conclusion and perspective
Increasing evidence supports the notion that impaired brain glucose metabolism is closely associated with abnormal astrocytic function in AD. Oxidative stress and neuroinflammation induced by metabolic dysfunction of astrocyte likely contribute to Aβ pathology. Specifically, metabolic dysfunction of astrocyte may: 1) regulate proteolytic processing of APP; 2) modulate APP post-translational modification; 3) lead to the failure of Aβ clearance; 4) cause AD indirectly by interacting with many other pathologic processes. However, a few studies also demonstrated that metabolic dysfunction of astrocytes could have neuroprotective properties that did not exhibit detrimental outcomes [9, 72, 111, 130, 131]. Therefore, the exact role of astrocytic metabolic dysfunction in Aβ pathology remains unclear at this time. Clinically, it is also unclear whether metabolic dysfunction of astrocytes would be indeed beneficial in AD intervention. Thus, it is necessary to further understand the mechanisms of metabolic dysfunction of astrocytes before it can be targeted for AD prevention and treatment.
Acknowledgments
This work was supported, in part, by the Anhui Provincial Nature Science Foundation (1308085MH158 to Z.C.) and by the National Institute of Neurological Disorders and Stroke, the National Institutes of Health (R01NS079792 to L.J.Y.). The content in this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: None declared.
References
- 1.Grimm MO, Zinser EG, Grosgen S, Hundsdorfer B, Rothhaar TL, Burg VK, Kaestner L, Bayer TA, Lipp P, Muller U, et al. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer's disease pathology with ganglioside metabolism. PLoS One. 2012;7(3):e34095. doi: 10.1371/journal.pone.0034095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mal'tsev AV, Davidchenko NV, Uteshev VK, Sokolik VV, Shtang OM, Iakushun MA, Sokolova NM, Suprin AK, Galzitskaia OV. Intensive protein synthesis in neurons and phosphorylation of beta-amyloid precursor protein and tau-protein are triggering factors of neuronal amyloidosis and Alzheimer's disease. Biomed Khim. 2013;59(2):144–170. doi: 10.18097/pbmc20135902144. [DOI] [PubMed] [Google Scholar]
- 3.Lo AC, Iscru E, Blum D, Tesseur I, Callaerts-Vegh Z, Buee L, De Strooper B, Balschun D, D'Hooge R. Amyloid and Tau Neuropathology Differentially Affect Prefrontal Synaptic Plasticity and Cognitive Performance in Mouse Models of Alzheimer's Disease. J Alzheimers Dis. 2013 doi: 10.3233/JAD-122296. [DOI] [PubMed] [Google Scholar]
- 4.Schrotter A, Pfeiffer K, El Magraoui F, Platta HW, Erdmann R, Meyer HE, Egensperger R, Marcus K, Muller T. The amyloid precursor protein (APP) family members are key players in S-adenosylmethionine formation by MAT2A and modify BACE1 and PSEN1 gene expression-relevance for Alzheimer's disease. Mol Cell Proteomics. 2012;11(11):1274–1288. doi: 10.1074/mcp.M112.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maloney B, Lahiri DK. The Alzheimer's amyloid beta-peptide (Abeta) binds a specific DNA Abeta-interacting domain (AbetaID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: characterizing a new regulatory motif. Gene. 2011;488(1-2):1–12. doi: 10.1016/j.gene.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14(6):724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 7.Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55(12):1263–1271. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- 8.Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006;48(6-7):586–595. doi: 10.1016/j.neuint.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34(2):76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci. 2010;30(9):3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavillet M, Allaman I, Magistretti PJ. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia. 2008;56(9):975–989. doi: 10.1002/glia.20671. [DOI] [PubMed] [Google Scholar]
- 12.Pope S, Land JM, Heales SJ. Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim Biophys Acta. 2008;1777(7-8):794–799. doi: 10.1016/j.bbabio.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Ling Y, Morgan K, Kalsheker N. Amyloid precursor protein (APP) and the biology of proteolytic processing: relevance to Alzheimer's disease. Int J Biochem Cell Biol. 2003;35(11):1505–1535. doi: 10.1016/s1357-2725(03)00133-x. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, Takeuchi M, Makita Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer's disease. Brain Res. 2001;888(2):256–262. doi: 10.1016/s0006-8993(00)03075-4. [DOI] [PubMed] [Google Scholar]
- 15.Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. doi: 10.1038/cddis.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks JB, Lai Y, Sheng W, Yang X, Zhu D, Sun GY, Lee JC. Amyloid-beta peptide induces temporal membrane biphasic changes in astrocytes through cytosolic phospholipase A2. Biochim Biophys Acta. 2008;1778(11):2512–2519. doi: 10.1016/j.bbamem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui H, Hung AC, Freeman C, Narkowicz C, Jacobson GA, Small DH. Size and sulfation are critical for the effect of heparin on APP processing and Abeta production. J Neurochem. 2012;123(3):447–457. doi: 10.1111/j.1471-4159.2012.07929.x. [DOI] [PubMed] [Google Scholar]
- 18.Tomita S, Kirino Y, Suzuki T. Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem. 1998;273(11):6277–6284. doi: 10.1074/jbc.273.11.6277. [DOI] [PubMed] [Google Scholar]
- 19.Mulder SD, Veerhuis R, Blankenstein MA, Nielsen HM. The effect of amyloid associated proteins on the expression of genes involved in amyloid-beta clearance by adult human astrocytes. Exp Neurol. 2012;233(1):373–379. doi: 10.1016/j.expneurol.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci. 2006;26(43):10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Racchetti G, D'Alessandro R, Meldolesi J. Astrocyte stellation, a process dependent on Rac1 is sustained by the regulated exocytosis of enlargeosomes. Glia. 2012;60(3):465–475. doi: 10.1002/glia.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein GW. Endothelial cell-astrocyte interactions A cellular model of the blood-brain barrier. Ann N Y Acad Sci. 1988;529:31–39. doi: 10.1111/j.1749-6632.1988.tb51417.x. [DOI] [PubMed] [Google Scholar]
- 23.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200(6):629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutly F, Schwab ME. Neurons and astrocytes influence the development of purified O-2A progenitor cells. Glia. 1991;4(6):559–571. doi: 10.1002/glia.440040603. [DOI] [PubMed] [Google Scholar]
- 25.Escartin C, Valette J, Lebon V, Bonvento G. Neuron-astrocyte interactions in the regulation of brain energy metabolism: a focus on NMR spectroscopy. J Neurochem. 2006;99(2):393–401. doi: 10.1111/j.1471-4159.2006.04083.x. [DOI] [PubMed] [Google Scholar]
- 26.Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, Mavroudis G, Ray DE. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45(4):325–337. doi: 10.1002/glia.10333. [DOI] [PubMed] [Google Scholar]
- 27.Tran ND, Schreiber SS, Fisher M. Astrocyte regulation of endothelial tissue plasminogen activator in a blood-brain barrier model. J Cereb Blood Flow Metab. 1998;18(12):1316–1324. doi: 10.1097/00004647-199812000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Li G, Simon MJ, Cancel LM, Shi ZD, Ji X, Tarbell JM, Morrison B, 3rd, Fu BM. Permeability of endothelial and astrocyte cocultures: in vitro blood-brain barrier models for drug delivery studies. Ann Biomed Eng. 2010;38(8):2499–2511. doi: 10.1007/s10439-010-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchetti B, L'Episcopo F, Morale MC, Tirolo C, Testa N, Caniglia S, Serapide MF, Pluchino S. Uncovering novel actors in astrocyte-neuron crosstalk in Parkinson's disease: the Wnt/beta-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. Eur J Neurosci. 2013;37(10):1550–1563. doi: 10.1111/ejn.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waak J, Weber SS, Waldenmaier A, Gorner K, Alunni-Fabbroni M, Schell H, Vogt-Weisenhorn D, Pham TT, Reumers V, Baekelandt V, et al. Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ-1. FASEB J. 2009;23(8):2478–2489. doi: 10.1096/fj.08-125153. [DOI] [PubMed] [Google Scholar]
- 31.Brosnan CF, Raine CS. The astrocyte in multiple sclerosis revisited. Glia. 2013;61(4):453–465. doi: 10.1002/glia.22443. [DOI] [PubMed] [Google Scholar]
- 32.Rajkowska G, Stockmeier CA. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. 2013 doi: 10.2174/13894501113149990156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scuderi C, Steardo L. Neuroglial roots of neurodegenerative diseases: therapeutic potential of palmitoylethanolamide in models of Alzheimer's disease. CNS Neurol Disord Drug Targets. 2013;12(1):62–69. doi: 10.2174/1871527311312010011. [DOI] [PubMed] [Google Scholar]
- 34.Episcopo FL, Tirolo C, Testa N, Caniglia S, Morale MC, Marchetti B. Reactive astrocytes are key players in nigrostriatal dopaminergic neurorepair in the mptp mouse model of Parkinson's disease: focus on endogenous neurorestoration. Curr Aging Sci. 2013;6(1):45–55. doi: 10.2174/1874609811306010007. [DOI] [PubMed] [Google Scholar]
- 35.Park SJ, Jung JM, Lee HE, Lee YW, Kim DH, Kim JM, Hong JG, Lee CH, Jung IH, Cho YB, et al. The memory ameliorating effects of INM-176, an ethanolic extract of Angelica gigas, against scopolamine- or Abeta(1-42)-induced cognitive dysfunction in mice. J Ethnopharmacol. 2012;143(2):611–620. doi: 10.1016/j.jep.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 36.Peake KB, Vance JE. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J Biol Chem. 2012;287(12):9290–9298. doi: 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomi M, Zhao Y, Thamotharan S, Shin BC, Devaskar SU. Early life nutrient restriction impairs blood-brain metabolic profile and neurobehavior predisposing to Alzheimer's disease with aging. Brain Res. 2013;1495:61–75. doi: 10.1016/j.brainres.2012.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamashiro-Duran JH, Squarzoni P, de Souza Duran FL, Curiati PK, Vallada HP, Buchpiguel CA, Lotufo PA, Wajngarten M, Menezes PR, Scazufca M, et al. Cardiovascular risk in cognitively preserved elderlies is associated with glucose hypometabolism in the posterior cingulate cortex and precuneus regardless of brain atrophy and apolipoprotein gene variations. Age (Dordr) 2013;35(3):777–792. doi: 10.1007/s11357-012-9413-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.La Joie R, Perrotin A, Barre L, Hommet C, Mezenge F, Ibazizene M, Camus V, Abbas A, Landeau B, Guilloteau D, et al. Region-specific hierarchy between atrophy, hypometabolism, and beta-amyloid (Abeta) load in Alzheimer's disease dementia. J Neurosci. 2012;32(46):16265–16273. doi: 10.1523/JNEUROSCI.2170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC, Clegg DJ. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol Aging. 2012;33(2):425, e419–427. doi: 10.1016/j.neurobiolaging.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF. Multiple defects in energy metabolism in Alzheimer's disease. Curr Drug Targets. 2010;11(10):1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- 42.Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, et al. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008;105(11):4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoyer S. Risk factors for Alzheimer's disease during aging Impacts of glucose/energy metabolism. J Neural Transm Suppl. 1998;54:187–194. doi: 10.1007/978-3-7091-7508-8_18. [DOI] [PubMed] [Google Scholar]
- 44.Pietrini P, Dani A, Furey ML, Alexander GE, Freo U, Grady CL, Mentis MJ, Mangot D, Simon EW, Horwitz B, et al. Low glucose metabolism during brain stimulation in older Down's syndrome subjects at risk for Alzheimer's disease prior to dementia. Am J Psychiatry. 1997;154(8):1063–1069. doi: 10.1176/ajp.154.8.1063. [DOI] [PubMed] [Google Scholar]
- 45.Hoyer S. Oxidative metabolism deficiencies in brains of patients with Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:18–24. doi: 10.1111/j.1600-0404.1996.tb05868.x. [DOI] [PubMed] [Google Scholar]
- 46.Hoyer S. Brain oxidative energy and related metabolism, neuronal stress, and Alzheimer's disease: a speculative synthesis. J Geriatr Psychiatry Neurol. 1993;6(1):3–13. doi: 10.1177/002383099300600101. [DOI] [PubMed] [Google Scholar]
- 47.Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis. 2006;9(1):1–12. doi: 10.3233/jad-2006-9101. [DOI] [PubMed] [Google Scholar]
- 48.Messier C, Gagnon M. Glucose regulation and cognitive functions: relation to Alzheimer's disease and diabetes. Behav Brain Res. 1996;75(1-2):1–11. doi: 10.1016/0166-4328(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 49.Peppard RF, Martin WR, Carr GD, Grochowski E, Schulzer M, Guttman M, McGeer PL, Phillips AG, Tsui JK, Calne DB. Cerebral glucose metabolism in Parkinson's disease with and without dementia. Arch Neurol. 1992;49(12):1262–1268. doi: 10.1001/archneur.1992.00530360060019. [DOI] [PubMed] [Google Scholar]
- 50.Bigl M, Bruckner MK, Arendt T, Bigl V, Eschrich K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J Neural Transm. 1999;106(5-6):499–511. doi: 10.1007/s007020050174. [DOI] [PubMed] [Google Scholar]
- 51.Yun SW, Hoyer S. Effects of low-level lead on glycolytic enzymes and pyruvate dehydrogenase of rat brain in vitro: relevance to sporadic Alzheimer's disease? J Neural Transm. 2000;107(3):355–368. doi: 10.1007/s007020050030. [DOI] [PubMed] [Google Scholar]
- 52.Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci U S A. 2010;107(41):17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zilberter M, Ivanov A, Ziyatdinova S, Mukhtarov M, Malkov A, Alpar A, Tortoriello G, Botting CH, Fulop L, Osypov AA, et al. Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer's disease. J Neurochem. 2013;125(1):157–171. doi: 10.1111/jnc.12127. [DOI] [PubMed] [Google Scholar]
- 54.Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, Siddiqui A, Tamura Y, Sesaki H, Wengenack TM, et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer's disease. PLoS One. 2012;7(2):e32737. doi: 10.1371/journal.pone.0032737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rocchi A, Valensin D, Aldinucci C, Giani G, Barbucci R, Gaggelli E, Kozlowski H, Valensin G. NMR metabolomic investigation of astrocytes interacted with Abeta(4)(2) or its complexes with either copper(II) or zinc(II) J Inorg Biochem. 2012;117:326–333. doi: 10.1016/j.jinorgbio.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 56.Jensen CJ, Massie A, De Keyser J. Immune Players in the CNS: The Astrocyte. J Neuroimmune Pharmacol. 2013 doi: 10.1007/s11481-013-9480-6. [DOI] [PubMed] [Google Scholar]
- 57.Wu X, Li J, Chen C, Yan Y, Jiang S, Shao B, Xu J, Kang L, Huang Y, Zhu L, et al. Involvement of CLEC16A in activation of astrocytes after LPS treated. Neurochem Res. 2012;37(1):5–14. doi: 10.1007/s11064-011-0581-4. [DOI] [PubMed] [Google Scholar]
- 58.Aronica E, Ravizza T, Zurolo E, Vezzani A. Astrocyte immune responses in epilepsy. Glia. 2012;60(8):1258–1268. doi: 10.1002/glia.22312. [DOI] [PubMed] [Google Scholar]
- 59.Ishida Y, Nagai A, Kobayashi S, Kim SU. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol. 2006;65(1):66–77. doi: 10.1097/01.jnen.0000195941.48033.eb. [DOI] [PubMed] [Google Scholar]
- 60.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 61.Arregui L, Benitez JA, Razgado LF, Vergara P, Segovia J. Adenoviral astrocyte-specific expression of BDNF in the striata of mice transgenic for Huntington's disease delays the onset of the motor phenotype. Cell Mol Neurobiol. 2011;31(8):1229–1243. doi: 10.1007/s10571-011-9725-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scheiber IF, Dringen R. Astrocyte functions in the copper homeostasis of the brain. Neurochem Int. 2013;62(5):556–565. doi: 10.1016/j.neuint.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 63.Konopaske GT, Bolo NR, Basu AC, Renshaw PF, Coyle JT. Time-dependent effects of haloperidol on glutamine and GABA homeostasis and astrocyte activity in the rat brain. Psychopharmacology (Berl) 2013 doi: 10.1007/s00213-013-3136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S, Torres C. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010;316(17):2961–2968. doi: 10.1016/j.yexcr.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 65.Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7(9):e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mattson MP, Chan SL. Dysregulation of cellular calcium homeostasis in Alzheimer's disease: bad genes and bad habits. J Mol Neurosci. 2001;17(2):205–224. doi: 10.1385/JMN:17:2:205. [DOI] [PubMed] [Google Scholar]
- 67.Schubert P, Ogata T, Marchini C, Ferroni S. Glia-related pathomechanisms in Alzheimer's disease: a therapeutic target? Mech Ageing Dev. 2001;123(1):47–57. doi: 10.1016/s0047-6374(01)00343-8. [DOI] [PubMed] [Google Scholar]
- 68.Quintanilla RA, Orellana JA, von Bernhardi R. Understanding risk factors for Alzheimer's disease: interplay of neuroinflammation, connexin-based communication and oxidative stress. Arch Med Res. 2012;43(8):632–644. doi: 10.1016/j.arcmed.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 69.Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease. Curr Pharm Des. 2010;16(25):2766–2778. doi: 10.2174/138161210793176572. [DOI] [PubMed] [Google Scholar]
- 70.Mhatre M, Floyd RA, Hensley K. Oxidative stress and neuroinflammation in Alzheimer's disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis. 2004;6(2):147–157. doi: 10.3233/jad-2004-6206. [DOI] [PubMed] [Google Scholar]
- 71.Cai Z, Zhao B, Ratka A. Oxidative stress and beta-amyloid protein in Alzheimer's disease. Neuromolecular Med. 2011;13(4):223–250. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- 72.Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer's disease. Biochim Biophys Acta. 2012;1822(5):650–656. doi: 10.1016/j.bbadis.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 73.Massaad CA. Neuronal and vascular oxidative stress in Alzheimer's disease. Curr Neuropharmacol. 2011;9(4):662–673. doi: 10.2174/157015911798376244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vijayan VK, Geddes JW, Anderson KJ, Chang-Chui H, Ellis WG, Cotman CW. Astrocyte hypertrophy in the Alzheimer's disease hippocampal formation. Exp Neurol. 1991;112(1):72–78. doi: 10.1016/0014-4886(91)90115-s. [DOI] [PubMed] [Google Scholar]
- 75.von Bernhardi R, Ramirez G. Microglia-astrocyte interaction in Alzheimer's disease: friends or foes for the nervous system? Biol Res. 2001;34(2):123–128. doi: 10.4067/s0716-97602001000200017. [DOI] [PubMed] [Google Scholar]
- 76.Gatta LB, Vitali M, Verardi R, Arosio P, Finazzi D. Inhibition of heme synthesis alters Amyloid Precursor Protein processing. J Neural Transm. 2009;116(1):79–88. doi: 10.1007/s00702-008-0147-z. [DOI] [PubMed] [Google Scholar]
- 77.Strazielle C, Jazi R, Verdier Y, Qian S, Lalonde R. Regional brain metabolism with cytochrome c oxidase histochemistry in a PS1/A246E mouse model of autosomal dominant Alzheimer's disease: correlations with behavior and oxidative stress. Neurochem Int. 2009;55(8):806–814. doi: 10.1016/j.neuint.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 78.Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J. Amyloid-beta reduction by memapsin 2 (beta-secretase) immunization. FASEB J. 2007;21(12):3184–3196. doi: 10.1096/fj.06-7993com. [DOI] [PubMed] [Google Scholar]
- 79.Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- 80.Sawmiller DR, Nguyen HT, Markov O, Chen M. High-energy compounds promote physiological processing of Alzheimer's amyloid-beta precursor protein and boost cell survival in culture. J Neurochem. 2012;123(4):525–531. doi: 10.1111/j.1471-4159.2012.07923.x. [DOI] [PubMed] [Google Scholar]
- 81.Yao J, Chen S, Mao Z, Cadenas E, Brinton RD. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS One. 2011;6(7):e21788. doi: 10.1371/journal.pone.0021788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng Y, Wang Z, Wang R, Zhang X, Zhang S, Wu Y, Staufenbiel M, Cai F, Song W. Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci. 2013;37(12):1962–1969. doi: 10.1111/ejn.12235. [DOI] [PubMed] [Google Scholar]
- 83.Cole SL, Vassar R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J Biol Chem. 2008;283(44):29621–29625. doi: 10.1074/jbc.R800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25(47):10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Zhou W, Qiao H. Bioenergetic homeostasis decides neuroprotection or neurotoxicity induced by volatile anesthetics: a uniform mechanism of dual effects. Med Hypotheses. 2011;77(2):223–229. doi: 10.1016/j.mehy.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 86.Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KS, Shohami E, Stein R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia. 2008;56(5):552–567. doi: 10.1002/glia.20638. [DOI] [PubMed] [Google Scholar]
- 87.Walter J, Haass C. Posttranslational modifications of amyloid precursor protein : ectodomain phosphorylation and sulfation. Methods Mol Med. 2000;32:149–168. doi: 10.1385/1-59259-195-7:149. [DOI] [PubMed] [Google Scholar]
- 88.Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76(1):173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 89.Johnson RJ, Xiao G, Shanmugaratnam J, Fine RE. Calreticulin functions as a molecular chaperone for the beta-amyloid precursor protein. Neurobiol Aging. 2001;22(3):387–395. doi: 10.1016/s0197-4580(00)00247-5. [DOI] [PubMed] [Google Scholar]
- 90.Perdivara I, Petrovich R, Allinquant B, Deterding LJ, Tomer KB, Przybylski M. Elucidation of O-glycosylation structures of the beta-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J Proteome Res. 2009;8(2):631–642. doi: 10.1021/pr800758g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Griffith LS, Mathes M, Schmitz B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J Neurosci Res. 1995;41(2):270–278. doi: 10.1002/jnr.490410214. [DOI] [PubMed] [Google Scholar]
- 92.Georgopoulou N, McLaughlin M, McFarlane I, Breen KC. The role of post-translational modification in beta-amyloid precursor protein processing. Biochem Soc Symp. 2001;(67):23–36. doi: 10.1042/bss0670023. [DOI] [PubMed] [Google Scholar]
- 93.Maccioni RB, Munoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001;32(5):367–381. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 94.Murakami-Sekimata A, Sato K, Takashima A, Nakano A. O-Mannosylation is required for the solubilization of heterologously expressed human beta-amyloid precursor protein in Saccharomyces cerevisiae. Genes Cells. 2009;14(2):205–215. doi: 10.1111/j.1365-2443.2008.01263.x. [DOI] [PubMed] [Google Scholar]
- 95.Sano Y, Nakaya T, Pedrini S, Takeda S, Iijima-Ando K, Iijima K, Mathews PM, Itohara S, Gandy S, Suzuki T. Physiological mouse brain Abeta levels are not related to the phosphorylation state of threonine-668 of Alzheimer's APP. PLoS One. 2006;1:e51. doi: 10.1371/journal.pone.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sodhi CP, Perez RG, Gottardi-Littell NR. Phosphorylation of beta-amyloid precursor protein (APP) cytoplasmic tail facilitates amyloidogenic processing during apoptosis. Brain Res. 2008;1198:204–212. doi: 10.1016/j.brainres.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 97.Chang KA, Kim HS, Ha TY, Ha JW, Shin KY, Jeong YH, Lee JP, Park CH, Kim S, Baik TK, et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol. 2006;26(11):4327–4338. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163(1):83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 100.Buccoliero R, Bodennec J, Van Echten-Deckert G, Sandhoff K, Futerman AH. Phospholipid synthesis is decreased in neuronal tissue in a mouse model of Sandhoff disease. J Neurochem. 2004;90(1):80–88. doi: 10.1111/j.1471-4159.2004.02457.x. [DOI] [PubMed] [Google Scholar]
- 101.Yang Y, Yang D, Tang G, Zhou C, Cheng K, Zhou J, Wu B, Peng Y, Liu C, Zhan Y, et al. Proteomics reveals energy and glutathione metabolic dysregulation in the prefrontal cortex of a rat model of depression. Neuroscience. 2013;247:191–200. doi: 10.1016/j.neuroscience.2013.05.031. [DOI] [PubMed] [Google Scholar]
- 102.Martinez E, Gerard N, Garcia MM, Mazur A, Gueant-Rodriguez RM, Comte B, Gueant JL, Brachet P. Myocardium proteome remodelling after nutritional deprivation of methyl donors. J Nutr Biochem. 2013;24(7):1241–1250. doi: 10.1016/j.jnutbio.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 103.Chen X, Wei S, Yang F. Mitochondria in the pathogenesis of diabetes: a proteomic view. Protein Cell. 2012;3(9):648–660. doi: 10.1007/s13238-012-2043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mrak RE, Sheng JG, Griffin WS. Glial cytokines in Alzheimer's disease: review and pathogenic implications. Hum Pathol. 1995;26(8):816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wallace MN, Geddes JG, Farquhar DA, Masson MR. Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp Neurol. 1997;144(2):266–272. doi: 10.1006/exnr.1996.6373. [DOI] [PubMed] [Google Scholar]
- 107.DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Neurol. 1998;149(2):329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 108.Mrak RE, Griffinbc WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22(6):915–922. doi: 10.1016/s0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- 109.Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer's disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. 1999;160(1):289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- 110.Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer's disease. Glia. 2010;58(3):300–314. doi: 10.1002/glia.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 2011;122(3):293–311. doi: 10.1007/s00401-011-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goos JD, Teunissen CE, Veerhuis R, Verwey NA, Barkhof F, Blankenstein MA, Scheltens P, van der Flier WM. Microbleeds relate to altered amyloid-beta metabolism in Alzheimer's disease. Neurobiol Aging. 2012;33(5):1011 e1011–1019. doi: 10.1016/j.neurobiolaging.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 113.Pflanzner T, Janko MC, Andre-Dohmen B, Reuss S, Weggen S, Roebroek AJ, Kuhlmann CR, Pietrzik CU. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol Aging. 2011;32(12):2323 e2321–2311. doi: 10.1016/j.neurobiolaging.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 114.Wong HE, Qi W, Choi HM, Fernandez EJ, Kwon I. A safe, blood-brain barrier permeable triphenylmethane dye inhibits amyloid-beta neurotoxicity by generating nontoxic aggregates. ACS Chem Neurosci. 2011;2(11):645–657. doi: 10.1021/cn200056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rutgers KS, Nabuurs RJ, van den Berg SA, Schenk GJ, Rotman M, Verrips CT, van Duinen SG, Maat-Schieman ML, van Buchem MA, de Boer AG, et al. Transmigration of beta amyloid specific heavy chain antibody fragments across the in vitro blood-brain barrier. Neuroscience. 2011;190:37–42. doi: 10.1016/j.neuroscience.2011.05.076. [DOI] [PubMed] [Google Scholar]
- 116.Jeynes B, Provias J. The case for blood-brain barrier dysfunction in the pathogenesis of Alzheimer's disease. J Neurosci Res. 2011;89(1):22–28. doi: 10.1002/jnr.22527. [DOI] [PubMed] [Google Scholar]
- 117.Banks WA. Drug delivery to the brain in Alzheimer's disease: consideration of the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):629–639. doi: 10.1016/j.addr.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou QH, Fu A, Boado RJ, Hui EK, Lu JZ, Pardridge WM. Receptor-mediated abeta amyloid antibody targeting to Alzheimer's disease mouse brain. Mol Pharm. 2011;8(1):280–285. doi: 10.1021/mp1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD. RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer's disease. Curr Mol Med. 2007;7(8):735–742. doi: 10.2174/156652407783220741. [DOI] [PubMed] [Google Scholar]
- 121.Askarova S, Yang X, Sheng W, Sun GY, Lee JC. Role of Abeta-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A(2) activation in astrocytes and cerebral endothelial cells. Neuroscience. 2011 doi: 10.1016/j.neuroscience.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Candela P, Gosselet F, Saint-Pol J, Sevin E, Boucau MC, Boulanger E, Cecchelli R, Fenart L. Apical-to-basolateral transport of amyloid-beta peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J Alzheimers Dis. 2010;22(3):849–859. doi: 10.3233/JAD-2010-100462. [DOI] [PubMed] [Google Scholar]
- 123.Pflanzner T, Janko MC, Andre-Dohmen B, Reuss S, Weggen S, Roebroek AJ, Kuhlmann CR, Pietrzik CU. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 124.Ito S, Ueno T, Ohtsuki S, Terasaki T. Lack of brain-to-blood efflux transport activity of low-density lipoprotein receptor-related protein-1 (LRP-1) for amyloid-beta peptide(1-40) in mouse: involvement of an LRP-1-independent pathway. J Neurochem. 2010;113(5):1356–1363. doi: 10.1111/j.1471-4159.2010.06708.x. [DOI] [PubMed] [Google Scholar]
- 125.Leake A, Morris CM, Whateley J. Brain matrix metalloproteinase 1 levels are elevated in Alzheimer's disease. Neurosci Lett. 2000;291(3):201–203. doi: 10.1016/s0304-3940(00)01418-x. [DOI] [PubMed] [Google Scholar]
- 126.Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer's disease. Curr Pharm Des. 2008;14(16):1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2007;4(2):191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- 128.Lee SH, Takahashi M, Honke K, Miyoshi E, Osumi D, Sakiyama H, Ekuni A, Wang X, Inoue S, Gu J, et al. Loss of core fucosylation of low-density lipoprotein receptor-related protein-1 impairs its function, leading to the upregulation of serum levels of insulin-like growth factor-binding protein 3 in Fut8-/- mice. J Biochem. 2006;139(3):391–398. doi: 10.1093/jb/mvj039. [DOI] [PubMed] [Google Scholar]
- 129.Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108(1):165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- 130.Sailasuta N, Harris K, Tran T, Ross B. Minimally invasive biomarker confirms glial activation present in Alzheimer's disease: a preliminary study. Neuropsychiatr Dis Treat. 2011;7:495–499. doi: 10.2147/NDT.S23721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jhala SS, Hazell AS. Modeling neurodegenerative disease pathophysiology in thiamine deficiency: consequences of impaired oxidative metabolism. Neurochem Int. 2011;58(3):248–260. doi: 10.1016/j.neuint.2010.11.019. [DOI] [PubMed] [Google Scholar]