

Abstract
The adsorption of even a single serum protein molecule on a gold nanosphere used in biomedical imaging may increase the size too much for renal clearance. Herein, we design charged ~5 nm Au nanospheres coated with binary mixed charge ligand monolayers that do not change in size upon incubation in pure fetal bovine serum (FBS). This lack of protein adsorption is unexpected given the Au surface is moderately charged. The mixed charge monolayers are comprised of anionic citrate ligands modified by place exchange with naturally-occurring amino acids: either cationic lysine or zwitterionic cysteine ligands. The zwitterionic tips of either the lysine or cysteine ligands interact weakly with the proteins and furthermore increase the distance between the “buried” charges closer to the Au surface and the interacting sites on the protein surface. The ~5 nm nanospheres were assembled into ~20 nm diameter nanoclusters with strong NIR absorbance (of interest in biomedical imaging and therapy) with a biodegradable polymer, PLA(1k)-b-PEG(10k)-b-PLA(1k). Upon biodegradation of the polymer in acidic solution, the nanoclusters dissociated into primary ~5 nm Au nanospheres, which also did not adsorb any detectable serum protein in undiluted FBS.
For Au nanoparticles of interest in biomedical imaging, the hydrodynamic diameter (Dh) must be less than ~6 nm for efficient renal clearance.1,2 As these nanoparticles are exposed to blood, the adsorption of even a single protein molecule on their surface, particularly the highly prevalent serum albumin (Dh = ~ 7 nm),3,4 may increase the size too much for clearance. The adsorption of serum proteins on flat surfaces5–9 and curved nanoparticles10–14 coated with nonionic, zwitterionic, or charged ligands depends in a complex manner on the ligand orientation on the surface, charge, and hydrophobicity.15–17 Remarkably, precisely defined experiments to study renal clearance indicated that protein adsorption from 10% fetal bovine serum (FBS) was fully prevented for neutral (PEG) or zwitterionic (cysteine) ligands, but was high for charged anionic and cationic ligands such as dihydrolipoic acid (DHLA) and cysteamine, respectively.1 In numerous other studies, serum protein adsorption has been found to be relatively low on zwitterionic and nonionic surface coatings with zero net charge.1,7,11,13,18–21 For example, zwitterionic peptide ligands on flat Au surfaces synthesized from equal amounts of lysine (q = +1) and glutamic acid (q = −1) were shown to adsorb minimal amounts (< 0.3 ng protein/cm2) of the model serum proteins lysozyme and fibrinogen.8 Similar low adsorption levels of these proteins were found for flat Au surfaces tailored with binary ligands with equal amounts of positive and negative charges.5,6 The close spacings between the positive and negative charges on single zwitterionic ligands favor hydration and essentially zero protein adsorption on nanoparticles, as measured by dynamic light scattering (DLS).11,22
For Au nanoparticles coated with charged ligands, electrostatic interactions, as well as charge-dipole interactions and specific interactions with hydrogen bond donor and acceptor sites raise adsorption, relative to nonionic and zwitterionic ligands.1,5,23,24 However, the roles of net charge and the topology of charge on the Au and protein surfaces on adsorption are not well understood. For nanoparticles coated with highly charged citrate, DHLA, or cysteamine ligands, adsorption of serum proteins has been found to increase the Dh significantly, on the order of 10 nm.1,25,26 For highly anionic citrate-capped Au nanoparticles with zeta potentials (ζ) of ~−40 mV, the Dh grew from 30 to ~80 nm upon incubation in undiluted human plasma.25 Interestingly, very small ~3 nm highly charged Au nanospheres coated with glutathione (GSH), with two negative and one positive charge at neutral pH, were shown to clear efficiently through the kidneys.2 While very low adsorption is typically measured with techniques such as gel electrophoresis,27,28 surface plasmon resonance sensing,6,29 and quartz crystal microbalance analysis,30 these techniques do not have the sensitivity to measure the adsorption at the single protein molecule level, as can be done by DLS2,10,11,22 or gel filtration chromatography.1
Although charged monolayers on nanoparticles composed of single ligands are not thought to resist protein adsorption,1,25,26,31 relatively little is known about the behavior for binary and multicomponent mixed charge monolayers. For binary zwitterionic mixtures with equal amounts of cationic and anionic ligands, adsorption is very low.5,6,8,29 However, mixed monolayers of charged ligands, such as peptides on Au composed of lysine and glutamic acid, bind significant amounts of proteins (> 50 ng/cm2) such as fibrinogen and lysozyme when the lysine to glutamic acid ratio deviates from unity and the surface becomes charged.8 Verma et al., however, reported that Au nanospheres with ordered “stripes” of anionic mercaptoundecanesulfonate (MUS) and nonionic octanethiol (OT) adsorbed nearly zero serum protein upon incubation in 10% serum, as shown by a negligible change in Dh via DLS, despite a highly negative ζ of ~−35 mV.10 Here, the inhibition of protein adsorption was attributed to the close proximities (~5 Å) of hydrophobic and hydrophilic groups on the nanosphere surface.10,32 However, Yang et al. have demonstrated that Au nanoparticles and flat Au surfaces which do not adsorb protein in 10% human blood serum may adsorb significant amounts of protein in 100% serum.7,11 Novel concepts are required to determine if it is possible to form charged mixed monolayers for essentially zero protein adsorption, even in undiluted serum.
Herein, we design charged ~ 5 nm Au nanospheres that adsorb essentially zero protein from undiluted fetal bovine serum, as shown by a negligible increase in the Dh by dynamic light scattering. The charged surfaces were tailored with binary ligand monolayers composed of two naturally occurring, relatively hydrophilic ligands, citrate (q = −3) and either cationic lysine (q = +1) or zwitterionic cysteine (q = 0). The Au surface charge was tuned by place exchange of the citrate ligands with each amino acid, as characterized by the zeta potential and X-ray photoelectron spectroscopy (XPS). Relatively hydrophilic ligands were used to attempt to limit hydrophobic interactions that may increase adsorption.12,23,33 For pure citrate or highly charged mixed charge monolayers with high citrate levels, the Dh increased ~3 nm or more with protein adsorption. However, the change in Dh was negligible for lower citrate fractions, even for a moderate ζ of −22 mV in undiluted fetal bovine serum. The zwitterionic tips of the lysine and cysteine ligands interact weakly with the protein and, furthermore, mitigate the interactions of the “buried” charges on the anchor groups at the Au surface. Upon assembly of the Au nanospheres into ~20 nm nanoclusters of closely spaced primary particles, following an earlier methodology,34–36 they exhibited intense NIR extinction that is of interest in biomedical applications including photoacoustic imaging.37 Upon biodegradation of PLA(1k)-b-PEG(10k)-b-PLA(1k) on the surface of the nanoclusters, they dissociated into the original ~5 nm constituent nanospheres, which will now be shown to totally resist adsorption of serum proteins.
In order to form the binary mixed charge monolayers on the surface of ~5 nm Au nanospheres, citrate-capped nanospheres were first synthesized and place exchange reactions were conducted with either lysine or cysteine ligands (schematically depicted in Figures 1a and 1b), with experimental procedures discussed in the supplemental section. In order to determine the final ligand ratio on the nanosphere surface, excess ligands were removed by ultracentrifugation, and XPS was conducted on the dried nanosphere pellet, as described in detail in the supplemental section. For lysine/citrate molar feed ratios from 4.5 to 9, place exchange led to final ligand ratios of 0.5 to 1.4 according to XPS (Table 1, Figure S1). The initial Dh value of 4.3 ± 0.8 nm (Table 1) increased only slightly for both lysine/citrate ratios after place exchange (Table 1, Figure 1c). This result is expected given the very small difference in the size of these two ligands relative to the diameter of the Au core. The increase in the amount of lysine from lysine/citrate ratios of 0.5 to 1.4 raised the ζ to −28.9 ± 3.9 mV and −16.1 ± 2.9 mV, compared with a strongly negative value of −58.4 ± 5.3 mV for pure citrate (Table 1). The ratios of each of these zeta potentials relative to that of citrate were 49 and 28% in good agreement of estimated ratios of 56 and 22%, respectively, from the number of charges on the ligands and known ratio from XPS. (Table S1, see Supporting Information for calculations and for reproducibilities in Dh and ζ, Tables S4–S7) incubation.
Figure 1.

Schematics of nanosphere surfaces coated with (a) citrate and lysine, and (c) citrate and cysteine. DLS distributions in water (green curve) and FBS (red curve) for (c) 1.4 lysine/citrate nanospheres, and (d) 1.6 cysteine/citrate nanospheres. Black curve in (c) is DLS distribution of citrate only-capped nanospheres after FBS incubation.
Table 1.
Properties of nanospheres capped with citrate or binary ligands before and after incubation in FBS.
| Ligand (s) | Ligand Ratio (feed) | Ligand Ratio (XPS) | Dh (nm) | ζ (mV) | Dh in FBS (nm) |
|---|---|---|---|---|---|
| Citrate | n/a | n/a | 4.3 ± 0.8 | −58.4 ± 5.3 | 19.9 ± 2.1 |
| Lysine/citrate | 4.5 | 0.5 | 5.0 ± 1.2 | −28.9 ± 3.9 | 7.7 ± 3.8 |
| Lysine/citrate | 9 | 1.4 | 4.6 ± 1.1 | −16.1 ± 2.9 | 3.9 ± 2.1 |
| Cysteine/citrate | 0.3 | 1.0 | 5.1 ± 3.9 | −28.8 ± 3.2 | 8.8 ± 5.8 |
| Cysteine/citrate | 0.7 | 1.6 | 3.4 ± 2.5 | −21.6 ± 1.7 | 3.4 ± 2.7 |
For the place exchange of citrate with zwitterionic cysteine, smaller feed ratios were used than for lysine, given the stronger binding of the thiol group on cysteine to Au relative to the amino group on lysine. Again the increase in Dh was negligible (Table 1, Figure 1d). The final cysteine/citrate ligand ratios, as determined by XPS, were 1.0 and 1.6 for feed ratios of 0.3 and 0.7, respectively. (Table 1, Figure S1), and the ζ values were −28.8 ± 3.2 mV to −21.6 ± 1.7 mV. The corresponding zeta potential ratios of 49 and 37% relative to pure citrate-coated nanospheres were in good agreement with the calculated ratios of 49 and 39%, respectively, from the stoichiometries via XPS (Table S1).
The resistance of the charged mixed monolayer nanospheres to serum protein adsorption was evaluated in 100% fetal bovine serum (FBS). Here, even adsorption of a single 7 nm BSA or 14 nm immunoglobulin G molecule,4 would produce a substantial change in Dh. The adsorption of one BSA molecule would correspond to ~ 0.1 μg/cm2 BSA, for a 5 nm Au nanosphere. For highly charged nanospheres with single ligands, the Dh increased significantly, by 16 nm for citrate-capped nanospheres (Table 1, Figure 1c) and 13 nm for glutathione-capped nanospheres (Figure S2). In control experiments with DLS reported in the supporting information, it was found that scattering from FBS solutions without added Au nanospheres was weak relative to the scattering by the Au nanospheres. For incubation in 100% FBS for lysine/citrate nanospheres with the lower ratio of 0.5, the Dh increased only modestly, by 3 nm as shown in Table 1. With a greater lysine/citrate ratio of 1.4 and ζ of only −16.1 mV, serum protein adsorption was completely inhibited, as the change in Dh was less than 1 nm, within experimental error (Table 1, Figure 1c). Similar behavior was observed in the case of the cysteine/citrate mixed monolayers. For the lower cysteine/citrate ratio of 1.0, Dh increased only 4 nm (Table 1). For a larger ratio of 1.6, however, protein adsorption was completely inhibited, as the nanosphere Dh change from 3.4 ± 2.5 nm to 3.4 ± 2.7 nm was within experimental error (Table 1, Figure 1d). Remarkably, not a single protein molecule was adsorbed, despite the substantial nanosphere surface charge (ζ = 21.6 ± 1.7 mV). If protein molecules adsorb they may produce aggregation of Au nanospheres; however, our DLS distributions did not reveal any aggregates in the 100 nm–1000 nm size range (Tables S8 and S9).
To support the results by DLS, nanosphere sedimentation was measured in a centrifugal field. The Au yield by mass was measured in the pellet, after centrifugation for 15 min at 10000 rpm as described in the Supporting Information. For Au nanospheres in deionized water, the yield was ~ 20% (Supporting Information) in each case. For the 1.6 cysteine/citrate level a similar yield of 21% was observed in FBS consistent with the lack of protein adsorption. However, for the ratio of 1.0 where the Dh increased to 8.8 nm, the yield in the pellet increased to 39% indicating the centrifugation technique was also highly sensitive to protein adsorption. A similar trend was observed for lysine/citrate nanospheres (see Supporting Information). Thus, the DLS and sedimentation techniques provide complimentary evidence that the protein adsorption was neglible for the Au nanospheres coated with either cysteine/citrate or lysine/citrate ligands at the higher ratios. To our knowledge, this study presents the first examples of moderately charged gold nanospheres coated with binary mixed charge ligands that completely prevent serum protein adsorption in undiluted serum. Furthermore, both ligands are naturally found in the body.
The significant level of protein adsorption on citrate and GSH-capped nanospheres can be partially attributed to overall electrostatic interactions between negatively charged nanosphere surfaces and positively-charged proteins, as well as local charges and hydrogen bonding sites on protein surfaces. Beurer et al., for example, found that protein adsorption on surfaces with a charge gradient from positively-charged aminoundecanethiol to negatively-charged mercaptoundecanoic acid was correlated with overall electrostatic attraction.24 Here, negatively charged BSA and fibrinogen adsorbed mostly on the cationic quaternary ammonium section and positively-charged lysozyme adsorbed mainly on the anionic carboxylates.24 The most prevalent protein in serum, BSA, with a pI of 4.7,24 is negatively charged at neutral pH and thus the overall electrostatic interaction with anionic Au surfaces is repulsive. However, interactions must be considered between the charged ligands and local charges and hydrogen bonding sites on the protein surface. For example, local attraction or salt bridges between cationic lysine residues on BSA and citrate ligands on Au nanospheres contribute to adsorption.31,38 Thus, serum protein adsorption observed on citrate and GSH-capped nanospheres may be caused by overall electrostatic interactions with positively-charged proteins, as well as local electrostatic and hydrogen bonding interactions for both positively and negatively charged proteins.
Our observation via DLS of essentially zero serum protein adsorption on a moderately charged binary monolayer is unexpected, relative to previous studies with single ligand monolayers1,31,38,39 as well as mixed charge monolayers with substantial net charge.24,29 For the 1.4 lysine/citrate monolayers, the lack of protein adsorption suggests that the lysine screens the strong interactions of the trivalent citrate with the proteins, similar to the 1.6 cysteine/citrate monolayers. The overall electrostatic interaction between the net negative charge of the binary monolayer and positively charged serum proteins is attractive. The difference in length of the citrate ligand versus either lysine or cysteine, however, may be expected to play an important role in resisting protein adsorption. For example, the zwitterionic tips of lysine or cysteine will interact weakly with protein surfaces given the lack of net charge and strong hydration, as is known for other zwitterions.5,6,29 Another important factor is that each of these amino acids is considerably longer than the citrate molecule, as shown in Figures 1a and 1b. The amino acids in the monolayers thus provide steric hindrance by increasing the distance between the three carboxylates on citrate and the protein surface. Thus, the local “buried” charges in the ligand monolayers will interact more weakly with the local charges and hydrogen bonding sites on the protein surface. In addition, delocalization of the charge with the gold electrons for the two carboxylate anions on citrate and the protonated amine on lysine will further reduce the strength of the electrostatic interactions with the proteins.
Lysine and cysteine, as well as citrate, are all highly hydrophilic unlike hydrophobic ligands that interact with hydrophobic segments of serum proteins and facilitate adsorption of serum proteins.33 For example, the hydrophilicity value is 3.0 for lysine, relative to 0.0 for glycine and −3.4 for highly hydrophobic tryptophan in the Hopp and Woods hydrophilicity index.40 Cysteine is more hydrophobic than lysine (hydrophilicity value of −1.040) but hydrophilic enough to resist protein adsorption when combined with citrate in our mixed monolayers. In summary, the tunability of the ligand ratio and thus surface charge for each of our mixed monolayers could be utilized to tailor the surfaces to resist protein adsorption even for moderate net charge.
Various techniques have been used to form nanoclusters with controlled properties from primary particles.41–44 The 1.4 lysine/citrate nanospheres were assembled into nanoclusters upon solvent evaporation in the presence of a weakly adsorbing polymer, PLA-b-PEG-b-PLA, following a previously reported procedure, as discussed in the supplemental section.34–36 The nanoclusters with a Dh of 21.7 ± 4.3 nm were composed of closely-spaced primary Au nanospheres (Figure 2), which shifted the extinction in the near infrared region (NIR) from 650 nm to 900 nm. Upon incubating the nanoclusters in pH 5 HCl for 24 h, the PLA(1k)-b-PEG(10k)-b-PLA(1k) on the surface was hydrolyzed, and consequently the nanoclusters dissociated to Au nanospheres with the original nanosphere size, as seen in the UV-Vis-NIR spectrum (Figure 2c) as well as the DLS size distribution (Figure 2d). The dissociated nanoclusters did not adsorb serum proteins, as the Dh remained only 4.2 ± 2.6 nm upon incubation in 100% FBS (Figure 2d), a desired size for kidney clearance.
Figure 2.
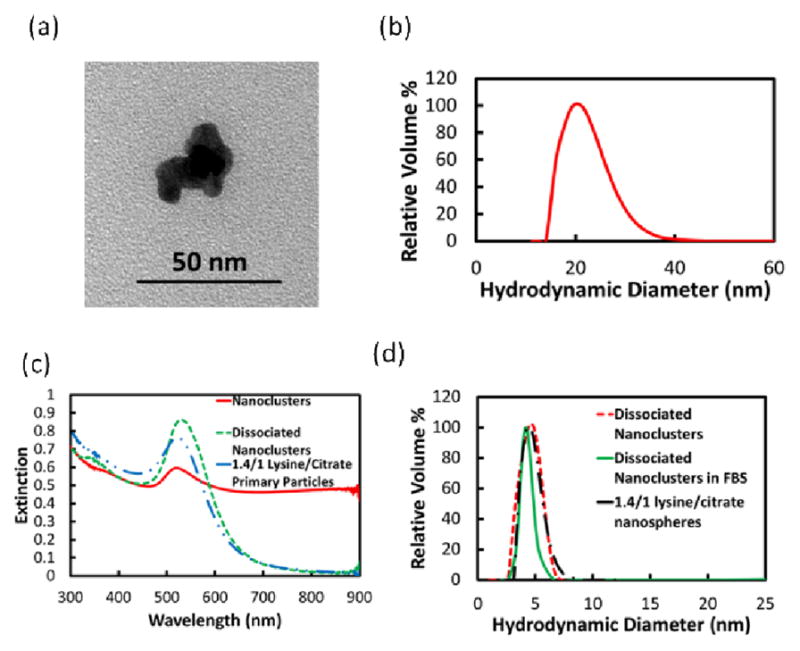
Lysine/citrate nanoclusters (a) TEM image, (b) DLS Dh distribution, and (c) UV-Vis-NIR extinction spectrum, with spectra of dissociated nanoclusters and nanospheres included, and (d) DLS Dh distributions of dissociated nanoclusters, dissociated nanoclusters in FBS, and lysine/citrate nanospheres.
In this robust colloidal assembly approach,34–36 the size of nanoclusters may be tuned as a function of the polymer and gold concentrations, chemical structure of the surface ligands, and the extent of solvent evaporation. The biodegradable polymer adsorbs on the surface of the nanoclusters and quenches the size via an equilibrium mechanism.36 In the current study, we show for the first time that these clusters may be formed from Au particles with a large enough charge for nanocluster dissociation upon biodegradation of the polymer coating, but yet the surface charge is small enough to fully resist protein adsorption.
In conclusion, incubation of charged ~5 nm Au nanospheres with binary natural and relatively hydrophilic ligands in undiluted serum protein does not increase the hydrodynamic diameter according to dynamic light scattering, indicating essentially zero protein adsorption. A secondary conclusion is that the Au nanospheres may be assembled into NIR-active nanoclusters which biodegrade in vitro to primary Au nanospheres, again with essentially zero protein adsorption.
Supplementary Material
Acknowledgments
K.P.J. and K.S. acknowledge support from NSF grant CBET-0968038 and the National Institute of Health grant CA143663. K.P.J. and T.M.T acknowledge support from the Welch Foundation (F-1319 and F1-1696, respectively). T.M.T. and K.P.J acknowledge support from the National Science Foundation (CBET-1247945). We thank a reviewer for suggesting the sedimentation experiments to support the DLS results for protein adsorption.
Footnotes
The authors declare no competing financial interests.
Experimental details, determination of ligand ratios via XPS, correlation of XPS and zeta potential results, GSH Dh distributions before and after FBS incubation, Dh distribution of pure FBS, XPS spectra, Dh distributions of 0.5 lysine/citrate and 1.0 cysteine/citrate nanospheres, Au yields by mass of mixed monolayer capped nanospheres in FBS and in water, reproducibility of mixed monolayer-capped nanospheres, and tabular full size distribution data for Au nanospheres in FBS. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou C, Long M, Qin Y, Sun X, Zheng J. Angew Chem Int Ed. 2011;50:3168. doi: 10.1002/anie.201007321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Molecular & Cellular Proteomics. 2003;2:1096. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Striemer CC, Gaborski TR, McGrath JL, Fauchet PM. Nature. 2007;445:749. doi: 10.1038/nature05532. [DOI] [PubMed] [Google Scholar]
- 5.Holmlin RE, Chen X, Chapman RG, Takayama S, Whitesides GM. Langmuir. 2001;17:2841. doi: 10.1021/la0015258. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Yu F, Yu Q, He Y, Jiang S. Langmuir. 2006;22:8186. doi: 10.1021/la061012m. [DOI] [PubMed] [Google Scholar]
- 7.Yang W, Xue H, Li W, Zhang J, Jiang S. Langmuir. 2009;25:11911. doi: 10.1021/la9015788. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Cao Z, Jiang S. Biomaterials. 2009;30:5893. doi: 10.1016/j.biomaterials.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Ostuni E, Chapman RG, Holmlin RE, Takayama S, Whitesides GM. Langmuir. 2001;17:5605. doi: 10.1021/la0015258. [DOI] [PubMed] [Google Scholar]
- 10.Verma A, Uzun O, Hu Y, Hu Y, Han HS, Watson N, Chen S, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang W, Zhang L, Wang S, White AD, Jiang S. Biomaterials. 2009;30:5617. doi: 10.1016/j.biomaterials.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 12.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, Dawson KA, Linse S. Proc Natl Acad Sci. 2006;104:2050. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW. J Am Chem Soc. 2012;134:2139. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 14.Larson TA, Joshi PP, Sokolov K. ACS Nano. 2012;6:9182. doi: 10.1021/nn3035155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S, Li L, Zhao C, Zheng J. Polymer. 2010;51:5283. [Google Scholar]
- 16.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Proc Natl Acad Sci. 2008;105:14265. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markarucha AJ, Todorova N, Yarovsky I. Eur Biophys J. 2011;40:103. doi: 10.1007/s00249-010-0651-6. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Chen S, Zheng J, Ratner BD, Jiang S. J Phys Chem B. 2005;109:2934. doi: 10.1021/jp0473321. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Choi HS, Zimmer JP, Tanaka E, Frangioni JV, Bawendi M. J Am Chem Soc. 2007;129:14530. doi: 10.1021/ja073790m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladd J, Zhang Z, Chen S, Hower JC, Jiang S. Biomacromolecules. 2008;9:1357. doi: 10.1021/bm701301s. [DOI] [PubMed] [Google Scholar]
- 21.Estephan ZG, Jaber JA, Schlenoff JB. Langmuir. 2010;26:16884. doi: 10.1021/la103095d. [DOI] [PubMed] [Google Scholar]
- 22.Jia G, Cao Z, Xue H, Xu Y, Jiang S. Langmuir. 2009;25:3196. doi: 10.1021/la803737c. [DOI] [PubMed] [Google Scholar]
- 23.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Chem Rev. 2011;111:5610. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 24.Beurer E, Venkataraman NV, Sommer M, Spencer ND. Langmuir. 2012;28:3159. doi: 10.1021/la203964a. [DOI] [PubMed] [Google Scholar]
- 25.Dobrovolskaia MA, Patri AK, Zheng J, Clogston JD, Ayub N, Aggarwal P, Neun BW, Hall JB, McNeil SE. Nanomedicine. 2009;5:106. doi: 10.1016/j.nano.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Paoli Lacerda SH, Park JJ, Meuse C, Pristinski D, Becker ML, Karim A, Douglas JF. ACS Nano. 2010;4:365. doi: 10.1021/nn9011187. [DOI] [PubMed] [Google Scholar]
- 27.Yu M, Zhou C, Liu J, Hankins JD, Zheng J. J Am Chem Soc. 2011;133:11014. doi: 10.1021/ja201930p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Huang H, Jin Q, Ji J. Langmuir. 2011;27:5242. doi: 10.1021/la2002223. [DOI] [PubMed] [Google Scholar]
- 29.Chen S, Zhen J, Li L, Jiang S. J Am Chem Soc. 2005;127:14473. doi: 10.1021/ja054169u. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman ED, Belyea J, Johnson MC, Nicholson ZM, Ricks JL, Shah PK, Bayless M, Pettersson T, Feldoto Z, Blomberg E, Claesson P, Franzen S. Langmuir. 2007;23:6053. doi: 10.1021/la063725a. [DOI] [PubMed] [Google Scholar]
- 31.Brewer SH, Glomm WR, Johnson MC, Knag MK, Franzen S. Langmuir. 2005;21:9303. doi: 10.1021/la050588t. [DOI] [PubMed] [Google Scholar]
- 32.Jackson AM, Myerson JW, Stellacci F. Nat Mater. 2004;3:330. doi: 10.1038/nmat1116. [DOI] [PubMed] [Google Scholar]
- 33.You CC, De M, Han G, Rotello VM. J Am Chem Soc. 2005;127:12873. doi: 10.1021/ja0512881. [DOI] [PubMed] [Google Scholar]
- 34.Tam JM, Murthy AK, Ingram DR, Nguyen R, Sokolov KV, Johnston KP. Langmuir. 2010;26:8988. doi: 10.1021/la904793t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tam JM, Tam JO, Murthy A, Ingram DR, Ma LL, Travis K, Johnston KP, Sokolov KV. ACS Nano. 2010;4:2178. doi: 10.1021/nn9015746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murthy AK, Stover RJ, Borwankar AU, Nie GD, Gourisankar S, Truskett TM, Sokolov KV, Johnston KP. ACS Nano. 2013;7:239. doi: 10.1021/nn303937k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon SJ, Mallidi S, Tam JM, Tam JO, Murthy A, Johnston KP, Sokolov KV, Emelianov SY. Opt Lett. 2010;35:3751. doi: 10.1364/OL.35.003751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dominguez-Medina S, McDonough S, Swanglap P, Landes CF, Link S. Langmuir. 2012;28:9131. doi: 10.1021/la3005213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casals E, Pfaller T, Duschl A, Oostingh GJ, Puntes V. ACS Nano. 2010;4:3623. doi: 10.1021/nn901372t. [DOI] [PubMed] [Google Scholar]
- 40.Hopp TP, Woods WR. Proc Natl Acad Sci. 1981;78:3824. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuang J, Wu H, Yang Y, Cao YC. Angew Chem Int Ed. 2008;47:2208. doi: 10.1002/anie.200705049. [DOI] [PubMed] [Google Scholar]
- 42.Ma LL, Feldman MD, Tam JM, Paranjape AS, Cheruki KK, Larson TA, Tam JO, Ingram DR, Paramita V, Villard JW, Jenkins JT, Wang T, Clarke GD, Asmis R, Sokolov K, Chandrasekar B, Milner TE, Johnston KP. ACS Nano. 2009;3:2686. doi: 10.1021/nn900440e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia YS, Nguyen TD, Yang M, Lee B, Santos A, Podsiadlo P, Tang ZY, Glotzer SC, Kotov NA. Nat Nanotechnol. 2011;6:580. doi: 10.1038/nnano.2011.121. [DOI] [PubMed] [Google Scholar]
- 44.Lu Z, Yin Y. Chem Soc Rev. 2012;41:6874. doi: 10.1039/c2cs35197h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.