

Abstract
Basic studies of human pluripotential stem cells have advanced rapidly and stem cell products are now seeing therapeutic applications. However, questions remain regarding the tumorigenic potential of such cells. Here, we report the tumorigenic potential of induced pluripotent stem cell (iPSC)-derived retinal pigment epithelium (RPE) for the treatment of wet-type, age-related macular degeneration (AMD). First, immunodeficient mouse strains (nude, SCID, NOD-SCID and NOG) were tested for HeLa cells’ tumor-forming capacity by transplanting various cell doses subcutaneously with or without Matrigel. The 50% Tumor Producing Dose (TPD50 value) is the minimal dose of transplanted cells that generated tumors in 50% of animals. For HeLa cells, the TPD50 was the lowest when cells were embedded in Matrigel and transplanted into NOG mice (TPD50 = 101.1, n = 75). The TPD50 for undifferentiated iPSCs transplanted subcutaneously to NOG mice in Matrigel was 102.12; (n = 30). Based on these experiments, 1×106 iPSC-derived RPE were transplanted subcutaneously with Matrigel, and no tumor was found during 15 months of monitoring (n = 65). Next, to model clinical application, we assessed the tumor-forming potential of HeLa cells and iPSC 201B7 cells following subretinal transplantation of nude rats. The TPD50 for iPSCs was 104.73 (n = 20) and for HeLa cells 101.32 (n = 37) respectively. Next, the tumorigenicity of iPSC-derived RPE was tested in the subretinal space of nude rats by transplanting 0.8–1.5×104 iPSC-derived RPE in a collagen-lined (1 mm×1 mm) sheet. No tumor was found with iPSC-derived RPE sheets during 6–12 months of monitoring (n = 26). Considering the number of rodents used, the monitoring period, the sensitivity of detecting tumors via subcutaneous and subretinal administration routes and the incidence of tumor formation from the iPSC-derived RPE, we conclude that the tumorigenic potential of the iPSC-derived RPE was negligible.
Introduction
Clinical cell therapy trials were recently initiated for treatment of Stargardt’s disease and the dry type of age-related macular degeneration (dry AMD). The trials have used human embryonic stem cell (hESC)-derived retinal pigment epithelium (RPE) [1]–[4]. In addition, several groups are planning clinical trials with autologous human induced pluripotent stem cell (hiPSC)-derived RPE for the wet type of AMD. Thus, cell therapy using human pluripotent stem cells (hPSCs) has reached clinical application. However, in contrast to tissue stem cells that have a limited proliferation potential, tumor formation from residual undifferentiated or incompletely differentiated hPSCs in hPSC-derived cell products is an issue that must be carefully analyzed. This issue is particularly important when transplanting autologous hiPSC-derived cells.
We recently reported a highly sensitive residual hiPSC detection method based on qRT-PCR using primers for the LIN28A transcript [5] in hiPSC-derived RPE. This method enables us to detect residual hiPSCs down to 0.002% of differentiated RPE cells. As we plan to transplant 4–8×104 hiPSC-derived RPE cells into the subretinal space of patients, this method is sensitive enough to detect a few residual hiPSCs, if any, in a clinical setting.
The tumorigenic potential of hiPSC-derived RPE cells is attributable to contamination by undifferentiated hiPSCs, intermediate products having proliferation potentials and/or tumorigenic transformed cells. Contamination by these cells should be assessed by nonclinical testing using suitable animal models [6], [7]. However, there is no internationally recognized guideline for tumorigenicity testing in cell therapy products. The most relevant guideline is the WHO TRS 878, “Recommendation for the evaluation of animal cell cultures as substrates for the manufacture of cell banks” [8], [9]. The guideline recommends transplanting 1×107 test cells subcutaneously to 10 nude mice and monitoring tumor formation for more than 16 weeks. Transplantation of the same dose of a well-known tumorigenic cell line such as HeLa in parallel is suggested as a tumor-forming positive control. The WHO guideline covers animal cell substrates for the production of biological medicinal products and specifically excludes viable animal cells that are intended for therapeutic transplantation into patients. To examine the tumorigenicity of hiPSC-derived cells intended for administration to patients, several teratoma-forming tests exploring dose and administration route were studied using immuno-deficient mice [6],[10]. However, discussions how we can interpret and extrapolate the results of tumorigenicity testing with immuno-deficient or immuno-suppressant animals to human patients continue [6], [7]. Recently a commentary report from FDA/CBER pointed out the issues to be considered for cell-based products and associated challenges for preclinical animal study [11]. The report stated that although the nature of cells used for cellular therapy is diverse, tumorigenic test results from the administration of cells through nonclinical routes would not be considered relevant as it would not track the behavior of transplanted cells in a micro-environment. When tumorigenicity testing of ESC-derived cellular products is undertaken, the study design should include groups of animals that have received undifferentiated ESCs, serial dilutions of undifferentiated ESCs combined with ESC-derived final products and the final intended clinical products. This approach would thereby address the tumor-forming potential of these cell groups in animal models.
Tumorigenicity testing via the clinical route of administration could recapitulate the fate of transplanted cells in a microenvironment of host tissue and could be fairly extrapolated to human application. However, elaborate surgical intervention requires skills that greatly influence the outcome of transplantation. For example, it is difficult to determine whether the cells were transplanted into the right location or organ in small rodents. These concerns can be overcome by conducting a subcutaneous tumorigenicity test in addition to testing via the clinical route.
In this report, we conducted 2 types of in vivo tumorigenicity tests by transplanting hiPSC-derived RPE cells into subcutaneous and sub-retinal spaces in immuno-deficient animals. The results and limits of these tests are discussed.
Results
Tumorigenicity Tests with Several Types of Immuno-deficient Mice
The tumor-forming potential of human iPSC-derived cell products should be examined using a suitable animal transplantation model. One should take into account the number of cells to be transplanted, the method of transplantation, the microenvironment of the transplantation site, the monitoring period and the status of the immune-deficient animals.
First, we checked the tumor-forming potential of several immune-deficient animals by subcutaneously transplanting HeLa cells over a wide range of doses (1 to 1×106 in 10-fold increments) with or without Matrigel (BD) and observed tumor formation every day for up to 36 weeks on a daily basis. Matrigel is known to enhance the tumor-forming potential of transplanted cells [16]. Recipient animals included immune-deficient nude, SCID [10], NOD-SCID [17], and NOG [18] mice. The minimal dose of transplanted cells that generated tumors in 50% of the transplanted animal (TPD50) was calculated statistically to evaluate the sensitivities of tumor formation in each animal model [19]. We found the NOG mouse was most susceptible to tumors. That is, when transplanted subcutaneously with Matrigel, tumors were generated by the lowest number of HeLa cells. The TPD50 for HeLa was 101.1 (n = 75), in agreement with a previous report [20], (Figure 1, Table 1). It is interesting to note that among the conditions tested, the highest number of HeLa cells was required to form tumors in nude mice without Matrigel. TPD50 for nude mice without Matrigel was 104.9 (n = 120), which is also in agreement with a previous report [19] (Figure 1, Table 1). Therefore, we selected NOG mice and Matrigel for embedding the test cells for further assays as it provided sensitive tumor detection using small numbers of transplanted cells. The tumor-formation potential of iPSCs was assessed by subcutaneously transplanting several doses of the iPSC cell line 201B7 with Matrigel into NOG mice. The TPD50 for iPSC was 102.12 (n = 30) over 12 months’ monitoring (Table 2). The TPD50 value for iPSCs in subcutaneous transplantation provided a reference cell number for the contamination of iPSCs in iPSC-derived RPE cells.
Figure 1. Tumorigenicity testing (TPD50 log10) by subcutaneous transplantation of HeLa cells.

Log10TPD50 values (minimal cell doses for 50% of animals to form a tumor) for HeLa cells when transplanted subcutaneously in various immuno-deficient mouse strains (nude, SCID, NOD-SCID, NOG) with or without Matrigel as indicated. Abscissa, weeks after transplantation. Ordinate, Log10 TPD50 values, logarithmic scale.
Table 1. Incidence of tumor formation after transplanting HeLa cells in various immunodeficient mice.
| strain | use of Matrigel | min.dose fortumor formation | weeks to observeTumor (first to last) | number of mice | Log10TPD50 |
| nude | with | 1×104 cells | 3 to 8 | 120 | 3.5 |
| nude | w/o | 1×104 cells | 4 to 12 | 120 | 4.9 |
| SCID | with | 1×103 cells | 3 to 11 | 24 | 2.5 |
| SCID | w/o | 1×103 cells | 3 to 11 | 24 | 3.83 |
| NOD-SCID | with | 1×102 cells | 3 to 16 | 24 | 2.17 |
| NOD-SCID | w/o | 1×103 cells | 3 to 14 | 24 | 3.5 |
| NOG | with | 1×101 cells | 5 to 18 | 75 | 1.1 |
| NOG | w/o | 1×104 cells | 3 to 13 | 105 | 3.97 |
Log10TPD50 values for HeLa cells transplanted subcutaneously into various immunodeficient mouse strains with or without Matrigel. Tumor-forming potentials of HeLa cells in nude mice without Matrigel and in NOG mice with Matrigel are highlighted in gray.
Table 2. Tumorigenicity testing by subcutaneous transplantation of hiPSC-derived RPE into NOG mice.
| hiPSC cell line | cell form | min.dose fortumor formation | weeks to observeTumor (first to last) | numberof mice | Log10TPD50 |
| 201B7 | Cell suspension in Matrigel | 1×101 cells | 5–40 | 30 | 2.12 |
| RPE cell line | cell form | number of cells transplanted | monitor period | number of mice | tumor formation |
| 59-G3(1) | RPE cell suspension in Matrigel | 1×106 cells | 26–84 weeks | 9 | none |
| K21-G18 | RPE cell suspension in Matrigel | 1×106 cells | 26–74 weeks | 8 | none |
| 101-G25 | RPE cell suspension in Matrigel | 1×106 cells | 23–70 weeks | 10 | none |
| 59-G3(1) | RPE cell sheet in Matrigel | 1×106 cells | 28–85 weeks | 5 | none |
| K21-G18 | RPE cell sheet in Matrigel | 1×106 cells | 13–79 weeks | 5 | none |
| 101-G25 | RPE cell sheet in Matrigel | 1×106 cells | 23–79 weeks | 5 | none |
| primary RPE | Cell suspension in Matrigel | 1×106 cells | 52 weeks | 3 | none |
| primary RPE | Cell suspension w/o Matrigel | 1×106 cells | 52 weeks | 2 | none |
| 59-G3(2) | RPE cell sheet in Matrigel | 1×106 cells | 26–50 weeks | 3 | none |
| RNT10 | RPE cell sheet in Matrigel | 1×106 cells | 26–46 weeks | 3 | none |
| RNT9 | RPE cell sheet in Matrigel | 1×106 cells | 26–38 weeks | 3 | none |
| 101-EV3 | RPE cell suspension in Matrigel | 1×106 cells | 39 weeks | 5 | none |
| K11-EV9 | RPE cell suspension in Matrigel | 1×106 cells | 39 weeks | 3 | none |
| K21-EV15 | RPE cell suspension w/o Matrigel | 1×106 cells | 39 weeks | 4 | none |
| K11-EV9 | RPE cell suspension w/o Matrigel | 1×106 cells | 39 weeks | 2 | none |
Log10TPD50 value for hiPSC 201B7 determined by subcutaneously transplanting cells in Matrigel into NOG was calculated by the Trimmed Spearman-Karber method (upper panel). Tumor formation from 1×106 hiPSC-derived RPE cells prior to making RPE sheets (cell suspension) or after making RPE sheets (cell sheet) transplanted subcutaneously in various conditions into NOG mice. Animals were monitored for 13–85 weeks (lower panel).
Characterization of Established hiPSCs and hiPSC-derived RPE
hiPSC lines 59-G3, 101-EV3, K11-EV9, K21-EV15 K21-G18, 101-G25, RNT9-2-8, and RNT10–24 were established from dermal fibroblasts of 6 patients (59, K11, K21, 101, RNT9, RNT10) with retinitis pigmentosa. Quality control tests for established iPSCs were as follows. (1) Cells form colonies and must show human ESC-like morphology by microscopic observation. (2) Cells must express SSEA-4, TRA-1-60, POU5F1 (OCT3/4) and NANOG proteins as determined by immunostaining. (3) Cells must not express EBNA plasmid fragment by PCR or qRT-PCR. (4). Cells must possess a normal karyotype by the G-band method.
Retinal differentiation was subsequently initiated. The resulting RPE cell lines were established as follows. 59-G3 RPE was derived from hiPSC clone 59-G3; 101-EV3 RPE was from 101-EV3; K11-EV9 RPE was from K11-EV9; K21-EV15 RPE was from K21-EV15; K21-G18 RPE was from K21-G18; 101-G25 RPE and RNT9 RPE were from RNT9-2-8; and, RNT10 RPE was from RNT10-24. The protocol for RPE differentiation from hiPSC was shown in our recent report [21]. It requires 3 months for RPE differentiation and another 2 months to prepare the RPE sheet. The following quality control tests for the hiPSC-derived RPE cell lines were conducted. (1) The EBNA plasmid fragment was not detectable by PCR. (2) The cells showed the characteristic morphology and pigmentation of RPE with a single or double layer cell structure. (3) BEST1 and PAX6 molecules were detected by immunohistochemistry in over 95% of final hiPSC-derived RPE cells. (4) RPE-specific markers RPE65, CRALBP, MERTK and BEST1 were confirmed by RT-PCR. (5) LIN28A was not detected by qRT-PCR. (6) Migration of non-RPE cells into the collagen layer lining the hiPSC-derived RPE cell sheet shall be below 0.1% of the total RPE cells. (7) The RPE cell sheet shall consist of over 70% viable cells with a density of over 4500 cells/mm2. Items (6) and (7) were quality control tests for the RPE cell sheet. All the cell culture processes including establishment of hiPSCs from a patient’s fibroblasts and differentiation to RPE were conducted in a GMP-grade cell processing facility. The morphology and immunostaining of hiPSC-derived RPE cell lines 59-G3 RPE, K21-G18 RPE and 101-G25 RPE are shown in Figure 2A, B. The other hiPSC-derived RPE cell lines showed the same phenotype. The gene expression patterns of these cell lines are shown in Figure 2C. Primary RPE was used as a reference. It is notable that neither LIN28A nor POU5F1 (OCT3/4) was detected above background levels in hiPSC-derived RPE cells5. This finding serves as a useful criterion to eliminate immature hiPSCs in hiPSC-derived RPE (Figure 2D, 2E).
Figure 2. Characterization of hiPSC-derived RPE.

A: Phase contrast images of hiPSC-derived RPE cell lines 59-G3 RPE, K21-G18 RPE and 101–G25 RPE. B: Expression of pluripotency-related molecules POU5F1 (OCT3/4, upper panels) and RPE-related molecules BEST1 (lower panels) in lines 59-G3 RPE, K21-G18 RPE and 101–G25 RPE as detected by immunostaining with specific antibodies. Nuclei were stained with DAPI. Magnified photos of BEST1 staining are appended in left lower corners. C: Gene expression profiles of hiPSC-derived RPE cell lines 59-G3-RPE, K21-G18 RPE, 101-G25 RPE. Expression of pluripotent stem cell-related gene markers LIN28A and POU5F1, or RPE-related makers BEST1 (bestrophin), CRALBP, PAX6 and TYR (tyrosinase) in hiPSC cell lines 201B7 and 836B1, primary RPE (hRPE-1) and hiPSC-derived RPE cell lines 59-G3 RPE, K21-G18 RPE, 101-G25 RPE as determined by RT-PCR (left panel). GAPDH was used as an internal control. 50 ng RNA was used for one RT reaction. Gene expression of LIN28A (D) or POU5F1 (E) in hiPSC-derived RPE cell lines 59-G3 RPE, K21-G18 RPE, 101-G25 RPE, hiPSC 836B1 and primary RPE was quantified by qRT-PCR. UD: undetectable level (D). *, P<0.005 (E). P values for primary RPE, 101-G25 RPE, 59-G3 RPE, or K21-G18 RPE versus 836B1 are 0.000153, 0.000177, 0.000432 or 0.000489, respectively.
Tumorigenicity Testing of hiPSC-derived RPE (Subcutaneous Transplantation)
To assess the tumorigenic potential of hiPSC-derived RPE cells with high sensitivity, subcutaneous transplantation of a large number of RPE cells would be ideal. However, the maximal number of RPE cells available for transplantation was limited by the culture capacity of the cell processing facility. We hypothesized that transplanting 1×106 hiPSC-derived RPE cells was an acceptable cell number to address the tumorigenic potential of the final cell product when embedded in Matrigel in NOG mice. This hypothesis was based on the facts that we expect to transplant 4–8×104 hiPSC-derived RPE in a clinical setting and as few as 10 undifferentiated hiPSCs or HeLa cells embedded in Matrigel could generate tumors in NOG mice (Table 1, Table 2).
Thus, we subcutaneously transplanted 1×106 hiPSC-derived RPE cells embedded in Matrigel into NOG mice [total n = 42; 59-G3 RPE (n = 14), K21-G18 RPE (n = 13), 101-G25 RPE (n = 15)]. Tumor formation was monitored for more than 70 weeks. Teratoma derived from subcutaneously transplanted iPSCs was analyzed as a positive control for tumor formation event (Figure 3A–3E). The proliferative status of living cells was assessed by HE, Hoechst 33258 and anti-Ki67 antibody staining (Figure 3G–3I). iPSC-derived neural rosette-like human cells were stained by anti-Lamin A antibody to check the specificity of anti-Lamin A antibody for human cells (Figure 3F). We assumed that anti-human Lamin A antibody could stain a wide range of human cell types and was not limited to human RPE. Transplantation of 1×106 primary RPE cells embedded in Matrigel (n = 3) was used as a transplantation control. No tumor formation was observed from transplanted 1×106 hiPSC-derived RPE of several origins in various administration forms. All of the subcutaneous tumorigenicity tests conducted for hiPSC-derived RPE under various conditions using NOG mice are shown in Table 2.
Figure 3. Histological analyses of hiPSCs subcutaneously transplanted into NOG mice.

Tumor (teratoma) in NOG mouse was detected 5 weeks after transplanting 1.0×104 hiPSCs embedded with Matrigel (A, B). HE staining of sectioned hiPSC-derived teratoma consisted of three germ layers: cartilage-like tissue (mesoderm) (C), intestinal epithelium-like tissue (endoderm) (D) and neural rosette-like tissue (ectoderm) (E). Anti-Lamin A antibody (F) staining of rosette-like tissue. HE (G), Hoechst 33258 (H) and anti-Ki-67 antibody (I) staining of cartilage-like tissue.
All subcutaneous transplants consisting of RPE cells embedded in Matrigel were excised and subjected to histological examination. The size of transplants in subcutaneous tissue (Figure 4A, 4B) was similar to that of Matrigel without RPE (Figure 4C). Histological and immunohistological study showed that Lamin A-, BEST1- and Hoechst-positively staining RPE cells were present in all the Matrigel transplants (Figure 4F–4M). None of the cells transplanted in Matrigel stained with anti-Ki67 antibody, suggesting the absence of active proliferation in these transplanted cells (Figure 4D, 4E). Human cells derived from transplanted iPSC-derived RPE could be detected by Alu PCR at a level of ≥0.1% in mouse cells. However, we could not detect human cells in subcutaneous mouse tissue just beneath the transplants, in liver, spleen, kidney or lung by this method (Figure 5A, 5B).
Figure 4. Histological analysis of hiPSC-derived RPE transplanted subcutaneously into NOG mice.

NOG mice were examined six months after transplantation of 1.0×106 hiPSC-derived RPE cells in Matrigel into subcutaneous tissue. No tumor was detected visually. Site of transplant (A), excised transplant (B), and excised Matrigel only transplant (Matrigel without RPE cells C). Transplants were sectioned and stained with HE (D) and anti-Ki67 antibody (E). Photomicrograph of unstained serial section (F), and section stained with Hoechst 33258 (G). Photomicrograph of unstained serial section (H) or stained with anti-Lamin A antibody (I). Photomicrograph of unstained serial section (J) or stained with anti-BEST1 antibody (K) and Hoechst 33258 (L) and merged (M). Ki-67 positive cells were not observed.
Figure 5. Detection of human cells in host mouse tissue by Alu PCR.
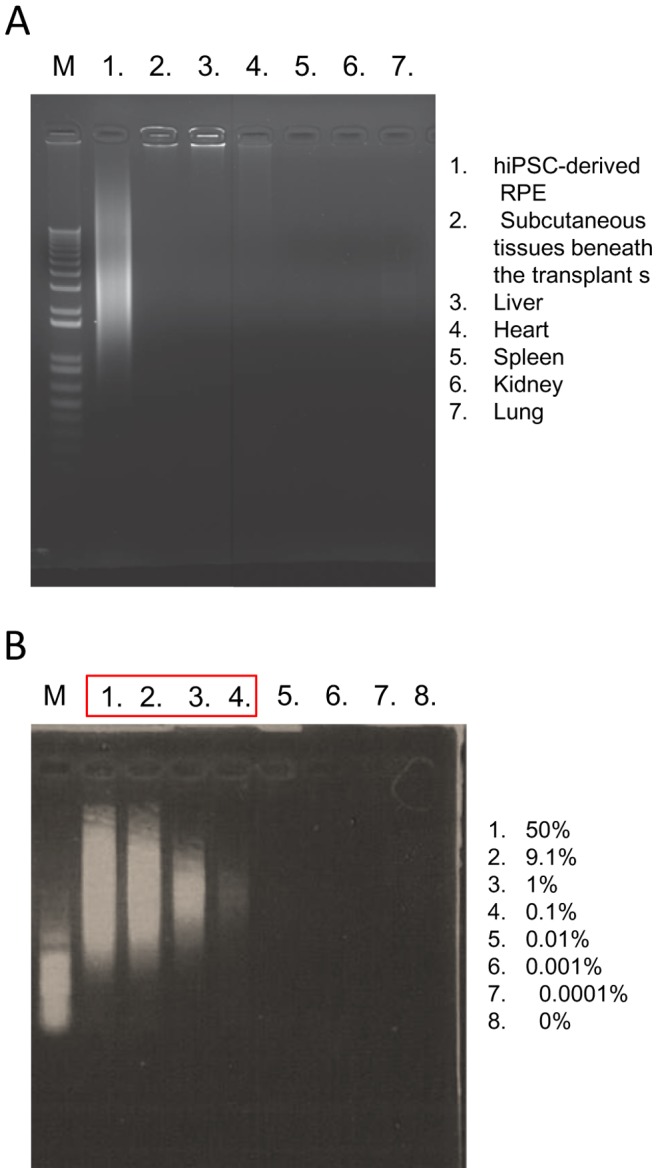
DNA from hiPSC-derived RPEs (positive control, Lane 1), NOG mouse subcutaneous tissue just beneath the transplants (2), mouse liver (3), mouse heart (4), mouse spleen (5), mouse kidney (6) and mouse lung (7) were used as PCR templates. M: 1 kb marker (A). Alu PCR detects ≥0.1% human cells included in mouse cells determined by visual assessment of PCR products generated from various ratios of human: mouse DNA template mixtures. Percentage of human DNA in DNA mixture is shown in a respective lane number (1–8) (B). M: 1 kb marker.
Tumorigenicity Test of hiPSC-derived RPE (Subretinal Transplantation)
Next, we conducted tumorigenicity tests by transplanting test cells into the subretinal space, a procedure that is technically demanding. We chose large albino nude rats to facilitate transplantation and minimize variability of test results. This choice also permitted us to transplant larger doses of human hiPSC-derived RPE cells to the subretinal spaces. First, we assessed the tumor-formation potential of HeLa and iPSC 201B7 via subretinal transplantation. The TPD50 for HeLa was 101.32 (n = 37) and 104.73 for iPSC (n = 20) (Table 3). Teratomas derived from subretinally transplanted iPSC or tumors derived from transplanted HeLa cells were analyzed as positive controls for tumor formation event in the subretinal space (Figure 6A–6I). The proliferative status of living cells was assessed by HE, Hoechst 33258 and anti-Ki-67 antibody staining (Figure 6J–6L). iPSC-derived human cells were stained by anti-Lamin A antibody to check the specificity of anti-human Lamin A antibody to human cells (Figure 6M–6O).
Table 3. Tumorigenicity testing by subretinal transplantation of hiPSC-derived RPE in nude rats.
| hiPSC cell line | cell form | min.dose fortumor formation | weeks to observeTumor (first to last) | numberof rats | Log10TPD50 |
| HeLa | Cell suspension w/o Matrigel | 1×101 cells | 5–33 | 37 | 1.32 |
| hiPSC 201B7 | Cell suspension w/o Matrigel | 1×104 cells | 7–33 | 20 | 4.73 |
| RPE cell line | cell form | number of cells transplanted | monitor period | number of rats | tumor formation |
| 59-G3 (1) | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 9–82 weeks | 4 | none |
| K21-G18 | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 9–82 weeks | 4 | none |
| 101-G25 | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 9–82 weeks | 3 | none |
| 59-G3 (2) | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 8–50 weeks | 5 | none |
| RNT10 | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 26–47 weeks | 5 | none |
| RNT9 | RPE cell sheet w/o Matrigel | 0.8–1.5×104 cells | 12–38 weeks | 5 | none |
Log10TPD50 values for HeLa cells or for hiPSC 201B7 cells following subretinal transplantation to nude rats (upper panel). Subretinal tumorigenicity tests conducted using nude rats under various conditions (lower panel).
Figure 6. Histological analyses of hiPSCs or HeLa cells transplanted into the subretinal space of nude rats.

Eye balls were excised from a nude rat 7 weeks after subretinal transplantation of hiPSC. Non-transplanted right eye ball (NT) and left eye ball transplanted with 1×104 hiPSCs (hiPSC) (A). HE staining of cross section of NT eye ball (B) and hiPSC-transplanted eye ball (C). HE staining of hiPSC-derived teratoma with three germ layers: cartilage-like tissue (mesoderm, D), intestinal epithelium-like tissue (endoderm, E) and neuron-like tissue (ectoderm, F) in hiPSC-transplanted eye ball. (G – O) Eye balls were excised from a nude rat 5 weeks after subretinal transplantation of HeLa cells. Non-transplanted right eye ball (NT) and left eye ball transplanted with 1×105 HeLa cells (HeLa) (G). HE staining of cross section of HeLa cell-transplanted eye ball (H) and HeLa-derived tumor tissue (I). Anti-Ki-67-antibody (J), Hoechst 33258 (K) and HE staining (L) of serial sections of hiPSC-derived teratoma. Anti-Lamin A antibody (M), Hoechst 33258 (N) staining and microscopic image (O) of serial cross sections containing a boundary of hiPSC-derived teratoma and host rat tissue. Anti-Lamin A antibody specifically recognizes human cells in rat tissue.
Next, we conducted tumorigenicity tests of iPSC-derived RPE by transplanting 1 mm2-sized (1 mm×1 mm) iPSC-derived RPE sheets consisting of 0.8–1.5×104 RPE cells into the subretinal space of nude rats (n = 26). The RPE cell number was assessed by cell density and sheet size transplanted. Considering the relative sizes of humans and rats, we estimated that transplanting 0.8–1.5×104 RPE cells into the subretinal space of nude rats would provide the information required to determine the incidence of tumor formation in humans, as we expect to transplant 4–8×104 RPE cells in a clinical setting. Thus, we transplanted 5 different hiPSC-derived RPE cell sheets from 5 different patients to minimize individual variations. hiPSC-derived RPE sheets [59-G3 RPE (n = 9), K21-G18 RPE (n = 4), 101-G25 RPE (n = 3), RNT9 RPE (n = 5), RNT10 RPE (n = 5)] were prepared and transplanted under various conditions. Transplanted nude rats were monitored for tumor formation and physical condition daily for 8 to 82 weeks.
No tumor was found during the period of observation (Table 3). All transplanted eye balls were excised and subjected to histological examination. The location of transplanted RPE sheet was shown by the brown color of the RPE sheet in albino nude rats (Figure 7A–7B). Histological and immunohistological studies showed that Lamin A- and Hoechst-positively staining or BEST1- and Hoechst-positively staining transplanted RPE cells were present in the subretinal space (Figure 7H–7O). Although we used serial sections for this staining, we believe more than half of Lamin A positive-human cells were stained with Hoechst, suggesting that these cells were live human transplanted cells at the end of the experiment (Figure 7H–7K). However, none of the cells in the sub-retinal space was stained with anti-Ki-67 antibody, suggesting that there was no ongoing proliferation in transplanted RPE cells (Figure 7D–7G). Histological analysis of serial sections showed that the shape of hiPSC-derived RPE sheet was maintained after transplantation and no evidence of tissue invasion or destruction of the vicinity of retinal structure was observed (Figure 7C, 7D).
Figure 7. Histological analysis of hiPSC-derived RPE sheets transplanted into the subretinal space of nude rats.

(A) Eye balls of nude rat 9 months after subretinal transplantation of 0.8–1.4×104 hiPSC-derived RPE (in a 1 mm×1 mm cell sheet). Left eye ball transplanted with hiPSC-derived RPE (RPE) and non-transplanted right eye ball (NT). (B) HE staining of cross section of left eye ball following transplantation of hiPSC-derived RPE. (C) HE staining of section of eye ball following transplantation of hiPSC-derived RPE, high magnification. HE- (D), anti-Ki67 antibody- (E), and Hoechst 33258-staining (F) and merged (G) images of serial sections of nude rat retina after transplantation of hiPSC-derived RPE. Anti-Lamin A antibody (H), Hoechst 33258 staining (I), merged (J) and micrographic image (K) of serial sections of nude rat retina after transplantation of hiPSC-derived RPE. Anti-BEST1 antibody (L), Hoechst 33258 (M), merged (N) staining and micrographic image (O) of serial sections of nude rat retina following transplantation of hiPSC-derived RPE.
Discussion
Here, we presented the results of nonclinical tests assessing the tumorigenic potential of hiPSC-derived RPE sheets. These studies represent a portion of the nonclinical testing of our scheduled clinical study for the use of autologous hiPSC-derived RPE sheets for the treatment of wet type AMD. The clinical study is scheduled to commence in 2014. The hiPSC-derived RPE cells used in this study were prepared in a GMP-grade cell processing facility using the same procedures that will be used for patient treatment.
Two types of tumorigenicity tests are summarized in this report. The first was a subcutaneous tumorigenicity test in NOG mice using Matrigel and the second was a subretinal tumorigenicity test in nude rats. It is intriguing to compare the objectives and the validity of the 2 tests in regard to hiPSC-derived RPE cell transplantation. Rationales for conducting subcutaneous tumorigenicity test are as follows:
Large numbers of test cells can be transplanted without difficulty, and bias-inducing variations in technical skills can be neglected. Moreover, the tumors are easy to detect. Therefore, statistical and endpoint analysis (TPD50 assessment) can be conducted in a timely and accurate manner.
It is possible to conduct comparison studies of the tumor-forming potential of different cellular products under the same transplantation conditions.
Above all, this test could serve as a substitute for in vitro soft agar assays of PSCs. PSCs cannot survive in soft agar, so that mode of testing is not feasible [5]. In contrast, PSCs or PSC-derived cells can survive long-term (more than 12 months) in Matrigel when subcutaneously transplanted in NOG mice. We can detect tumors derived from as few as 10 iPSCs or HeLa cells in this system (Table 1, Table 2). Thus, it provides a highly sensitive tumorigenic test for detecting both residual hiPSCs and tumorigenic transformed cells in hiPSC-derived cell products.
In this context, subcutaneous transplantation testing can be considered a quality control test of final cell products to ensure the absence of tumorigenic cells rather than characterizing the tumorigenic potential of the final cell products at a clinical transplantation site.
Next, we conducted tumorigenicity tests of hiPSC-derived RPE via clinical administration route. Under physiological conditions, the RPE is a monolayer that secretes various cytokines to maintain its structure in the retina. Diniz et al [3] reported that RPE transplanted in sheets retain better survival than when transplanted in suspension. For clinical application, we plan to transplant hiPSC-derived RPE in a sheet form and our preclinical testing was designed accordingly. Although we do not have RPE cell survival data in suspension form, it would be logical to presume that the transplantation of RPE in a sheet form exerts physiological function more effectively than in suspension, which may also facilitate the adaption of transplanted cells to the subretinal tissues.
With regard to the tests’ ability to detect immature (undifferentiated) hiPSCs in the subretinal space as a growing tumor, we demonstrated the trans-effects of RPE on hiPSC in our recent studies. We reported that RPE secreted pigment epithelium derived-factor (PEDF) that markedly induced apoptosis in hiPSC and hESC [21]. hiPSCs or ESCs in culture inserts ceased to survive when co-cultured with RPEs. Further addition of hPEDF induced apoptosis in hiPSCs or ESCs drastically. In fact, when hiPSCs were transplanted into the subretinal space of nude rats, the log10TPD50 value was 4.73 (n = 20), whereas the value was only 2.12 (n = 30) when hiPSCs alone were transplanted subcutaneously into NOG mice with Matrigel. The 400-fold difference in the TPD50 values under these conditions are at least partly explained by an environmental effect related to the subretinal space, besides the difference in the status of immunodeficiency in these species or use of Matrigel. We suggest that the environmental effects of the subretinal space are mediated by PEDF secreted by RPE. The close protein sequence identity between human PEDF and the rat counterpart support this idea.
As many as 1×104 hiPSC cells were required to form tumors in the subretinal space of nude rats. Similar numbers (0.8–1.5×104) of hiPSC-derived RPE were transplanted into the subretinal space in the tumorigenicity test. Note that tumorigenicity testing of iPSC-derived RPE via clinical administration route will always give “negative” results, if we aim to detect a tumor from remaining small number of undifferentiated hiPSCs in final product. In this context, tumorigenicity tests conducted by transplanting serial dilutions of hiPSCs combined with hiPSC-derived RPEs into the subretinal space might not be informative. We suggest that tumorigenicity testing via clinical administration route might be useful to detect tumors from intermediate or incompletely differentiated RPE cells, but not for those relatively rare remaining undifferentiated iPSC. For these reasons, we conducted high dose (1×106) subcutaneous RPE transplantation in parallel to examine tumor-forming events from rare hiPSC in hiPSC-derived RPE and full dose hiPSC-derived RPE subretinal transplantation without diluting them with hiPSCs. This was the basis for our rationale in designing multiple tumorigenicity tests for iPSC-derived RPE. As the FDA commentary report11 stated, the design of tumorigenicity tests should be tailored for each specific product. We hope our approach will facilitate a further discussion related to tumorigenicity testing of iPSC-derived cell products.
Considering the number of rodents used, the duration of the monitoring period, the sensitivity to detect tumors in immuno-deficient rodents via both subcutaneous and subretinal administration routes and the overall incidence of tumor formation from iPSC-derived RPE final cell products in these rodents, we conclude that the tumorigenic potential of the hiPSC-derived RPE cells produced by our methods is negligible. Of course, in considering the overall safety of the procedure in humans, discussion should include the site of transplantation as well as the source of the cells (autologous or allogeneic) and the immune-suppression status of the patients.
Materials and Methods
All the experiments using human samples and animal studies were reviewed and approved by the IRB of the Foundation for Biomedical Research and Innovation (FBRI) and Riken Center for Developmental Biology (Riken CDB), and the committee for animal experiments of the FBRI.
Cell Culture
The human iPSC (hiPSC) line 201B7 [12] established from dermal fibroblast with retroviruses pMXs-POU5F1, -Sox2, -c-Myc, and -Klf4 (Riken Bio Resource Center, Tsukuba, Japan) was maintained on feeder layers (SNL [13]) in ReproFF2 (ReproCELL) and 5 ng/mL bFGF (Peprotech). Cell line 836B1 (supplied by CiRA Kyoto University) was established from dermal fibroblast of a healthy donor, and 59, K11, K21, 101, RNT9 or RNT10 lines were derived from dermal fibroblast of 6 patients with retinitis pigmentosa (with a photoreceptor-specific gene mutation) after obtaining informed consent from the patients. These fibroblasts were reprogrammed with episomal EBNA vectors carrying integrated POU5F1, SOX2, KLF4, MYCL, LIN28A and GLIS1 (59-G, K21-G, 101-G, RNT9 and RNT10) or POU5F1, SOX2, KLF4, MYCL, LIN28A and p53shRNA (101-EV, K11-EV and K21-EV). They were established on autologous fibroblast-derived feeders and were maintained in primate ES medium (ReproCELL) with 5 ng/mL bFGF (Wako) [14]. iPSCs were differentiated into retinal pigment epithelium (RPE) as reported previously [15]. iPSC-derived RPE cell clones (59-G3 RPE, K21-G18 RPE, 101-G25 RPE, RNT9 RPE, RNT10 RPE, 101-EV RPE, K11-EV9 RPE or K21-EV15 RPE) were differentiated from the following parental iPSC clones: 59-G3, K21-G18, 101-G25, RNT9-2-8, RNT10-24, 101-EV3, K11-EV9 or K21-EV15, respectively. They were maintained in RPE maintenance medium [5],[15] [DMEM:F12 (7∶ 3) (Sigma-Aldrich) containing B-27 supplement (Invitrogen), 2 mM L-glutamine (Sigma), 0.5 nM SB431542 (Sigma-Aldrich) and 10 ng/mL bFGF (Wako)]. Human primary RPE (Lonza) was maintained in Retinal Pigment Epithelial Cell Basal Medium (Lonza Biologics, Basel, Switzerland) containing supplements [L-glutamine, GA-1000, and bFGF (Lonza)]. For transplant studies, hiPSC-derived RPE cells in suspension were collected for subcutaneous transplantation or seeded on collagen gel (collagen gel culture kit, Nitta Gelatin) to make a collagen-lined RPE monolayer or double layer cell sheet. The RPE cell sheet was maintained in F10 culture medium (Sigma) and 10% FBS for 4 weeks and RPE maintenance medium for 3 weeks and detached from the collagen gel with collagenase-1 (Roche). The RPE cell sheet was then pipetted and mixed with Matrigel for subcutaneous transplantation or dissected with laser micro dissection (LMD, Carl Zeiss) just before retinal transplantation into animals.
Animal Studies
Mouse subcutaneous transplantation
Various doses of HeLa cells either embedded in 200 µL of Matrigel™ (BD Biosciences) or suspended in 200 µL of PBS (without Matrigel) were injected into subcutaneous tissue of 7- to 8-week-old female nude mice (BALB/cA, JCl-nu/nu; Clea Japan, Inc. Tokyo), SCID mice (C.B-17/Icr-scid/scid, Jcl; Clea), NOD-SCID mice (NOD/ShiJic-scid, Jcl; Clea) or NOG mice (NOD/ShiJic-scid, IL-2R γOD/S KO Jic; Clea) using a 1 mL syringe (TERMO) with a 26 G needle. Animals were monitored for 36 weeks. At the end of the experiments, mice were sacrificed and tumors were removed and fixed with 4% PFA. Paraffin sections were stained with hematoxylin and eosin (HE) for histological observation. Various doses of hiPSC 201B7 cells or 1×106 hiPSC-derived RPE cells were embedded in 200 µL of Matrigel™ (BD Bioscience) or suspended in 200 µL of PBS (without Matrigel) and injected subcutaneously into 7- to 8-week-old female NOG mice using a 1 mL syringe (TERMO) with a 26 G needle and monitored for 6–15 months. At the end of the experiments, mice were sacrificed and all the transplants including RPE embedded in 200 µL Matrigel were removed with tweezers and fixed with 4% paraformaldehyde (PFA).
Rat subretinal transplantation
Three-week-old female nude rats (F344/NJcl-rnu/rnu; Clea) were anesthetized by intraperitoneal administration of a mixture of ketamine 100 mg/kg: xylazine 10 mg/kg (Daichi-Sankyo). The pupil of the right eye was dilated with mydriatics (0.5% tropicamide and 0.5% phenylephrine hydrochloride, Santen Pharma). A small incision was made at the right eye corner of the sclera with a 27 G needle. Then, various doses of HeLa cells, hiPSCs or 1 mm×1 mm hiPSC-RPE cell sheets in 2 µL DMEM/F12 medium were injected (Hamilton syringe with 33 G needle) into the subretinal space through the previously made incision in the sclera. The cells or the RPE sheet was transplanted just above the subretinal capillary plexus by observing the position of the Hamilton syringe needle through the dilated pupil under a surgical microscope. The subretinal capillary plexus was readily observed in albino nude rats and was used as a landmark of the subretinal space. The transplanted nude rats were monitored for 8–82 weeks. At the end of the experiments, rats were sacrificed and transplanted whole eye balls were removed and fixed with 4% PFA.
RT-PCR and qRT-PCR
Total RNA was isolated with the RNeasy plus Mini Kit (Qiagen) in accordance with the manufacturer’s instructions. Contaminating genomic DNA was removed using a gDNA Eliminator spin column. cDNA was generated from 50 ng of total RNA using PrimeScript RT Master Mix (Takara Bio) and PrimeSTAR MAX DNA Polymerase (TaKaRa Bio). Real-time PCR was then performed with an ABI 7000 Sequence Detection System (Applied-Biosystems) and SYBR-green in accordance with the manufacturer’s instruction. Gene expression levels were normalized to that of GAPDH. qRT-PCR was performed using the QuantiTect Probe one-step RT-PCR Kit (Qiagen). The expression levels of target genes were normalized to those of the RNase P transcript, which were quantified using TaqMan human RNase P control reagents (Applied Biosystems). All qRT-PCR reactions were run for 45 cycles. The sequences of primers and probes used in the present study are listed in Table S1.
Alu PCR
Alu sequences specific to human cells were used to design the primers. The Alu primer 5′-AAGTCGCGGCCGCTTGCAGTGAGCCGAGAT-3′ and 50 ng of DNA template, PrimeSTAR Max DNA Polymerase (Takara) were used for PCR reactions (28 cycles). The DNA templates in various ratios (human HeLa DNA: mouse NIH3T3 DNA) were used to determine the human cell detection sensitivity by Alu PCR. PCR products were separated by electrophoresis (MyRun, Cosmobio) with 1% agarose gel (Nacalai), and the image was digitally captured (Bio-Pyramid, Mecan).
Immunohistochemistry
Transplanted tissues were fixed with 4% paraformaldehyde. Paraffin embedded tissue sections were stained with haematoxylin/eosin. Then, the paraffin sections were deparaffinized with xylene and sequential 100%, 95%, 80%, 70% ethanol treatments for 5 min each. The sections were treated with 10 mM citric acid (pH 6) at 95°C for 50 min followed by permeation with 0.4% Triton-X in PBS at room temperature for 30 min. The deparaffinized sections were stained with antibodies against human Lamin-A (1∶200; ab108595; Abcam), BEST1 (1∶200; ab2182; Abcam) and Ki-67 (1∶400; #9449; Cell Signaling). Nuclei were stained with Hoechst 33258 (Dojindo) and DAPI (Dojindo). hiPSC-derived RPE cells were collected in suspension and fixed with 4% paraformaldehyde followed by staining with antibodies against POU5F1 (OCT3/4) (1∶ 100; sc-5279; Santa Cruz), or BEST1 (1∶ 200; ab2182; Abcam). Antibodies were visualized with Alexa Fluor 488 goat anti-mouse (1∶ 1,000; Invitrogen) or Alexa Flour 488 goat anti-rabbit (1∶ 1,000; Invitrogen). Fluorescent microscopic images were captured with a fluorescent microscope (Olympus BX51, IX71, Tokyo, Japan).
Conclusion
We tested the tumorigenic potential of hiPSC-derived RPE using immuno-deficient rodents. These preclinical tests laid the foundation for upcoming clinical studies using autologous hiPSC-derived RPE sheets for treatment of wet type age-related macular degeneration (AMD). One million hiPSC-derived RPE cells were transplanted subcutaneously into 65 NOG mice and 0.8–1.5×104 hiPSC-derived RPE cells were transplanted into the subretinal space of 26 nude rats. No tumors were found after 6–15 months of monitoring. Considering the number of rodents used, the duration of the monitoring period, the sensitivity to detect tumors in immuno-deficient rodents, we conclude that the tumorigenic potential of the hiPSC-derived RPE cells prepared by our method is negligible.
Supporting Information
Primers for RT-PCR and Alu PCR, Probes and Primers for qRT-PCR are listed.
(DOCX)
Acknowledgments
We thank Yoji Sato of National Institute of Health Sciences, Tokyo for critical reading of the manuscript, Shin-Ichi Nishikawa of JT BRH and Takao Hayakawa of Kinki University for scientific discussions and Mamoru Ito of CIEA for supplying NOG mice.
Funding Statement
This study was supported by funding from JST research grant “Safety Tests for Pluripotent Stem Cell (2010–2014)” Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lu B, Malcuit C, Wang S, Girman S, Francis P, et al. (2009) Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cell 27: 2126–2135. [DOI] [PubMed] [Google Scholar]
- 2. Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, et al. (2012) Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379: 713–720. [DOI] [PubMed] [Google Scholar]
- 3. Diniz B, Thomas P, Thomas B, Ribeiro R, Hu Y, et al. (2013) Subretinal implantation of retinal pigment epithelial cells derived from human embryonic stem cells: improved survival when implanted as a monolayer. Invest Ophthalmol Vis Sci. 54: 5087–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu Y, Liu L, Lu B, Zhu D, Ribeiro R, et al. (2012) A novel approach for subretinal implantation of ultrathin substrates containing stem cell-derived retinal pigment epithelium monolayer. Ophthalmic Res. 48: 186–191. [DOI] [PubMed] [Google Scholar]
- 5. Kuroda T, Yasuda S, Kusakawa S, Hirata N, Kanda Y, et al. (2012) Highly sensitive in vitro methods for detection of residual undifferentiated cells in retinal pigment epithelial cells derived from human iPS cells. PloS One 7: e37342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Müller FJ, Goldmann J, Löser P, Loring JF (2010) A call to standardize teratoma assays used to define human pluripotent cell lines. Cell Stem Cell 6: 412–414. [DOI] [PubMed] [Google Scholar]
- 7. Prockop DJ (2010) Defining the probability that a cell therapy will produce a malignancy. Mol Ther. 18: 1249–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.World Health Organization (2010) “Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks. Proposed replacement of TRS 878, Annex 1.”: <http://www.who.int/biologicals/BS2132-CS_Recommendations_CLEAN_19_July_2010.pdf>.
- 9.World Health Organization (2008) “Requirements for the use of animal cells as in vitro substrates for the production of biologicals.WHO Technical Report Series No.878, Annex 1.”: <http://whglibdoc.who. int/trs/WHO _TRS_878.pdf>.
- 10. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, et al. (2009) Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. (Amst.) 2: 198–210. [DOI] [PubMed] [Google Scholar]
- 11. Bailey AM (2012) Balancing Tissue and Tumor Formation in Regenerative Medicine. Sci Transl Med. 4: 147fs28. [DOI] [PubMed] [Google Scholar]
- 12. Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, et al. (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol 26: 101–106. [DOI] [PubMed] [Google Scholar]
- 13. Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448: 313–317. [DOI] [PubMed] [Google Scholar]
- 14. Maekawa M, Yamaguchi K, Nakamura T, Shibukawa R, Kodanaka I, et al. (2011) Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474: 225–229. [DOI] [PubMed] [Google Scholar]
- 15. Osakada F, Jin ZB, Hirami Y, Ikeda H, Danjyo T, et al. (2009) In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J. Cell. Sci 122: 3169–3179. [DOI] [PubMed] [Google Scholar]
- 16. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, et al. (2008) Efficient tumour formation by single human melanoma cells. Nature 456: 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okane H (2009) Strategies toward CNS-regeneration using induced pluripotent stem cells. Genome Informatics 217–220. [PubMed]
- 18. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, et al. (2005) Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain (null) mice. Blood 106: 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis AM (2005) “Regulatory Implications of Neoplastic Cell Substrat Tumorigenicity.”: <http://www.fda.gov/ohrms/dockets/ac/05/slides/5-4188S1_2.ppt>.
- 20. Machida K, Suemizu H, Kawai K, Ishikawa T, Sawada R, et al. (2009) Higher susceptibility of NOG mice to xenotransplanted tumors. J. Toxicol. Sci 34: 123–127. [DOI] [PubMed] [Google Scholar]
- 21. Kanemura H, Go JM, Nishishita N, Sakai N, Kamao H, et al. (2013) Pigment Epithelium-Derived Factor Secreted from Retinal Pigment Epithelium Facilitates Apoptotic Cell Death of iPSC. Scientific Reports 3: 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers for RT-PCR and Alu PCR, Probes and Primers for qRT-PCR are listed.
(DOCX)