

Abstract
Inhibition of DNA repair is a recognized mechanism for arsenic enhancement of ultraviolet radiation-induced DNA damage and carcinogenesis. Poly(ADP-ribose) polymerase-1 (PARP-1), a zinc finger DNA repair protein, has been identified as a sensitive molecular target for arsenic. The zinc finger domains of PARP-1 protein function as a critical structure in DNA recognition and binding. Since cellular poly(ADP-ribosyl)ation capacity has been positively correlated with zinc status in cells, we hypothesize that arsenite binding-induced zinc loss from PARP-1 is equivalent to zinc deficiency in reducing PARP-1 activity, leading to inhibition of DNA repair. To test this hypothesis, we compared the effects of arsenite exposure with zinc deficiency, created by using the membrane-permeable zinc chelator TPEN, on 8-OHdG formation, PARP-1 activity and zinc binding to PARP-1 in HaCat cells. Our results show that arsenite exposure and zinc deficiency had similar effects on PARP-1 protein, whereas supplemental zinc reversed these effects. To investigate the molecular mechanism of zinc loss induced by arsenite, ICP-AES, near UV spectroscopy, fluorescence, and circular dichroism spectroscopy were utilized to examine arsenite binding and occupation of a peptide representing the first zinc finger of PARP-1. We found that arsenite binding as well as zinc loss altered the conformation of zinc finger structure which functionally leads to PARP-1 inhibition. These findings suggest that arsenite binding to PARP-1 protein created similar adverse biological effects as zinc deficiency, which establishes the molecular mechanism for zinc supplementation as a potentially effective treatment to reverse the detrimental outcomes of arsenic exposure.
Keywords: zinc deficiency, arsenite, PARP-1, DNA damage repair, zinc finger
Introduction
Arsenic enhances the genotoxicity of UV radiation and other DNA damaging agents through inhibition of DNA repair. Arsenic has been reported to interact with certain zinc finger DNA repair proteins such as PARP-1, xerodermapigmentosum group A (XPA), and bacterial formamido-pyrimidine [fapy]-DNA glycosylase (FPG) (Ding et al., 2009; Hu et al., 1998; Lynn et al., 1997). PARP-1 catalyzes polymerization of ADP-ribose (PAR) units from donor NAD+ molecules onto target proteins and recruits additional proteins such as XRCC1, DNA ligase III and others to the DNA repair complex (Hassa et al., 2006; Rouleau et al., 2010). PARP activity is highly sensitive to arsenic inhibition, detectable at 100–200 nM (Ding et al., 2009; Hartwig et al., 2003). Inhibition of PARP-1 is predicted to broadly influence DNA repair processes based on its central role in DNA damage response. It has been reported that arsenic inhibits PARP-1 by direct (Zhou et al., 2011) and indirect (Wang et al., 2013) mechanisms. The three zinc fingers in the structure of PARP-1 are critically important for its DNA repair function, but have also been proposed as the targets for arsenic interaction.
In a zinc finger structure, the zinc ion is bonded tetrahedrally to cysteine thiolates and/or histidine imidazole groups, which maintains the functional three-dimensional structure (Kuwahara and Coleman, 1990). Under normal conditions, the three zinc fingers on PARP-1 are responsible for DNA-binding and DNA binding-dependent activation of catalytic activity (Gradwohl et al., 1990; Ikejima et al., 1990; Langelier et al., 2008). It has been reported that zinc deficiency leads to increased oxidative DNA damage, decreased DNA repair and increased cancer risk (Ho and Ames, 2002; Song et al., 2009). Zinc finger protein function can also be compromised by the substitution of zinc with other metals or the oxidation of thiolate donors (Nagaoka et al., 1993; Wilcox et al., 2001).
We reported previously that arsenite could bind with a synthetic peptide representing the first zinc finger of PARP-1 at the expense of zinc. Addition of zinc abolished the arsenite enhancement of UVR-stimulated 8-OHdG formation and restored PARP-1 activity in cellular systems (Cooper et al., 2013; Ding et al., 2009). These findings indicated a functional interaction/competition between arsenite and zinc within a specific target protein. The interaction of arsenic with PARP-1 zinc finger motifs may represent the underlying mechanism for arsenite-dependent PARP-1 inhibition. Since a positive correlation has been observed between cellular poly(ADP-ribosyl)ation capacity and zinc status in human peripheral blood mononuclear cells (Kunzmann et al., 2008), we hypothesize that arsenic binding-induced zinc loss from PARP-1 is equivalent to zinc deficiency in reducing PARP-1 activity, leading to inhibition of DNA repair. To test this hypothesis, we compared the effect of arsenic with zinc deficiency on 8-OHdG formation, PARP-1 activity and zinc binding to PARP-1 in a human keratinocyte cell line. Results show that arsenic exposure and zinc deficiency has similar effects on PARP-1 protein. Furthermore, we demonstrated arsenic binding and occupation of a peptide representing the first zinc finger of PARP-1 and showed that arsenic binding as well as zinc loss altered the conformation of zinc finger structure which functionally leads to PARP-1 inhibition.
Materials and methods
Materials
The N-terminally acetylated and C-terminally amidated 29-residue peptide GRASCKKCSESIPKDKVPHWYHFSCFWKV derived from the first zinc finger of human PARP-1(PARPzf) was custom-synthesized by Genmed Synthesis Inc., San Antonio TX. The purity was assessed by HPLC to exceed 95%. Sodium arsenite, cobalt chloride and zinc chloride were obtained from FlukaChemie (Buchs, Germany) and the purity is ≥ 99%. N,N,N’,N’-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN), tris (2-carboxyethyl) phosphine (TCEP) and 4-(2-Pyridylazo)-resorcinol monosodium salt (PAR) were from Sigma-Aldrich (St. Louis, MO).
Cell culture and treatments
The human keratinocyte cell line (HaCaT) cells were maintained as described previously (Qin et al., 2008). Stock solutions of sodium arsenite, zinc chloride and TPEN at 10 mM were prepared in double-distilled water and sterilized by passing through a 0.22 μm syringe filter. The working concentrations were diluted with DME/F-12 medium. For all experiments involving incubation with chemicals, cells were rinsed with PBS and placed into DME/F-12 medium containing concentrations of chemicals as indicated in the figure legends. For UVR exposure, the cell culture medium was removed, rinsed three times with PBS, covered with a thin layer of PBS and placed on ice in the dark. Cells were maintained on ice during UVR exposure and were exposed to 8 J/cm2 solar-simulated light using a 1000-watt Solar Ultraviolet Simulator (Oriel Corp., Stratford, CT). This solar simulator produces a high intensity UVR beam in both the UVA (320–400 nm) and UVB (280–320 nm) spectra. After UVR exposure, PBS was removed and replaced with a serum-free medium.
Cytotoxicity study
Potential cellular damage induced by TPEN was evaluated by monitoring the release of LDH in the culture medium. Cells were plated at 5 x 105 cells/well in 6-well culture plates and cultured for 2 days. After incubation with TPEN for 24h, culture medium was collected and centrifuged at 800x g for 10 min to obtain cell-free supernatant. The activity of LDH in the supernatant was determined using a Cytotoxicity Detection Kit (LDH) (Roche, Indianapolis, IN) according to the manufacturer’s instructions.
Immunocytochemistry staining for 8-OHdG formation
Immunoperoxidase staining for 8-OHdG was performed according to the procedure described previously (Qin et al., 2008). Briefly, cells grown on coverslips were fixed with methanol and then treated with RNase (100 μg/ml) (Sigma) and Proteinase K (10 μg/ml) (Sigma). DNA was denatured by treatment with 4 N HCl. Endogenous peroxidase was blocked by treating the cells with 3% H2O2 in methanol. After washing with PBS, the cells were treated with 10% normal horse serum and then incubated with primary antibody (anti-8-OHdG) (Trevigen, Gaithersburg, MD) (1:200 dilution). Then cells were treated with goat anti-mouse IgG conjugated with biotin (Vector Laboratories, Burlingame, CA). ABC reagent, avidin conjugated with horseradish peroxidase (Vector Laboratories) was added. To localize peroxidase, cells were treated with diaminobenzidine (Sigma). Finally, coverslips were washed with PBS, dehydrated and mounted to microscope slides using Permount. Images were obtained using an Olympus BH2-RFCA fluorescence microscope (Melville, NY) and Optronics MacroFire camera with PictureFrame 2.1 picture software (Optronix, Goleta, CA). The 8-OHdG level was represented by relative intensity of nucleus staining. It was quantified by measuring mean density of the nucleus using the Image-Pro Plus software (Media Cybernetics, Inc., Silver Spring, MD). Fifty to seventy randomly selected nuclei on each slide were measured and a minimum of 3 independent slides in each group were analyzed. The relative density was expressed by the fold of density of treated groups to that of the control.
Measurement of poly(ADP-ribosyl)ation capacity of PARP-1
HaCaT cells were treated with arsenite, TPEN, or arsenite with zinc as described in the figure legends. Cells were collected and the protein was extracted to measure PARP activity using the HT Colorimetric PARP/Apoptosis Assay kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s instructions. Protein was collected in the cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% triton X-100, 1 mM PMSF, 1×protease inhibitor cocktail), and the concentration was determined with the BCA protein detection kit (Sigma). Extracts containing 200 ng/25 μl protein were prepared and added to histone coated strip wells. PARP enzyme standards were prepared and assayed as the test samples. PARP substrate cocktail was added and the strip wells then incubated for 30 min at room temperature. The kit measures the incorporation of biotinylatedpoly (ADP-ribose) onto histone protein in the strip well format by the addition of the TACS-Sapphire colorimetric substrate and the absorbance was read with a SpectraMax 340 plate reader at 450 nm after stopping the reactions by adding 50 μl per well of 0.2M HCl. Absorbance values were averaged and activity was calculated from a standard curve. The relative activity was presented using the activity of treatment group divided by the activity of the control.
Isolation of PARP-1 by immunoprecipitation
PARP protein was isolated by immunoprecipitation as described before (Zhou et al., 2011). Briefly, protein from treated cells was extracted in 500 μl of lysis buffer. Protein (~2 mg in 500 μl) was incubated with 5 μl of rabbit polyconal antibody (Cell Signaling, #9542) for at least 1 hour at 4ºC, then Protein A beads (Invitrogen, Carlsbad, CA) were added in a 1:1 slurry and samples were incubated for an additional 1hour at 4ºC. The beads were recovered by centrifugation at 2,000 rpm for 5 min at 4ºC and washed five times with 1 ml of lysis buffer. To elute protein, the pellets were incubated with 100 μl of 0.1 M citric acid (pH 3.0) for 30 min followed by centrifugation at 2,000 rpm for 5 min at 4ºC. The resultant supernatant was adjusted to pH 7.0 with 10 N sodium hydroxide, then incubated with 10 mM hydrogen peroxide for 3 hours at 25ºC to release zinc from protein.
Measurement of zinc content by PAR chelation assay
Free zinc was measured by 4,(2-pyridylazo)-resorcinol (PAR) chelation assay according to the procedure described by Crow et al. (Crow et al., 1997). Briefly, 50 μM PAR was added to approximately 40 μg of isolated protein in a cuvette. The absorption spectrum was scanned at 25ºC on a Cary 50 spectrophotometer (Varian, Australia) in the range of 550 to 350 nm. The resorcinol indicator absorbance shifts from 411 to 492 nm in the presence of zinc and the 492 nm peak is recorded and compared to a standard curve for zinc content. The relative zinc content was normalized to protein concentration and expressed by the ratio to the zinc content of the control.
Preparation of peptide and metal solutions for the metal binding experiments
Weighed lyophilized peptide (grasckkcsesipkdkvphwyhfscfwkv, PARPzf) was suspended at a concentration of 2 mM in 10 mM tris buffer, pH 7.4 containing 1mM TCEP to allow the Cys residues remain in the reduced state. Stock solutions of metal ions were prepared at a concentration of 1 M in 10 mM Tris buffer, pH 7.4. For experimental analysis, both metal ions and peptide were diluted to working concentrations in 10 mM Tris, pH 7.4/1mM TCEP. All solutions were prepared freshly before each experiment.
Measurement of metal binding to the peptide by ICP/AES analysis
PARPzf (1.5 mM) were incubated with 3 mM NaAsO2 or ZnCl2 in the presence of 1 mM TCEP for 30 min. Free arsenic and zinc were removed by a 1800 molecular-weight cutoff (MWCO) polyacrylamide desalting columns (Thermo scientific, Rockford, IL). A mutant of peptide (mPARPzf) in which two cys were replaced by two histones was used as a control. The fractions containing peptide were determined by detecting A280 and were collected. 3 ml of nitric acid (HNO3) was added to digest the samples for about 2 hours by increasing the temperature slowly to 95°C. After digestion was completed, the acids were evaporated to 1 ml. The samples were diluted with 18 mega Ohms water to a total volume of 10 ml, and analyzed for the interested metal elements (arsenic and zinc) using PerkinElmer Optima 5300 DV ICP-AES following a method as described in US EPA 200.7 (US. Environmental Protection Agency, 2001). The elements’ peaks were examined, the peaks detection and background points were set, and the data was reprocessed. The data was expressed by means ± SD from three experiments.
Measurement of metal binding to the peptide by near UV spectroscopic study
Three-hundred microliters of 200 μM PARPzf peptide in 10 mM Tris buffer pH 7.4, with 1mM TCEP, was titrated with 1μl of 6 mM NaAsO2. An absorption spectrum was collected after each addition from 240 to 350 nm on a Cary 50 spectrophotometer (Varian, Australia) at 25 °C. The absorption spectrum of apo-PARPzf was subtracted from each spectrum collected during the course of titration. This process was repeated until the ligand-field absorption bands no longer increased in intensity, indicating that the protein was saturated with arsenite. Zinc competition with arsenic for the binding site of PARPzf was assessed by immediately back-titrating zinc chloride at the range of 20~180 μM with increment of 20 μM. The value of OD296nm was recorded at each addition.
Fluorescence Measurements
Fluorescence measurements were conducted with a SpectraMax M2 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) at room temperature. The excitation wavelength was 280 nm and the emission scans were made between 300 and 400 nm. Arsenite or zinc was added as sodium arsenite or chloride zinc, respectively, to a cuvette containing 200 μM peptide in tris buffer consisting of 10 mM tris, pH 7.4, 1 mM TCEP to the final concentration of 1 mM.
Circular Dichroism spectrometry
Circular dichroism (CD) spectra were recorded at 25 °C on a Jasco spectropolarimeter (Jasco analytical instruments, Easton, MD.). The wavelength range was 190–250 nm. Peptide at 100 μM final concentration in water was put in 1 cm cuvet for scan. The average of 3 scans was presented as final spectra.
Statistical analysis
The data is presented as means ± SD. Statistical analysis was carried out using one-way ANOVA followed by post-hoc test for two groups comparison. Differences between means were regarded as significant if p < 0.05, and significant differences are indicated by the sign *, # or &.
Results
Both arsenite and zinc chelator TPEN enhance UVR-induced 8-OHdG formation
Evidence shows that dietary and intracellular zinc deficiency has strong correlation with oxidative DNA damage in animals and cells (Ho, 2004). Ho and others’ studies demonstrated that zinc deficiency created by TPEN, a membrane-permeable zinc chelator, induced single-strand breaks, Fpg-sensitive lesions and oxidant production in IMR90 cells (Ho et al., 2003), and induced testicular oxidative stress and damage in mice (Zhao et al., 2011). In this study, to explore whether zinc deficiency impacts UV-induced oxidative DNA damage, we treated cells with TPEN and compared the cell response to TPEN or arsenite under UV exposure. 8-OHdG formation was used as an indicator for UV-stimulated oxidative DNA damage. Cell survival study indicated that 5 μM TPEN was non-cytotoxic (data not shown), and was selected for our zinc deficiency experiments. Treatment of cells with arsenite at 2 μM did not generate any observable increase in DNA damage as compared to control, whereas treatment with UV or TPEN (5 μM) produced significant increase in 8-OHdG formation (Fig. 1). More importantly, both TPEN and arsenite significantly increased UV-stimulated oxidative DNA damage. In cells treated with arsenite and TPEN simultaneously, only a slight further increase in 8OHdG formation over each individual treatment alone was observed. These results suggest that arsenic has very similar effect to zinc chelator TPEN in enhancing UVR-stimulated oxidative DNA damage.
Fig 1.
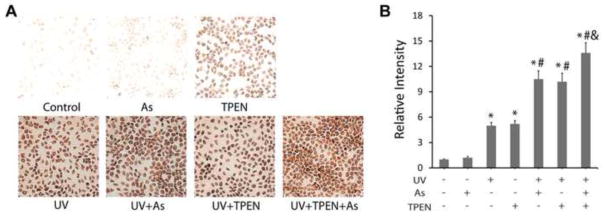
The effect of arsenic and TPEN on enhancing UVR-generated 8-OHdG formation. (A) HaCat cells were incubated with the 2μM arsenite and/or 5 μM TPEN for 24h, then exposed to 8 J/cm2 UV radiation on ice. 8-OHdG was measured by immunocytochemistry. (B) The results of (A) were quantified by measuring the optical density of the staining (n ≥ 3). Relative intensity was indicated as the density of treated group divided by that of the control and presented as the means ± SD from three experiments. *, p < 0.01 vs. Control; #, p<0.01 vs. UV alone; &, p<0.05 vs. Arsenic or TPEN alone.
Arsenite and zinc deficiency similarly induced zinc loss and decreased poly(ADP-ribosyl)ation capacity of PARP-1
Next we focused on PARP-1 as a target zinc finger protein and investigated whether treatment with arsenite and TPEN has similar effects in a functional study. Firstly, we measured the zinc content in PARP-1 immunoprecipitated from arsenite or TPEN exposed HaCaT cells. Total zinc content in PARP-1 was determined by releasing zinc by incubating protein extracts with 10 mM H2O2 for at least 2 hours at 25°C. Higher concentration of H2O2 did not release additional zinc (data not shown). Both TPEN and arsenite treatment led to dramatic zinc loss in PARP-1 protein. Zinc supplement significantly restored zinc content in arsenite-exposed cells (Fig. 2A). We then examined the effects of zinc loss on PARP-1 function. Our previous paper reported that the DNA binding capability of PARP-1 was decreased with zinc loss (Zhou et al., 2011). Here we compared arsenite and TPEN in altering PARP-1 activity in the cell extracts through detecting poly(ADP-ribosyl)ation initiated by extraneous DNA strand breaks. Both arsenite and TPEN exposure caused very similar declines of PARP-1 poly(ADP-ribosyl)ation capacity, whereas zinc supplementation partially reversed arsenite-dependent inhibition of PARP-1 activity (Fig. 2B). These findings suggest that zinc occupation is a critical factor for maintaining PARP-1 activity, and that both arsenite and zinc chelator TPEN decrease PARP-1 activity by reducing the zinc content in the PARP-1 protein.
Fig 2.

Arsenite and TPEN induced loss of zinc content and activity in PARP-1 protein. HaCat cells were treated with 5 μM TPEN, 2 μM arsenite, or 2 μM arsenite + 2 μM zinc for 24 hours. After treatment, the cells were collected and PARP-1 was isolated by immunoprecipitation from the cell extracts. (A) 10 mM H2O2 were incubated with the isolated PARP-1 to release the zinc. Released zinc was determined by measuring the absorbance at 492 nm after the addition of 50 μM PAR. The data were presented as the zinc content of treatment group divided by that of the control and expressed as the means±SD from three experiments. (B) The activity of PARP-1 in the cell extracts was detected by immunochemical detection of poly(ADP-ribose). The relative activity was indicated as the activity of treatment group divided by that of the control and presented as the means±SD from three experiments. *, p < 0.01 vs. Control; #, p<0.01 vs. arsenic alone.
Arsenite caused zinc loss by coordinating with PARP-1 zinc finger motif
While it is apparent that TPEN reduces zinc content of PARP-1 protein by chelating the metal, it is much less clear whether arsenite can replace zinc to bind with the zinc finger, and whether arsenite-bond zinc finger has altered structural conformation from that of zinc-bond. Earlier mass spectroscopy study from our lab has shown that arsenite could coordinate with the peptide derived from zinc finger 1 of PARP-1 (Ding et al., 2009; Zhou et al., 2011). To further investigate the molecular mechanism of zinc loss induced by arsenite, we conducted a quantitative analysis of arsenite and zinc binding to PARPzf peptide using ICP-AES. After incubation of PARPzf for 30 min with excess arsenite or zinc in the presence of TECP (a disulfide-reducing agent which maintains a reducing environment but does not coordinate with arsenic), free arsenite or zinc was removed by a desalting column. The amount of arsenite or zinc bound to the PARP-1 zinc finger peptide was found to be 4.45±0.18 μg and 5.00±0.23 μg, respectively. During the control peptide incubation no residual metal was detected in the samples. These results demonstrate that both arsenite and zinc could form a stable covalent bond with PARPzf in solution under reductive conditions.
Near UV spectrometry is another method that can provide the evidence of arsenite binding to the zinc finger. Optical absorbance in the range of 250 to 350 nm is attributed to a covalent bond between arsenite and sulfur in the peptide (Spuches et al., 2005). With the addition of arsenite to the PARPzf peptide solution, the observed optical absorbance increased as OD296nm increased to the maximal saturation (Fig. 3A). Together with the previous mass spectra showing that arsenite bound to the peptide with loss of three hydrogen ions (Zhou et al., 2011), these results indicate that arsenite coordinated with the three thiols of the zinc finger of the peptide. Additionally, during the back-titration of zinc to the complex solution of arsenite and peptide, the arsenic binding signal gradually diminished with the addition of zinc (Fig. 3B). These results suggest a competition between zinc and arsenite for the same binding site on PARPzf, and are consistent with the cellular phenomenon showing that supplemental zinc reversed arsenite-induced PARP-1 activity inhibition and zinc loss (Fig. 2).
Fig 3.

UV-vis spectrum of arsenite binding to the peptide. (A) 200 μM PARPzf was titrated with arsenite at the range of 20~140 μM with increment of 20 μM. (B) 200 μM PARPzf was saturated with 200 μM arsenite, then immediately back-titrated with zinc at the range of 20~180 μM with increment of 20 μM. The absorption spectrum of peptide addition was collected during the course of the metal-binding titration after being subtracted by the spectrum of apo-peptide shown as the inset of panel A and B. The value of OD296nm was recorded at each addition shown as panel A and B respectively.
Arsenite binding and zinc loss both lead to conformational change of PARP-1zf
Zinc is known to maintain the correct conformation of zinc finger motifs, allowing for DNA binding and other related activities. Here we studied the structural and conformational consequences of arsenite binding or zinc loss on PARP-1zf. Firstly, fluorescence analysis was used to investigate the tertiary structure of PARPzf with coordination to arsenic or zinc. Metal-free PARPzf exhibited a single fluorescence band, with a maximum at 348 nm, upon excitation at 280 nm. Fluorescence of PARPzf increased upon addition of excess zinc and decreased upon addition of excess arsenite (Fig. 4A). These results indicate that zinc binding helps PARPzf to form a tighter structure while arsenite binding leads to a looser structure compared to apo-PARPzf. CD spectrum showed that apo-peptide or zinc bound peptide formed a secondary structure (Fig. 4B), while arsenite treatment induced partial secondary structure loss of PARP-1 zinc finger 1 peptide, suggesting that arsenite compromised the folding pathway of PARP-1. Together, these findings demonstrate that the occupation by arsenite, or loss of zinc (such as chelation by TPEN), lead to conformational destruction of secondary and tertiary structures of PARP-1zf.
Fig 4.
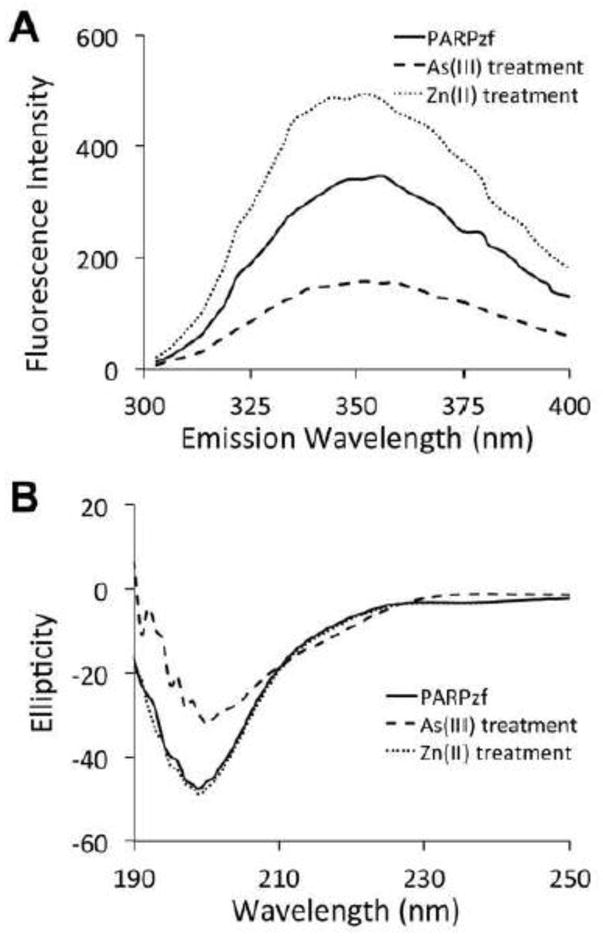
Differential conformation of vacant vs. zinc and arsenite bonded PARPzf. (A) Fluorescence measurement of the coordination of PARPzf with arsenite or zinc. Excess arsenite or zinc was added to a solution of PARPzf (200 μM in 10mM Tris buffer, pH 7.4, 1 mM TCEP) at 25°C. The excitation wavelength was 280 nm and the emission scans were made between 300 and 400 nm. (B) Circular Dichroism spectrums of PARP-1 zinc finger 1, coordination with zinc or arsenite. 200 μM zinc chloride or 200 μM sodium arsenite was added into 200 μM PARP-1 zinc finger 1 peptide in distilled water. CD spectrums were collected after 15 min incubation.
Discussion
The importance of zinc in zinc finger proteins and the DNA repair system has been demonstrated by zinc deficiency studies. Previous works showed that zinc deficiency causes oxidative DNA damage through a combined effect of increased oxidative stress and an impairment of the DNA repair signal pathway as evidenced by epidemiologic and cellular studies (Ho et al., 2003). Zinc-binding domains are frequently present in proteins required for maintaining genomic integrity. In the present study, we show that treatment with zinc chelator TPEN, or arsenite, can similarly lead to loss of zinc from PARP-1 protein, or “zinc deficiency”, resulting in PARP-1 activity inhibition. There has been accumulating evidence that PARP-1 may be a particularly sensitive target, not only for intracellular zinc deficiency caused by dietary inadequacy or aging, but also for toxic metal ions. In maintaining the protein structure, zinc loss disrupts the zinc finger structure and leads to improper folding of the protein, which is associated with loss of correct protein function (Hartwig et al., 2002). For example, Hartwig’s works showed trivalent antimony interacted with the zinc finger domain of XPA, which resulted in a concentration dependent release of zinc from a peptide consistent with this domain. In the cellular system, XPA interaction with damaged DNA was diminished in the presence of SbCl3 (Hartwig et al., 2002). Another study showed mutations in the zinc finger motif eliminated the ability of XPA to confer a protective mechanism of action on the endonucleases involved in NER (Bartels and Lambert, 2007). These reported studies provide a strong link between zinc depletion/deficiency and zinc finger protein XPA function. In other words, zinc serves an essential and critical function in DNA repair system and protein activity.
It has been shown that zinc finger proteins are direct molecular targets of arsenic (Ding et al., 2009; Zhou et al., 2011). Enhanced DNA damage is found in arsenic co-carcinogenesis studies (Ding et al., 2009; Qin et al., 2008). Although the expression of some DNA repair proteins were induced at low intracellular zinc status or arsenic exposure, the binding with important downstream signals leading to proper DNA repair was lost. Exploring the link between zinc deficiency and arsenite exposure is helpful for discovering the mechanism of DNA repair protein dysfunction. In the present study, we demonstrated that zinc loss induced by arsenite is a critical process in impairment of PARP-1 function, at similar level and in a similar manner to that induced by zinc deficiency created by zinc chelator TPEN. Cells treated with arsenite lead to inhibition of PARP-1 activity, accompanied with zinc loss from PARP-1 in a dose and time dependent manner (data not shown), which was almost identical to the effect of TPEN. This indicates that loss of protein activity is not only associated with global zinc deficiency, but can also be induced by zinc loss from a specific site of the target protein. Furthermore, we investigated the mechanism and consequence of arsenite-dependent zinc loss from PARP-1 by presenting arsenic and zinc coordination with the peptide derived from the first zinc finger of PARP-1. Arsenite or zinc coordinated with the peptide was shown in a mass spectrum (Zhou et al., 2011). Here ICP-AES and near UV spectrum demonstrated arsenic bound to thiols forms a stable complex in reductive conditions and competition of zinc with arsenic for the same binding site in PARPzf. Arsenite binding with target protein leads to zinc loss, thus creating a consequence similar to zinc deficiency. Our results offer the direct evidence, at the molecular level, of arsenic competing with zinc for zinc finger occupation. This explains why arsenic behaves similarly to a zinc chelator in the inhibition of the DNA repair system. In addition, we further investigated the structural change of the zinc finger motif as a consequence of arsenic induced zinc loss. Since the correct conformation is critical for PARP-1 to bind with specific sites on DNA that need to be repaired. The fluorescence spectrum of metal-free PARPzf is typical of an “incompletely ordered” peptide. Zinc binding is accompanied by the proper folding of the peptide. In contrast, arsenic binding strongly quenches the protein fluorescence of peptide, indicating that a loose “unordered” peptide has formed. CD spectrum also showed less secondary structure with arsenic bound peptide. Similarly, another study showed that arsenic can compromise protein folding pathway by coordination to the cysteine residues of unfolding reduced protein (RfBP, lysozyme and RNase) in vivo contributing to oxidative and protein folding stresses (Ramadan et al., 2009). All these evidence support the potential of arsenic leading to the same effect as zinc loss from PARP-1, as related to protein conformational changes. This is the direct consequence of arsenic induced zinc loss in terms of structure, and this is always tightly related to functional consequences, i. e. protein dysfunction or activity inhibition. As we have shown, arsenite decreased DNA binding activity of PARP-1 in cells (Zhou et al., 2011), which can be explained by arsenic induced zinc loss and improper folding of the zinc finger domain.
Since zinc deficiency is demonstrated as a key event in inhibition of DNA repair by arsenic, zinc supplementation is naturally considered as a treatment to reverse the effect of arsenic exposure (Cooper et al., 2013). In a cellular study, we observed that the supplemental zinc could restore the zinc content in PARP-1 proteins as well as the protein’s activity (Fig. 2). The mechanism of zinc supplement was also confirmed by a back-titration experiment (Fig. 3B). These findings suggest that zinc supplement would be an efficient approach for prevention of DNA damage from arsenic exposure.
In summary, our findings suggest that protein-associated zinc loss is a key event involved in arsenite-induced PARP-1 activity inhibition. Based on our understanding of the mechanism for arsenite-dependent zinc loss leading to inhibition of PARP-1 mediated DNA repair damage, zinc supplementation is a useful therapeutic approach to counteract the arsenic effect on UV-induced carcinogenesis.
Highlights.
Arsenite binding is equivalent to zinc deficiency in reducing PARP-1 function
Zinc reverses arsenic inhibition of PARP-1 activity & enhancement of DNA damage
Arsenite binding and zinc loss alters the conformation of zinc finger structure
Acknowledgments
This study was funded by grants from the U.S. National Institutes of Health (R01ES15826, R01ES15826-03S1, and R01ES021100). Support was also provided by the UNM Cancer Center P30CA118100. We would like to thank Dr. Mehdi Ali for his work of ICP/AES.
Abbreviations
- UVR
ultraviolet radiation
- PARP-1
poly(ADP-ribose) polymerase-1
- 8-OHdG
8-hydroxyl-2’-deoxyguanine
- TPEN
N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine
- TECP
tris (2-carboxyethyl) phosphine
- PAR
4-(2-Pyridylazo)-resorcinol monosodium salt
- ICP/AES
inductively coupled plasma atomic emission spectroscopy
- PMSF
phenylmethylsulfonyl fluoride
- zf
zinc finger
- CD spectrum
Circular Dichroism spectrum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartels CL, Lambert MW. Domains in the XPA protein important in its role as a processivity factor. Biochem Biophys Res Commun. 2007;356:219–225. doi: 10.1016/j.bbrc.2007.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KL, King BS, Sandoval MM, Liu KJ, Hudson LG. Reduction of arsenite-enhanced ultraviolet radiation-induced DNA damage by supplemental zinc. Toxicol Appl Pharmacol. 2013;269:81–88. doi: 10.1016/j.taap.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Ding W, Liu W, Cooper KL, Qin XJ, de Souza Bergo PL, Hudson LG, Liu KJ. Inhibition of poly(ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J Biol Chem. 2009;284:6809–6817. doi: 10.1074/jbc.M805566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Ménissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc Natl Acad Sci USA. 1990;87:2990–2994. doi: 10.1073/pnas.87.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig A, Asmuss M, Blessing H, Hoffmann S, Jahnke G, Khandelwal S, Pelzer A, Bürkle A. Interference by toxic metal ions with zinc-dependent proteins involved in maintaining genomic stability. Food Chem Toxicol. 2002;40:1179–1184. doi: 10.1016/s0278-6915(02)00043-1. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Pelzer A, Asmuss M, Bürkle A. Very low concentrations of arsenite suppress poly(ADP-ribosyl)ation in mammalian cells. Int J Cancer. 2003;104:1–6. doi: 10.1002/ijc.10911. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E. Zinc deficiency, DNA damage and cancer risk. J Nutr Biochem. 2004;15:572–578. doi: 10.1016/j.jnutbio.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Ho E, Ames BN. Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFkappa B, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc Natl Acad Sci USA. 2002;99:16770–16775. doi: 10.1073/pnas.222679399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E, Courtemanche C, Ames BN. Zinc deficiency induces oxidative DNA damage and increases p53 expression in human lung fibroblasts. J Nutr. 2003;133:2543–2548. doi: 10.1093/jn/133.8.2543. [DOI] [PubMed] [Google Scholar]
- Hu Y, Su L, Snow ET. Arsenic toxicity is enzyme specific and its affects on ligation are not caused by the direct inhibition of DNA repair enzymes. Mutat Res. 1998;408:203–218. doi: 10.1016/s0921-8777(98)00035-4. [DOI] [PubMed] [Google Scholar]
- Ikejima M, Noguchi S, Yamashita R, Ogura T, Sugimura T, Gill DM, Miwa M. The zinc fingers of human poly(ADP-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. Other structures recognize intact DNA. J Biol Chem. 1990;265:21907–21913. [PubMed] [Google Scholar]
- Kunzmann A, Dedoussis G, Jajte J, Malavolta M, Mocchegiani E, Bürkle A. Effect of zinc on cellular poly(ADP-ribosyl)ation capacity. Exp Gerontol. 2008;43:409–414. doi: 10.1016/j.exger.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Kuwahara J, Coleman JE. Role of the zinc(II) ions in the structure of the three-finger DNA binding domain of the Sp1 transcription factor. Biochemistry. 1990;29:8627–8631. doi: 10.1021/bi00489a019. [DOI] [PubMed] [Google Scholar]
- Langelier MF, Servent KM, Rogers EE, Pascal JM. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J Biol Chem. 2008;283:4105–4114. doi: 10.1074/jbc.M708558200. [DOI] [PubMed] [Google Scholar]
- Lynn S, Lai HT, Gurr JR, Jan KY. Arsenite retards DNA break rejoining by inhibiting DNA ligation. Mutagenesis. 1997;12:353–358. doi: 10.1093/mutage/12.5.353. [DOI] [PubMed] [Google Scholar]
- Nagaoka M, Kuwahara J, Sugiura Y. Alteration of DNA binding specificity by nickel (II) substitution in three zinc (II) fingers of transcription factor Sp1. Biochem Biophys Res Commun. 1993;194:1515–1520. doi: 10.1006/bbrc.1993.1996. [DOI] [PubMed] [Google Scholar]
- Qin XJ, Hudson LG, Liu W, Timmins GS, Liu KJ. Low concentration of arsenite exacerbates UVR-induced DNA strand breaks by inhibiting PARP-1 activity. Toxicol Appl Pharmacol. 2008;232:41–50. doi: 10.1016/j.taap.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C. Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry. 2009;48:424–432. doi: 10.1021/bi801988x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Leonard SW, Traber MG, Ho E. Zinc deficiency affects DNA damage, oxidative stress, antioxidant defenses, and DNA repair in rats. J Nutr. 2009;139:1626–1631. doi: 10.3945/jn.109.106369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuches AM, Kruszyna HG, Rich AM, Wilcox DE. Thermodynamics of the As(III)-thiol interaction: arsenite and monomethylarsenite complexes with glutathione, dihydrolipoic acid, and other thiol ligands. Inorg Chem. 2005;44:2964–2972. doi: 10.1021/ic048694q. [DOI] [PubMed] [Google Scholar]
- US. Environmental Protection Agency. Trace Elements in Water, Solids, and Biosolids by Inductively Coupled Plasma-atomic Emission Spectrometry. 2001. Method 200.7. [Google Scholar]
- Wang F, Zhou X, Liu W, Sun X, Chen C, Hudson LG, Liu KJ. Arsenite-induced ROS/RNS generation causes zinc loss and inhibits the activity of poly(ADP-ribose) polymerase-1. Free Radic Biol Med. 2013;61:249–256. doi: 10.1016/j.freeradbiomed.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox DE, Schenk AD, Feldman BM, Xu Y. Oxidation of Zinc-Binding Cysteine Residues in Transcription Factor Proteins. Antioxid Redox Signal. 2001;3:549–564. doi: 10.1089/15230860152542925. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Tan Y, Dai J, Li B, Guo L, Cui J, Wang G, Shi X, Zhang X, Mellen N, Li W, Cai L. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol Lett. 2011;200:100–106. doi: 10.1016/j.toxlet.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, Hudson LG. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. 2011;286:22855–22863. doi: 10.1074/jbc.M111.232926. [DOI] [PMC free article] [PubMed] [Google Scholar]