

Abstract
Hardklör and Krönik are software tools for feature detection and data reduction of high resolution mass spectra. Hardklör is used to reduce peptide isotope distributions to a single monoisotopic mass and charge state, and can deconvolve overlapping peptide isotope distributions. Krönik filters, validates, and summarizes peptide features identified with Hardklör from data obtained during liquid chromatography mass spectrometry (LC-MS). Both software tools contain a simple user interface and can be run from nearly any desktop computer. These tools are freely available from http://proteome.gs.washington.edu/software/hardklor.
Keywords: proteomics, mass spectrometry, liquid chromatography, high resolution, feature detection, deisotoping, peptide isotope distribution
Introduction
Deconvolution of mass spectra of complex mixtures into a list of component masses is a difficult task that is best performed with the aid of computational tools. These tools employ many analytical techniques, including separating signal from background noise, calculation of ionic charge state, modeling of isotope distributions, and solving multiple signals (Senko et al., 1995a; Senko et al., 1995b; Rockwood et al., 1995; Kubinyi, 1991; Mann et al., 1989). ZSCORE and THRASH were some of the first algorithms to integrate these many techniques into comprehensive software tools for the automated deconvolution of mass spectra into component signals (Zhang and Marshall, 1998; Horn et al., 2000). Although powerful, these algorithms were limited in their computational speed and ability to solve some overlapping isotope distributions, paving the way for further algorithm development.
Hardklör (Hoopmann et al., 2007) and Krönik were designed to rapidly identify peptide and protein features in high resolution spectra of complex mixtures. These algorithms were developed to meet the demands of modern mass spectrometers that are capable of generating large amounts of robust mass spectrometry data. Spectra containing tens to hundreds of overlapping ion signals are reduced to a relevant list of observed monoisotopic mass values, charge states, and signal intensity values when using Hardklör. These features are then filtered, validated, and summarized by using Krönik for the analysis of liquid chromatography mass spectrometry (LC-MS) data. The modest computational specifications and simple user interfaces make Hardklör and Krönik ideal software tools for analysis of high resolution mass spectra on virtually any desktop computer.
This unit contains two protocols that describe how to operate Hardklör and its support software tool, Krönik, for the analysis of shotgun LC-MS data. Basic Protocol 1 describes in detail how to set up and operate Hardklör for the analysis of high resolution mass spectra. Once spectra are reduced to observed peptide features, Basic Protocol 2 instructs in the use of Krönik to organize, validate, and visualize the Hardklör results. The Support Protocol explains how to install Hardklör and Krönik. Hardklör and Krönik are actively developed and it is recommended that the user periodically check the Hardklör web site (http://proteome.gs.washington.edu/software/hardklor) for updates.
Basic Protocol 1: Identifying peptide features in precursor spectra using Hardklör
Mass spectra in shotgun proteomics often contain a complex set of overlapping isotope distributions representing many different ions measured simultaneously in a mass analyzer. Data reduction and feature detection are ways to simplify complex spectrum data to a list of observed masses and their charge states. Hardklör is a software tool designed to perform these tasks on high resolution mass spectra.
A sample data file, Data.mzXML, is provided for the protocols in this unit. The data are a subset of spectra from a shotgun LC-MS experiment analyzed on a Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific). The sample contains peptides from a tryptic digest of Saccharomyces cerevisiae. The scan cycle consists of a high resolution precursor scan event acquired at 60000 resolution, followed by ten high resolution higher-energy collision dissociation (HCD) MS/MS scan events acquired at 7500 resolution.
Necessary Resources
Hardware
A computer capable of running Windows XP (Service Pack 2 or later), Windows Vista, or Windows 7.
A minimum 2 GB RAM is recommended.
Software
Windows XP (Service Pack 2 or later), Windows Vista, or Windows 7
Hardklör (See Support Protocol 1)
Krönik (See Support Protocol 1)
Microsoft Excel
Files
Data.mzXML
Create a directory on the hard drive named C:\Hardklor. Install Hardklör and Krönik into this folder, as described in Support Protocol 1.
Copy Data.mzXML to C:\Hardklor.
-
Open C:\Hardklor\Hardklor.conf in Notepad, the simple text editor provided with Windows.
Hardklor.conf is a Hardklör configuration file. Configuration files can have any name and extension as long as they contain simple text. Hardklör configuration files contain the list of parameters to use during analysis and the file names to be analyzed. Using configuration files is the preferred method when running Hardklör instead of the alternative method of typing out the parameters on the command line.
The Hardklor.conf file that is packaged with Hardklör contains the default parameter settings for operating Hardklör. Most of these parameter settings do not need to be changed. The following steps explain how to change the parameters that most commonly need to be customized to ensure optimum Hardklör performance. A complete list of parameters is provided in Table 1.
-
Change the -res parameter to read ‘-res 60000 orbitrap’.
Resolution in Hardklör is assumed to be the resolution at 400 m/z. For instruments such as the LTQ-Orbitrap or LTQ-FT hybrid mass analyzers, the resolution is set to this value at the time of acquisition. For other instruments, the resolution may be reported differently and must be converted to the convention used in Hardklör. Additionally, the type of mass analyzer must be specified when setting the -res parameter. Valid analyzer codes are orbitrap, FTICR (Fourier Transform Ion Cyclotron Resonance), TOF (Time Of Flight), and QIT (Quadrupole Ion Trap).
-
Set the -win parameter to read ‘-win 6.0’.
A typical spectrum is a complex mixture of peptide signals and other peaks. To efficiently identify individual peptide isotope distributions from a spectrum, the spectrum is first divided into smaller pieces (referred to as windows) that are easier to solve than the entire spectrum at once. The -win parameter sets the maximum size, in m/z, of each of these windows. To minimize the likelihood of splitting a peptide isotope distribution into two windows, the Hardklör algorithm allows the window to be flexible so that division points between windows occur in spectrum regions of low signal or noise, instead of fixed intervals. Ultimately, most windows are smaller than the value specified by the -win parameter when the peptide isotope distributions are solved.
Setting a -win value too large or too small has implications that can affect Hardklör performance. Setting a value too small will force Hardklör to divide peptide isotope distributions into different windows, especially in regions of a spectrum with overlapping peptide signals. Setting the -win value too large will create windows that have too many peaks for Hardklör to solve efficiently, which increases computation time. A -win value between 5.0 and 6.0 is sufficiently wide enough to adequately contain peptide distributions at charge state 1+ while still dividing the spectrum into sufficiently small windows.
-
Change the -d parameter to read ‘-d 2’.
The -d parameter sets the depth of deconvolution that Hardklör must try to solve when encountered with multiple overlapping peptide isotope distributions in a window. A value of 1 instructs Hardklör to solve the peaks in the window without deconvolution. Each increase in depth could potentially increase computation time exponentially for that window. The Hardklör algorithm is designed to return the first combination of peptide isotope distributions that explains the observed peaks in a window. However, for cases where a window contains noise peaks and cannot be solved, Hardklör will continue searching for an answer through all possible theoretical peptide combinations, which is an exponentially large problem defined by the value of -d. In most cases, depth should be set to 2 or 3. Although there is no upper limit, the recommended maximum value for depth is 5.
-
Change the -corr parameter to read ‘-corr 0.92’.
-corr is the correlation threshold, or the minimum score at which a set of peaks is determined to be a peptide feature or set of features. When a window is analyzed, Hardklör will first try to solve the peaks in the window by fitting a single theoretical peptide isotope distribution. Many different theoretical distributions are fitted one at a time to the data, and once all have been tested, the theoretical distribution with the highest correlation score above the threshold is accepted. If none of theoretical distributions exceed the threshold, combinations of two distributions are tested. This pattern continues until the threshold is exceeded, or the depth of combinations (set by -d above) is reached. If the correlation threshold is not exceeded by any combination of theoretical distributions, then no peptide features are reported for that window of the spectrum.
Setting a -corr value that is too high will reduce the efficiency of Hardklör and will cause Hardklör to miss many detectible peptide features. This is because the data and the theoretical distributions rarely are a perfect match, and thus some discrepancy must be tolerated. When setting the -corr value too low, Hardklör will miss many overlapping peptide features because it could match a single peptide well enough to exceed the threshold. Thus, it will not continue to search out to the user-defined depth and will return fewer results. For the best performance, -corr values between 0.85 and 0.95 are recommended.
-
Add the following line to Hardklor.conf at the end of the parameter lines: ‘Data.mzXML Data.hk’.
At the bottom of the configuration file is the list of files to be analyzed with Hardkör. The syntax is to provide an input file name and output file name. In this tutorial, only one file will be analyzed. However, it is possible to analyze multiple files at once by listing each of them on new line.
Save Hardklor.conf and close Notepad.
Open a command prompt and navigate to the C:\Hardklor directory.
-
Type ‘Hardklor.exe -conf Hardklor.conf’ and press enter. Figure 1 illustrates the Hardklör user interface.
Computation times will vary based on data complexity and parameter settings. A counter displays the progress of the analysis as a percentage.
Table 1.
Summary of common Hardklör parameters.
| Parameter | Brief Description |
|---|---|
| -c [true|false] | Indicates whether the input spectra are centroided or not. |
| -chMax [num] | Confines the spectrum search space to maximum charge state. Setting an appropriate charge state range for the data improves algorithm speed and accuracy. |
| -chMin [num] | Confines the spectrum search space to minimum charge state. |
| -corr [num] | Sets the correlation score threshold that an observed peptide must exceed to be reported in the output file. |
| -d [num] | Sets the depth of deconvolution which is the maximum number of peptide isotope distributions to solve from a set of overlapping isotope peaks. |
| -da [true|false] | Toggles reporting of distribution area or base peak intensity for peptide features identified with Hardklör. |
| -m [formula] | Specifies a differential isotope modification that may exist in the observed peptide isotope distributions. Refer to the Hardklör website documentation for formula details. |
| -mF [code] | Filters files with mixed scan modes to a specific mode. Valid codes are MS1 for precursor spectra, MS2 for MS/MS spectra, and MS3 for MS3 spectra. |
| -p [num] | Sets the maximum number of peptides to test in each analysis window. |
| -res [num] [code] | Specifies the resolution and mass analyzer used to acquire the spectra. Refer to the Hardklör website documentation for additional details. |
| -s [num] | Sets the window size of Savitsky-Golay smoothing of peaks. |
| -sc [num] [num] | Narrows the Hardklör analysis to a range of scan numbers. |
| -sl [num] | Sets the sensitivity level of the analysis. |
| -sn [num] | Sets a signal-over-noise threshold for processing spectrum peaks prior to analysis. |
| -win [num] | Sets the maximum analysis window size in Thomsons. |
| -xml [true|false] | Alternatively outputs results in an XML document. The Hardklör support software, HKViewer and Krönik, do not support the XML output. It is provided for users who prefer XML when processing Hardklör results. |
Figure 1.
Image of the Hardklör user interface.
Alternate protocol: Identifying features in high-resolution MS/MS spectra using Hardklör
Typical shotgun proteomics data acquisition on a hybrid mass analyzer such at the Orbitrap or LTQ-FT is performed with low resolution acquisition of MS/MS in the linear ion trap. However, MS/MS acquisition is possible at high resolution in the orbitrap or ion cyclotron resonance (ICR) cell. High-resolution MS/MS spectra have the advantage of improved mass accuracy and resolution of fragment ion isotope distributions. Because isotope distributions are fully resolved, it is possible to perform “deisotoping”. Deisotoping is a method by which isotope distributions are reduced to a single peak and charge state, which reduces spectrum complexity and may provide several advantages in downstream analysis.
Hardklör is capable of analyzing MS/MS spectra with a few simple modifications to the parameter settings. Many of the steps are identical to Basic Protocol 1 for analysis of precursor spectra, and detailed explanations of each step can be found there.
Perform steps 1 and 2 as described in Basic Protocol 1.
-
Change the -mF parameter to read ‘-mF MS2’.
By default, Hardklör finds peptide features in precursor spectra, skipping any tandem mass spectra (MS/MS). Setting the mzXML filter to MS2 inverts this behavior: Hardklör will find features in MS/MS spectra and skip precursor spectra.
Change the -res parameter to read ‘-res 7500 orbitrap’.
Perform steps 4 to 6 as described in Basic Protocol 1.
Add the following line to Hardklor.conf at the end of the parameter lines: ‘Data.mzXML Data-MSMS.hk’. Remove any other lines instructing file analysis.
-
Perform steps 8 to 10 as described in Basic Protocol 1.
Hardklör output is in ASCII text (Figure 2).
Figure 2.
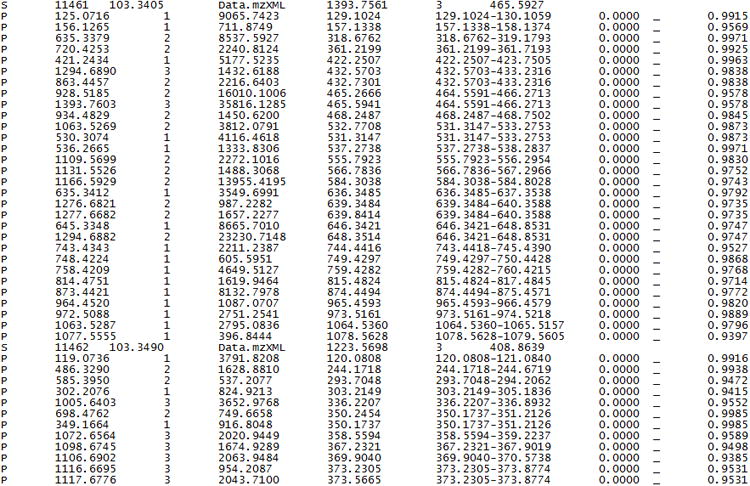
Sample of Hardklör output. The output is in tab-delimited ASCII text. Each line is labeled as either spectrum (S) information or peptide feature (P) information. Details of the output file contents can be found on the Hardklör website at: http://proteome.gs.washington.edu/software/hardklor/tutorial.html#results
Basic Protocol 2: Visualizing Hardklör results using HKViewer and performing additional analysis using Krönik
The file structure and contents of Hardklör output are described in detail on the Hardklör website (referenced in the introduction). The output is in ASCII text that can be imported into other applications or viewed in text editors and spreadsheet applications. However, file size often prohibits effectively navigating the Hardklör results files with these applications. The HKViewer was written specifically to quickly load, visualize, and navigate the Hardklör results.
When analyzing LC-MS data, sometimes it is more useful to summarize the data in terms of chromatographically unique peptide features rather than visualizing data scan-by-scan. The software tool, Krönik, was designed to filter, validate, and summarize Hardklör results by identifying peptide features that persist over a user-defined number of consecutive scans.
-
Open HKViewer.exe by clicking on its icon in the Windows file explorer. Once open, select ‘File’ then ‘Open’ from the pull-down menu. Open C:\Hardklor\Data.hk.
Figure 3 illustrates the HKViewer graphical user interface. A pull-down menu at the top is used to select Hardklör results files. Once loaded, results can be navigated using the toolbar below the menu, as demonstrated in the next step. In the upper right corner is a graph displaying the distribution of peptide features across all spectra in the results file. The text box on the left indicates the scan number being viewed and the number of peptide features detected. The table on the right provides a list of all peptide features for the current spectrum.
Peptide features are listed in each row of the table. The columns indicate the properties of each feature. ‘Mass’ is the monoisotopic, zero-mass of the feature in Daltons. ‘Charge’ is the charge state of the feature. ‘Monoisotopic Peak’ and ‘Base Isotope Peak’ are the first peak and the tallest peak in the feature's isotope distribution, respectively. Often, these values are the same (with minor decimal rounding differences) for peptides of lower mass. ‘Modifications’ shows an underscore if no feature modifications were detected with Hardklör (See Table 1 and Advanced Parameters). ‘Correlation Score’ is the Hardklör correlation score for that feature.
-
Navigate the spectra using the forward and backward buttons in the toolbar. Jump to scan number 13405 by typing the number in the navigation window and pressing enter.
The spectra are identified by the scan event number from the Data.mzXML file. Because Data.mzXML contained both precursor and MS/MS scan events interleaved, the precursor spectra do not have consecutive scan event numbers.
Close HKViewer, open a command prompt and navigate to C:\Hardklor
-
Type ‘Kronik Data.hk Data.kro’ and press enter.
Execution of this command runs Krönik with the default parameters, which are presented to the user on the screen. Peptide features are filtered by their persistence in at least three of four consecutive scans (persistence of three scans with a gap tolerance of one scan). Additionally, peptide features are filtered to exclude masses below 600 Daltons and above 8000 Daltons. Contaminants are also filtered out of the results. Contaminants are features that persist for longer than a user-defined amount of time. By default, the contaminants threshold is set to 5.0, with an assumption that the unit of time is minutes. The output for Krönik indicates that 3,267 peptide features were found to pass the filtering parameters.
-
Type ‘Kronik -c 2.0 Data.hk Data-strict.kro’ and press enter.
Changing the command line parameters (Table 2) will filter a different set of results to the output file. Here, the time threshold for contaminants is set to 2.0 minutes. Notice that the number of peptide features passing the filtering criteria is reduced to 3,226 peptide features. Also notice that the output was directed to a different file name so that the results from Step 4 are not overwritten.
-
Type ‘Kronik -n 1200.0 -m 1250.0 Data.hk Data-MassFilter.kro’ and press enter.
Multiple parameters can be set at the same time. This time the peptide features were filtered to a narrow mass range. The number of persistent peptide features is now 145.
-
Open Excel and load C:\Hardklor\Data.kro.
Krönik output is in tab-delimited text that is readable by most spreadsheet software (Figure 4). Peptide features are listed one-per-line with column headings listed on the first line. ‘First Scan’ and ‘Last Scan’ indicate the scan number boundaries over which the peptide feature was observed. ‘Num of Scans’ is the number of unique scan events over which the feature was observed, and is not the difference between the ‘First Scan’ and ‘Last Scan’ if the data were acquired using mixed mode data acquisition. ‘Charge’ is the charge state of the feature. The ‘Monoisotopic Mass’ is the zero-charge mass of the feature in Daltons. ‘Base Isotope Peak’ is the observed tallest isotope peak (in Thomsons) of the feature. ‘Best Intensity’ indicates the intensity value at the apex of the feature elution profile. ‘Intensity’ is represented either at maximum peak height or distribution area, depending on which was requested in the Hardklör parameters (Table 1). ‘Summed Intensity’ is the sum of all intensities for the feature over its observed scan events and is a proxy for chromatographic peak area. ‘First RTime’ and ‘Last RTime’ are the retention time boundaries of the feature observation, and ‘Best RTime’ is the retention time of the apex of the feature elution profile. ‘Best Correlation’ is the correlation score at the apex of the feature elution profile. An underscore for ‘Modifications’ indicate no peptide feature modifications were identified with Hardklör (see Table 1 and Advanced Parameters).
-
While still in Excel, load C:\Hardklor\Data-strict.kro.
Notice that much of the results are the same, with the exception that the retention time span (‘Last RTime’ – ‘First RTime’) never exceeds two minutes.
-
While still in Excel, load C:\Hardklor\Data-MassFilter.kro.
In this set of results, the peptide features have been filtered to span a mass range of 1,200 to 1,250 Daltons. Checking the ‘Monoisotopic Mass’ column confirms this.
Figure 3.
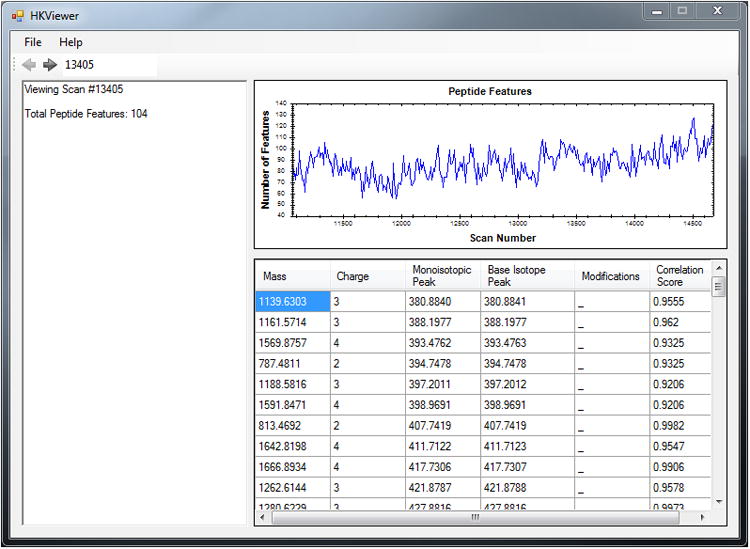
The HKViewer. This Windows-based graphical user interface presents Hardklör results to the user on a scan-by-scan basis using the navigation toolbar at the top left of the window. The graph in the upper right displays the distribution of peptide features identified with Hardklör across the input scan data.
Table 2.
Summary of Krönik parameters.
| Parameter | Brief Description |
|---|---|
| -c [num] | Sets a threshold for contaminants, defined by signals that persist too long. The unit of time used depends on the data file. |
| -d [num] | Sets the match tolerance, or the number of consecutive spectra (allowing for gaps, if specified) over which a feature must be observed. |
| -g [num] | Sets the gap tolerance when counting features over consecutive spectra. |
| -m [num] | Filters the features by maximum mass, in Daltons. |
| -n [num] | Filters the features by minimum mass in Daltons. |
| -p [num] | Sets the mass tolerance (in ppm) when matching features over consecutive spectra. |
Figure 4.
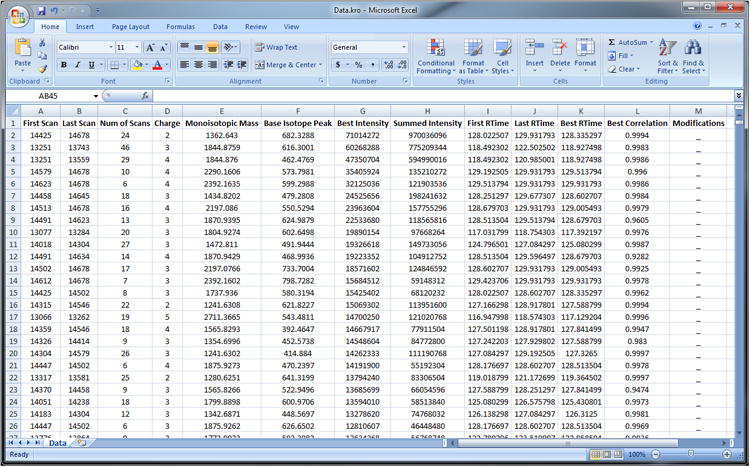
Screen capture of Krönik results visualized with Microsoft Excel.
Support Protocol: Installing Hardklör and Krönik
Hardklör and Krönik are provided in pre-compiled binaries for several operating systems that include Windows, Mac OS X, and Linux. There is no installer file. Installation is performed by extracting the binary that matches the operating system to that computer, with the recommended location of C:\Hardklor on Windows computers. Optionally, the user may wish to extract the files to a folder that resides in the system path or append the system path to include C:\Hardklor.
Necessary Resources
Hardware
A computer capable of running of Windows XP (Service Pack 2 or later), Windows Vista, or Windows 7.
Software
Windows XP (Service Pack 2 or later, 32-bit), Windows Vista (32-bit), or Windows 7 (32-bit).
A modern web browser such as Firefox, Chrome, or Internet Explorer.
Files
None
Create a folder on the C-drive called C:\Hardklor
-
Download Hardklör by navigating in a web browser to http://proteome.gs.washington.edu/software/hardklor and select the “Download” link from the navigation menu.
Before downloading Hardklör, the user is asked to agree to the Hardklör license. The Hardklör license allows free access to Hardklör for both academic and commercial institutions. After agreeing to the license, an e-mail message with a link and password is sent to the user to download the software.
-
Open the downloaded file, hardklor.zip, and extract the contents of win32 to C:\Hardklor.
For Basic Protocols 1 and 2, Hardklör should be extracted into C:\Hardklor. Advanced users may prefer to extract the files to a different location of their choosing.
Download Krönik from a web browser at http://proteome.gs.washington.edu/software/hardklor/public/kronik.zip
Open the downloaded file, kronik.zip, and extract the contents of win32 to C:\Hardklor
-
Navigate to C:\Hardklor using the Windows file explorer and confirm all files are present.
Nine files should reside in C:\Hardklor: Hardklor.conf, Hardklor.dat, Hardklor.exe, HKViewer.exe, ISOTOPE.dat, Kronik.exe, README_HARDKLOR.txt, README_KRONIK.txt, ZedGraph.dll
Guidelines for Understanding Results
Hardklör dramatically simplifies mass spectra from a complex set of overlapping ion signals to a smaller set of observed masses and their corresponding charge states, known as peptide features. The accuracy of the results is partially dependent on setting the correct parameters for the data. The most critical parameters are the resolution and mass analyzer settings (Basic Protocol 1, step 4). Setting an incorrect resolution or mass analyzer code results in a fewer detected features and poor mass accuracy measurements. These settings must match the resolution and acquisition instrument of the data being analyzed.
Peptide features in Hardklör are identified by calculating the similarity of observed spectrum peaks to theoretical peaks from a peptide model. This measure of similarity is made using the dot product of observed peaks and a peptide model that uses averagine (Senko et al., 1995b), which estimates the atomic composition of a peptide when given a mass. The atomic composition of the averagine model rarely matches the unknown atomic composition of the observed ion. For this reason, and also because observed peaks contain inherent noise irregularities, there is almost never a perfect correlation between the averagine model and the observed data. For each set of observed peaks, many averagine models are correlated, and the model or combination of models that produces the highest correlation score above the user-defined threshold is reported in the results. Regions of a spectrum with low signal and an abundance of noise peaks contain artifacts that will result in low correlation scores. Yet, in this context a peptide feature correlation score of 0.85 may be as trustworthy as a correlation score of 0.99 for a feature in a different portion of the spectrum. As a corollary, in a region of strong signal with little noise interference, a correlation score of 0.85 would be unacceptable. Thus, the correlation score is interpreted as how well the features fit the observed data rather than as a measure of accuracy. Guidelines for selecting an appropriate correlation threshold are detailed in Basic Protocol 1, step 7.
Evaluation of peptide features in LC-MS data analyzed with Hardklör is best performed using Krönik. Because peptides elute over a time span in which multiple scan events are acquired, features identified in Hardklör corresponding to peptides are found in multiple consecutive scan events. Features resulting from spurious or random events do not persist over consecutive scan events. Thus, Krönik results are features that exhibit chromatographic persistence, as would be expected of peptides. Furthermore, while the Hardklör results contain redundant peptide features across consecutive scan events, Krönik summarizes the features into a single result. This functionality simplifies the visualization and interpretation of Hardklör results.
The Hardklör results obtained from Alternate Protocol 1 differ in context from the results in Basic Protocol 1. Because these results were obtained from spectra acquired by data dependent acquisition, there is not a chromatographic relationship between adjacent MS/MS scan events. Thus, Krönik is not an applicable tool for additional data analysis. The results can still be navigated from the HKViewer. Comparison of the results from Data.hk (Basic Protocol 1) and Data-MSMS.hk (Alternate Protocol 1) with HKViewer will show that the scan numbers skipped when navigating Data.hk are the scan numbers present in Data-MSMS.hk. Thus, Hardklör is used to not only analyze all the spectra from a mixed-mode data file, but also separate the results into different files according to spectrum type.
Commentary
Background Information
Deconvolution of signals in the mass spectra of complex mixtures is an important aspect in mass spectrometry data analysis. Some of the first algorithms to make such analyses broad-reaching and routine were ZSCORE and THRASH (Zhang and Marshall, 1998; Horn et al., 2000). Other algorithms have since followed that improve upon these algorithms through variations in modeling (Kaur and O'Connor, 2006; Jaitly et al., 2009) or deconvolution strategies (Du and Angeletti, 2006; Renard et al. 2008). Hardklör, in particular, was developed to automate the detection of peptide and protein features from high resolution mass spectra, with particular focus on shotgun proteomics data (Hoopmann et al., 2007).
Accurate identification of peptide features has become an integral part of improving the performance of shotgun proteomics. Peptide features are used to target peptides of interest (Hoopmann et al., 2009), improve database searching results (Beausoleil et al., 2006;Hsieh et al., 2010;Shin et al., 2008), and perform quantitative comparisons of peptides (Cox and Mann, 2008;Yang et al., 2011). Recently, interest has increased in the acquisition of high resolution MS/MS (Jedrychowski et al., 2011; Nagaraj et al., 2010; Olsen et al., 2009). Alternative Protocol 1 provides step-by-step instruction in the use of Hardklör for the analysis of high resolution MS/MS data. This functionality also illustrates the versatility of Hardklör, which can be used for the analysis of high resolution mass spectra in virtually any proteomic context.
Critical Parameters and Troubleshooting
Tables 1 and 2 summarize the critical parameters to adjust when optimizing Hardklör and Krönik. More detailed explanations can be found in the documentation on the Hardklör website.
Troubleshooting too few results or incorrect mass values
If the Hardklör results contain few features or poor mass accuracy, a common solution is to correct the resolution and mass analyzer code to match the input spectra. In particular, the orbitrap and FTICR mass analyzer codes should not be confused despite both instruments being Fourier Transform mass spectrometers.
Troubleshooting long computation times
Users are sometimes tempted to use very relaxed parameters with regard to charge state, window size, depth, and number of peptide models. The intention is to perform the most rigorous and thorough examination of the data; however, this approach often results in excessive (and exponential) increases in computation time with little increase in peptide features identified. Instead, it is best to use parameters most appropriate for the data that were acquired. For example, analysis of data from a tryptic sample of peptides should be limited to a maximum charge state of 5, instead of 20. Similarly, the depth parameter (-d) should be set according to sample complexity (typically 2 or 3). Detailed explanations of the parameters and acceptable ranges can be found in Basic Protocol 1.
Troubleshooting Krönik filters
Krönik can be used to identify and eliminate contaminant features from Hardklör results. Contaminants are defined features that persist beyond a reasonable time given the chromatography conditions. The unit of time depends on the unit used in the data file. Sometimes the scan event time stamp is recorded in minutes and other times it is recorded in seconds. If a three minute cutoff is desired when using Krönik, specify -c 3.0 if the time is recorded in minutes, or -c 180.0 if the time is recorded in seconds.
Advanced Parameters
Users with data containing a specific atomic (Goodlett et al., 2000) or isotopic enrichment (Hoopmann et al., 2007;Zelter et al., 2010) label can identify peptide features with or without the label using Hardklör with the variable modification parameter: -m. Modifications must conform to a specific syntax for either the addition of atoms to the averagine model, or the enrichment of heavy isotopes in the averagine model. When searching for peptides with a single chlorine atom, the parameter line would be ‘-m Cl’. In this case, Hardklör would generate a normal averagine model and an averagine model with a single chlorine atom when analyzing each set of observed spectrum peaks. The two models would be competitively compared to the data and the model with the highest correlation score accepted. When searching for 25% 15N enrichment, the parameter line would be ‘-m 0.25N1’, which indicates 25 atom percent excess (APE) of the first nitrogen isotope. Two or more labels can be combined into a single modification: ‘-m Cl2 0.50O2’ indicates a label that contains two chlorine atoms and 50 APE of the second oxygen isotope. Furthermore, the modification parameter can be specified multiple times in the Hardklör configuration file to search for more than one modification in the data.
Bioniformatics terms.
Averagine: A computational model representing an average amino acid. Averagine is used to approximate the atomic composition of a molecule where mass is known, but not the molecular formula.
Acknowledgments
The authors would like to acknowledge Richard Johnson from the Institute for Systems Biology for the data used in this unit.
Literature Cited
- Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Du P, Angeletti RH. Automatic Deconvolution of Isotope-Resolved Mass Spectra Using Variable Selection and Quantized Peptide Mass Distribution. Anal Chem. 2006;78:3385–3392. doi: 10.1021/ac052212q. [DOI] [PubMed] [Google Scholar]
- Goodlett DR, Bruce JE, Anderson GA, Rist B, Pasa-Tolic L, Fiehn O, Smith RD, Aebersold R. Protein identification with a single accurate mass of a cysteine-containing peptide and constrained database searching. Anal Chem. 2000;72:1112–1118. doi: 10.1021/ac9913210. [DOI] [PubMed] [Google Scholar]
- Hoopmann MR, Finney GL, MacCoss MJ. High-speed data reduction, feature detection, and MS/MS spectrum quality assessment of shotgun proteomics data sets using high-resolution mass spectrometry. Anal Chem. 2007;79:5620–5632. doi: 10.1021/ac0700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann MR, Merrihew GE, von Haller PD, Maccoss MJ. Post Analysis Data Acquisition for the Iterative MS/MS Sampling of Proteomics Mixtures. J Proteome Res. 2009;8:1870–1875. doi: 10.1021/pr800828p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn DM, Zubarev RA, McLafferty FW. Automated Reduction and Interpretation of High Resolution Electrospray Mass Spectra of Large Molecules. J Am Soc Mass Spectrom. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- Hsieh EJ, Hoopmann MR, MacLean B, MacCoss MJ. Comparison of database search strategies for high precursor mass accuracy MS/MS data. J Proteome Res. 2010;9:1138–1143. doi: 10.1021/pr900816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitly N, Mayampurath A, Littlefield K, Adkins JN, Anderson GA, Smith RD. Decon2LS: An open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics. 2009;10:87. doi: 10.1186/1471-2105-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrychowski MP, Huttlin EL, Haas W, Sowa ME, Rad R, Gygi SP. Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, O'Connor PB. Algorithms for automatic interpretation of high resolution mass spectra. J Am Soc Mass Spectrom. 2006;3:459–468. doi: 10.1016/j.jasms.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. Calculation of isotope distributions in mass spectrometry. A trivial solution for a non-trivial problem. Anal Chim Acta. 1991;247:107–119. [Google Scholar]
- Nagaraj N, D'Souza RC, Cox J, Olsen JV, Mann M. Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J Proteome Res. 2010;9:6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, Lange O, Remes P, Taylor D, Splendore M, Wouters ER, Senko M, Makarov A, Mann M, Horning S. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics. 2009;8:2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard BY, Kirchner M, Steen H, Steen JA, Hamprecht FA. NITPICK: peak identification for mass spectrometry data. BMC Bioinformatics. 2008;9:355. doi: 10.1186/1471-2105-9-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood AL, Van Orden SL, Smith RD. Rapid Calculation of Isotope Distributions. Anal Chem. 1995;67:2699–2704. doi: 10.1021/ac951158i. [DOI] [PubMed] [Google Scholar]
- Senko MW, Beu SC, McLafferty FW. Automated Assignment of Charge States from Resolved Isotopic Peaks for Multiply Charged Ions. J Am Soc Mass Spectrom. 1995a;6:52–56. doi: 10.1016/1044-0305(94)00091-D. [DOI] [PubMed] [Google Scholar]
- Senko MW, Beu SC, McLafferty FW. Determination of Monoisotopic Masses and Ion Populations for Large Biomolecules from Resolved Isotopic Distributions. J Am Soc Mass Spectrom. 1995b;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- Shin B, Jung HJ, Hyung SW, Kim H, Lee D, Lee C, Yu MH, Lee SW. Postexperiment monoisotopic mass filtering and refinement (PE-MMR) of tandem mass spectrometric data increases accuracy of peptide identification in LC/MS/MS. Mol Cell Proteomics. 2008;7:1124–1134. doi: 10.1074/mcp.M700419-MCP200. [DOI] [PubMed] [Google Scholar]
- Yang L, Vaitheesvaran B, Hartil K, Robinson AJ, Hoopmann MR, Eng JK, Kurland IJ, Bruce JE. The fasted/fed mouse metabolic acetylome: n6-acetylation differences suggest acetylation coordinates organ-specific fuel switching. J Proteome Res. 2011;10:4134–4149. doi: 10.1021/pr200313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelter A, Hoopmann MR, Vernon R, Baker D, MacCoss MJ, Davis TN. Isotope signatures allow identification of chemically cross-linked peptides by mass spectrometry: a novel method to determine interresidue distances in protein structures through cross-linking. J Proteome Res. 2010;9:3583–3589. doi: 10.1021/pr1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Marshall AG. A Universal Algorithm for Fast and Automated Charge State Deconvolution of Electrospray Mass-to-Charge Ratio Spectra. J Am Soc Mass Spectrom. 1998;9:225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]