

Abstract
Existing evidence implicates regulatory roles for protein phosphatase 2A (PP2A) in a variety of cellular functions, including cytoskeletal remodeling, hormone secretion, and apoptosis. We report here activation of PP2A in normal rat islets and insulin-secreting INS-1 832/13 cells under the duress of hyperglycemic (HG) conditions. Small interfering RNA-mediated knockdown of the catalytic subunit of PP2A (PP2Ac) markedly attenuated glucose-induced activation of PP2A. HG, but not nonmetabolizable 3-O-methyl glucose or mannitol (osmotic control), significantly stimulated the methylation of PP2Ac at its C-terminal Leu-309, suggesting a novel role for this posttranslational modification in glucose-induced activation of PP2A. Moreover, knockdown of the cytosolic leucine carboxymethyl transferase 1 (LCMT1), which carboxymethylates PP2Ac, significantly attenuated PP2A activation under HG conditions. In addition, HG conditions, but not 3-O-methyl glucose or mannitol, markedly increased the expression of LCMT1. Furthermore, HG conditions significantly increased the expression of B55α, a regulatory subunit of PP2A, which has been implicated in islet dysfunction under conditions of oxidative stress and diabetes. Thapsigargin, a known inducer of endoplasmic reticulum stress, failed to exert any discernible effects on the carboxymethylation of PP2Ac, expression of LCMT1 and B55α, or PP2A activity, suggesting no clear role for endoplasmic reticulum stress in HG-induced activation of PP2A. Based on these findings, we conclude that exposure of the islet β-cell to HG leads to accelerated PP2A signaling pathway, leading to loss in glucose-induced insulin secretion.
Glucose-induced insulin secretion (GSIS) from pancreatic β-cells involves generation of second messengers, such as ions, cyclic nucleotides, and lipid hydrolytic products of phospholipases. Some of the known actions of these second messengers include regulation of various protein kinases indigenous to pancreatic β-cells. Indeed, extant studies have demonstrated localization of several kinases in normal rat islets as well as clonal β-cells; these include Ca2+-, Ca2+/calmodulin-, cAMP-, and phospholipid-dependent protein kinases (1, 2). The phosphorylation status of proteins is tightly regulated by the balance of the activities of protein kinases and phosphatases. Although several earlier studies were focused on the identification and characterization of protein kinases in islets, relatively little is known about the localization and regulation of protein phosphatases (PPs) in β-cells. Along these lines, original studies from our laboratory have suggested novel roles for okadaic acid-sensitive PP2A in islet function and GSIS (3, 4).
Existing evidence implicates regulatory roles for PP2A in a variety of cellular functions, including cell cycle progression, proliferation, cytoskeletal remodeling, hormone secretion, and even cellular dysfunction (4–6). The PP2A family of enzymes represents a major class of phospho-serine/threonine phosphatases, which have been implicated in the regulation of many cellular events. Several holoenzyme complexes have been isolated and characterized from a variety of tissues. The PP2A heterodimer complex is comprised of a scaffolding A subunit with an apparent molecular mass of 65 kDa and the 36-kDa catalytic subunit (catalytic subunit of PP2A [PP2Ac]). This A/C subunit heterodimer interacts with a regulatory B subunit yielding the PP2A heterotrimeric holoenzyme. Two different genes of A (Aα and Aβ) and C (Cα and Cβ), and several families of the B subunits (B, B', B”, and B”'), have been identified. The binding of B subunit to the A/C heterodimer provides further stability to the holoenzyme. It is also suggested that the variable B subunit(s) influences substrate specificity and/or subcellular localization of a given PP2A holoenzyme. It is estimated that the combination of all subunits (A, B, and C) could produce more than 75 different trimeric holoenzymes, although the precise number of the possible holoenzyme complexes that actually exist in cells still needs to be determined. Although the A and C subunits are ubiquitously expressed, certain B subunits are expressed in a tissue-specific manner and at various stages of cellular development. Recent years have witnessed significant progress in the area of functional regulation of PP2A, specifically via posttranslational modifications (4–9).
Unlike the regulatory B subunits, the PP2Ac is highly conserved among different species. The 6 C-terminal amino acid residues (TPDYFL) are conserved in all known PP2Ac subunits, and the 3 C-terminal residues (YFL) are also conserved in the other 2 PP2A family members, PP4 and PP6, implicating a role for these residues in the catalytic function of these enzymes (4–6). Previous work had suggested the C-terminal leucine residue in PP2Ac (Leu-309) undergoes reversible carboxymethylation (CML), a step catalyzed by leucine carboxymethyl transferase 1 (LCMT1). Several lines of evidence suggest that the CML of PP2Ac culminates in the functional activation of PP2A (3, 7, 8). In addition to the CML step, published evidence also suggests that the Tyr-307 residue of PP2Ac undergoes phosphorylation, which is linked to functional inactivation of the enzyme (6, 9, 10). Together, posttranslational modification of PP2Ac plays key regulatory roles in the catalytic function of PP2A.
It is well established that chronic exposure of pancreatic β-cells to hyperglycemia (HG) leads to metabolic dysfunction and demise (11, 12). Several intracellular signaling events have been identified as causal to HG-induced metabolic dysregulation of the islet. These include endoplasmic reticulum (ER) and oxidative stress. It has been suggested that both oxidative and ER stress lead to mitochondrial dysfunction, cytochrome C release, and caspase activation resulting in the demise of the β-cell (11, 13, 14). Despite this compelling evidence, little is known about potential detrimental effects of HG on phosphatases function in the islet β-cell. With this in mind, we undertook the current investigation to examine functional consequences of HG exposure on the catalytic activation of PP2A in the islet β-cell. We also investigated regulatory effects of HG on the CML of PP2Ac in an attempt to delineate the underlying mechanisms involved in the regulation of PP2A under these conditions. We present the first evidence to indicate that HG conditions cause activation of PP2A in normal rat islets and insulin-secreting INS-1 832/13 β-cells via increased expression and activation of LCMT1, which catalyzes the CML of PP2Ac (15).
Materials and Methods
Materials
Small interfering RNA (siRNA), designed to knockdown gene expression of PP2Acα and LCMT1, scrambled-siRNA (negative control), and Dharmafect transfection reagent were from Dharmacon. PP2A assay kit was from Millipore. Antisera directed against methylated-PP2Acα and total PP2A were from Abcam. Antisera against B55α and LCMT1 were from Santa Cruz Biotechnology, Inc. Antiserum against C/EBP homology protein (CHOP) was from Cell Signaling. Antimouse, antirabbit IgG-horseradish peroxidase conjugates, and enhanced chemiluminescence kits were from Amersham Biosciences. Antigoat and antirat IgG-horseradish peroxidase conjugates were from Santa Cruz Biotechnology, Inc. 3-O-methyl glucose (3MG) and mannitol were from Sigma.
Cell culture and treatments
INS-1 832/13 cells were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum supplemented with 100-IU/mL penicillin and 100-IU/mL streptomycin, 1mM sodium pyruvate, 50μM 2-mercaptoethanol, and 10mM HEPES (pH 7.4). Islets were isolated from normal 6 week-old male Sprague-Dawley rats (Harlan Laboratories) as we described in Refs. 16, 17. All protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Wayne State University (protocol number A-08–05-12). Human islets were procured from PRODO Laboratories, Inc. INS-1 832/13 cells, isolated rat islets, or human islets were treated with media consisting of glucose (2.5mM, low glucose [LG] or 20mM, HG) for 48 hours at 37°C. In select studies, INS-1 832/13 cells were treated with LG without or with thapsigargin (TG) (0.25μM) for 6 hours. Cell lysates were used for PP2A activity or Western blotting.
Isolation of subcellular fractions from INS-1 832/13 cells
INS-1 832/13 cells were rinsed once in PBS (pH 7.5), trypsinized, and spun at 100g for 5 minutes to remove the medium. The cellular pellet was washed once with an isotonic medium containing 230mM mannitol, 70mM sucrose, 5mM HEPES buffer (pH 7.4), protease inhibitor cocktail, 1mM phenylmethylsulfonylfluoride and 1mM EGTA and twice more with the same buffer without EGTA. Cells were then homogenized manually using a Potter-Elvehjem tissue homogenizer (20–30 strokes) in the same buffer containing 1mM dithiothreitol. Subcellular fractions were isolated from the homogenates, and their purity was assessed according to our published methods (18–20). In select experiments, rat islets or INS-1 832/13 cells were lysed in ice-cold radioimmunoprecipitation assay lysis buffer containing protease inhibitor cocktail (as above). The band intensity was quantified densitometrically using Kodak imaging software.
Quantitation of methylated PP2Ac
INS-1 832/13 cells, normal rat islets, or human islets were treated with LG or HG for different time intervals as indicated in the text. Relative abundance of total or methylated PP2Ac was determined by Western blotting using antisera directed against these proteins (21). The band intensity was quantified densitometrically using Kodak imaging software.
Quantitation of PP2A activity
PP2A was assayed using an immunoprecipitation phosphatase assay kit according to the manufacturer's instructions. Briefly, the cells were lysed in phosphatase extraction buffer containing 20mM imidazole-HCl, 2mM EDTA, and 2mM EGTA (pH 7.0), with 1mM phenylmethylsulfonylfluoride, 10 μg/mL each of aprotinin, leupeptin, and soybean trypsin inhibitor. The lysates were sonicated for 10 seconds and centrifuged at 20 000g for 5 minutes. Equal amount of supernatant protein was incubated with anti-PP2A antibody and protein A-agarose for 2 hours at 4°C. The beads were washed 3 times and incubated for 10 minutes at room temperature with phosphopeptide (750μM). After addition of the malachite green phosphate detection solution, PP2A activity was determined by measuring the absorbance at 650 nm.
siRNA-mediated knockdown of PP2Acα and LCMT1
siRNA against the PP2Acα and LCMT1 were from siGENOME SMARTpool (Dharmacon). The siRNA was transfected into INS-1 832/13 cells using Lipofectamine-RNAiMAX transfection reagent according to the manufacturer's instructions. After 24 hours of transfection, cells were subsequently treated with LG or HG for an additional 24 hours.
Statistical analysis of the experimental data
Data are expressed as means ± SEM. All statistical analyses were performed with Sigma Stat version 3.5. Statistical significance of the differences between the control and experimental conditions was determined by Student's t test, and P < .05 was considered significant.
Results
HG conditions activate PP2A in INS-1 832/13 cells and normal rat islets
One of the major objectives of the current study is to investigate the functional status of PP2A in pancreatic β-cells exposed to HG conditions. To assess this, INS-1 832/13 cells or normal rat islets were cultured in the presence of LG or HG for 48 hours, and the PP2A activity was quantitated in cell lysates. Data shown in Figure 1 demonstrate that HG causes a significant increase in the PP2A activity in INS-1 832/13 cells (∼3-fold) (Figure 1A) and normal rat islets (∼1.8-fold) (Figure 1B) relative to LG conditions. Furthermore, we noticed complete inhibition of GSIS (Figure 1C) in INS-1 832/13 cells under conditions in which HG activated PP2A.
Figure 1.

HG conditions activate PP2A and abolish GSIS in pancreatic β-cells. INS-1 832/13 cells (A) or rat islets (B) were incubated with LG (2.5mM) or HG (20mM) for 48 hours. Cell lysates were prepared in imidazole-HCl buffer (pH 7.0), and PP2A activity was quantified (20-μg lysate protein) using a kit according to manufacturer's instructions. Data are expressed as mean ± SEM of PP2A activity from 3 independent experiments. *, P < .05 vs LG. C, INS-1 832/13 cells were treated with LG (2.5mM) and HG (20mM) for 48 hours, after which they were stimulated with either LG or HG for 45 minutes. Insulin released into the medium was quantified using an insulin ELISA kit (15, 16). Data are expressed as fold change over basal and are means ± SEM from 3 independent experiments. *, P < .05 vs LG under 48 hours low-glucose treatment. #, P < .05 vs HG under 48 hours LG glucose treatments. We observed no significant difference in basal insulin secretion from INS-1 832/13 cells between normal vs HG conditions (7.97 ± 0.7 vs 5.63 ± 0.15 ng/mL). However, GSIS was completely abolished (to near basal values) in these cells exposed to glucotoxic conditions (18.29 ± 1.9 vs 6.87 ± 0.44 ng/mL).
HG increases the CML of PP2Ac in INS-1 832/13 cells and normal rodent and human islets
Several earlier studies in multiple cell types, including our own in pancreatic islet β-cells, have demonstrated that PP2Ac undergoes reversible methylation at its C-terminal leucine (Leu-309). In support of this, we have demonstrated expression of PP2Ac methyl transferase and esterase activities in the islet β-cell (3, 4). Furthermore, it has been proposed that the CML step promotes the catalytic activation of PP2A by facilitating the interaction between the structural A subunit, regulatory B subunit, and the catalytic C subunits (3, 4, 6, 7, 22, 23). Therefore, we asked whether activation of PP2A in pancreatic β-cells that we observed under HG conditions (Figure 1) is due to increased CML of PP2Ac. Data shown in Figure 2 demonstrate a significant increase in the CML of PP2Ac under the duress of HG in INS-1 832/13 cells (Figure 2A) and normal rat islets (Figure 2C). Pooled data from multiple experiments along these lines in INS-1 832/13 cells and normal rat islets are provided in Figure 2, B and D, respectively. Furthermore, we observed that the CML of PP2Ac is significantly augmented in human islets under HG conditions (Figure 2E). Availability and requirement of large number of human islets for PP2A activity measurements precluded us from additional investigations in human islets. Stimulatory effects of glucose on the CML of PP2Ac were seen at 10mM and 20mM glucose (2- to 2.3-fold over 2.5mM glucose; 24 hours exposure, n = 2 experiments). No significant differences between the degrees of CML of PP2Ac were demonstrable in INS-1 832/13 cells incubated with 2.5mM vs 5mM glucose (data not shown). Taken together, our findings suggest that HG conditions promote catalytic activation of PP2A in clonal β-cells and primary islet preparations and that such an activation may, in part, be due to increased CML of PP2Ac at Leu-309.
Figure 2.
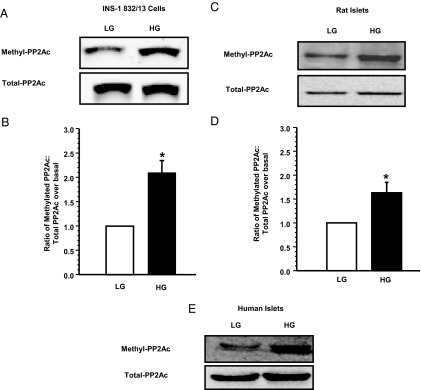
HG increases the CML of PP2Ac in INS-1 832/13 cells and rat and human islets. INS-1 832/13 cells, normal rat islets, or human islets were treated with LG or HG for 48 hours. Relative abundance of total or methylated PP2Ac was determined by Western blotting. Representative blots indicating increase in PP2Ac methylation in INS-1 832/13 cells, rat islets, and human islets are shown in A, C, and E, respectively. The human islet data are accrued using islet preparation from a single donor. Densitometric quantitation of PP2Ac methylation in INS-1 832/13 cells and rat islets is provided in B and D, respectively. These data are mean ± SEM from 3 independent experiments in each cell type. *, P < .05 vs LG.
Time course for HG-induced CML of PP2Ac and PP2A activity
To further test our hypothesis that HG-induced PP2A activation is due to increased CML of PP2Ac, we quantified both of these indices in INS-1 832/13 cells exposed to HG at 6, 12, and 24 hours. First, as shown in Figure 3, we noticed no significant change in the basal CML of PP2Ac at 6 hours of incubation with HG (Figure 3A). However, a marked increase in the CML of PP2Ac was seen after exposure to HG at 12 hours (∼2.54-fold) and 24 hours (∼2.9-fold). PP2A activity was modestly, but significantly, increased (116 ± 2.3% over LG; P < .05, n = 3 determinations) in INS-1 832/13 cells after 12 hours of exposure to HG (data not shown). HG conditions increased PP2A activity more significantly after 24 hours of exposure to glucose (∼2.5-fold; see below for additional data). Furthermore, glucose-induced effects on the CML of PP2Ac are derived from its metabolism, because incubation of INS-1 832/13 cells with glucose, but not 3MG, a metabolically inactive analog of glucose (24, 25), failed to increase the CML of PP2Ac (Figure 3C). In addition to 3MG, equimolar concentrations of mannitol (used as osmotic control) failed to exert any stimulatory effects on the CML of PP2Ac (Figure 3C), further suggesting the specificity of glucose-induced effects on the CML of PP2Ac. Together, these data suggest that HG exposure increases CML of PP2Ac and promotes activation of PP2A in rodent islets and INS-1 832/13 cells. We further validated this model in subsequent experiments.
Figure 3.

HG conditions promote CML of PP2Ac in INS-1 832/13 cells in a time-dependent fashion. INS-1 832/13 cells were treated with LG or HG for 6 hours (HG-6), 12 hours (HG-12), and 24 hours (HG-24) as indicated in the figure. Cell lysates were separated by SDS-PAGE, and relative abundance of methylated PP2Ac and total PP2Ac were determined by Western blotting followed by densitometry. Representative blot indicating a time-dependent effect of glucose on Leu-309 methylation (A) is shown here. Densitometric quantitation of PP2Ac leucine methylation data is provided in B. These data are expressed as mean ± SEM (n = 3). *, P < .05 vs LG. C, INS-1 832/13 cells were incubated with basal glucose (LG) (2.5mM), HG (10mM), 3MG (10mM), or mannitol (M) (10mM) for 24 hours. Relative abundance of methylated PP2Ac and total PP2Ac are quantitated by Western blotting followed by densitometry. These data are expressed as mean ± SEM (n = 3). *, P < .05 vs LG.
siRNA-mediated knockdown of PP2Acα attenuated activation of PP2A under HG conditions in INS-1 832/13 cells
At least 2 variants of PP2Ac, namely PP2Acα and PP2Acβ, have been reported in the literature; and the PP2Acα is known to be the predominant variant (6). Therefore, as a logical extension to the studies described above, and to further assess the contributory roles of PP2Acα in the activation of PP2A under HG treatment conditions (Figure 1), we quantified the PP2A activity in INS-1 832/13 after knockdown of expression of PP2Acα using siRNA-PP2Acα according to the method we described earlier (26). Our findings (Figure 4) demonstrated HG (20mM, 24 hours)-mediated activation of PP2A in INS-1 832/13 cells transfected with the scrambled siRNA (bar 3 vs bar 1). However, transfection of siRNA-PP2Acα in these cells totally abolished the activation of PP2A seen under HG conditions (bar 4 vs bar 3). Together, based on the data accrued thus far, we conclude that glucose-induced CML of PP2Ac is requisite for increased PP2A activity seen under these conditions.
Figure 4.

siRNA-mediated knockdown of PP2Ac alleviates activation of PP2A under HG exposure conditions in INS-1 832/13 cells. INS-1 832/13 cells were transfected with siRNA-PP2Ac (100nM) for 24 hours (22) and subsequently stimulated with LG or HG for 24 hours. PP2A activity was quantitated in cell lysates as described in Materials and Methods. Data are expressed as mean ± SEM from 3 independent experiments. *, P < .05 vs LG in the absence of siRNA, and **, P < .01 vs LG or +, P < .01 vs HG in the absence of siRNA (mock treatment condition).
Expression, subcellular distribution, and functional regulation of LCMT1 under HG conditions
It has been shown in other cell types that LCMT1 mediates CML of PP2Ac (15). However, this has not been studied in insulin-secreting cells thus far. Therefore, we attempted to study the expression and regulation of LCMT1 in pancreatic β-cells. Data depicted in Figure 5A indicate that LCMT1 is expressed in INS-1 832/13 cells, normal rat and human islets. We next isolated individual subcellular fractions (ie, nuclear, mitochondrial, insulin secretory granules, microsomes, and cytosolic fractions) from INS-1 832/13 cells to further determine the subcellular distribution of this enzyme in the β-cell. Data shown in Figure 5B indicated that LCMT1 is predominantly a cytosolic protein, compatible with observations of Longin et al (15).
Figure 5.
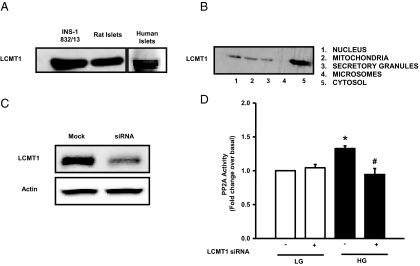
LCMT1 regulates activation of PP2A by HG conditions in INS-1 832/13 cells. Lysate proteins from INS-1832/13 cells, rat islets, and human islets were separated by SDS-PAGE followed by transfer to a nitrocellulose membrane. Representative blot showing the expression of LCMT1 in INS-1 832/13 cells, rat islets, and human islets is provided (A). Subcellular fractions were isolated by the differential centrifugation method (see Materials and Methods). Proteins from these fractions were separated by SDS-PAGE followed by transfer to a nitrocellulose membrane. Relative abundance of LCMT1 in different subcellular fractions was assessed by Western blotting and a representative blot is provided (B). In another set of studies, INS-1 832/13 cells were transfected with siRNA-LCMT1 (80nM) for 48 hours and subsequently stimulated with LG or HG for 24 hours. Extent of knockdown of LCMT1 was determined by Western blotting (C). PP2A activity was quantitated in cell lysates derived from scrambled or siRNA-LCMT1-transfected cells treated with LG and HG as indicated in D. Data are expressed as mean ± SEM from 3 independent experiments. *, P < .05 vs LG in scrambled siRNA, and #, P < .01 vs HG in scrambled siRNA.
We next assessed the role of LCMT1 in glucose-induced activation of PP2A. This was accomplished by selectively knocking down the endogenous expression of LCMT1 in INS-1 832/13 cells followed by quantitation of glucose-induced PP2A activation in cell lysates. We achieved greater than 75% knockdown of LCMT1 under our experimental conditions (Figure 5C). In addition, we observed a complete inhibition of glucose-induced PP2A activity in LCMT1-depleted cells (Figure 5D). Importantly, we also noted a marked increase in the expression of LCMT1 in INS-1 832/13 cells under HG conditions (Figure 6A). Pooled data from multiple studies are provided in Figure 6B. In a manner akin to its effects on CML of PP2Ac (Figure 3A), HG increased the expression of LCMT1 in a time-dependent manner. Significant stimulatory effects of glucose on LCMT1 were demonstrable within 12 hours of exposure to glucose (Figure 6C). Additional studies revealed comparable degrees of stimulation by glucose on LCMT1 expression at both 10mM and 20mM glucose (1.4-fold over basal 2.5mM glucose, 24 hours exposure; n = 2 experiments). Furthermore, no significant differences were noted in the expression levels of LCMT1 in INS-1 832/13 cells incubated at 2.5mM or 5mM glucose (data not shown). Lastly, neither 3MG nor mannitol affected the expression of LCMT1, suggesting that metabolic events of glucose are requisite to promote LCMT1 expression (Figure 6D). Based on these data, we conclude that glucose-induced activation of PP2A in pancreatic β-cells may, in part, be due to increased LCMT1 expression and, consequential, increased LCMT1-mediated CML of PP2Ac.
Figure 6.
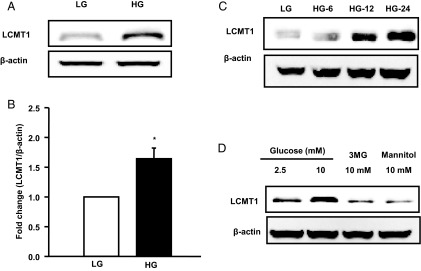
HG conditions increase LCMT1 expression in INS-1 832/13 cells. INS-1 832/13 cells were cultured in the presence of LG (2.5mM) and HG (20mM) for 48 hours, after which expression of LCMT1 was determined in cell lysates by Western blotting. Actin was used as a loading control. Representative blot indicating increased expression of LCMT1 under HG conditions is shown in A. Densitometric quantitation of LCMT1 is provided in B. These data are expressed as mean ± SEM. *, P < .05 vs LG (n = 3 determinations). In another set of experiments (C), INS-1 832/13 cells were treated with either LG or HG for 6 hours (HG-6), 12 hours (HG-12), and 24 hours (HG-24) as indicated in the figure. Lysate proteins were separated by SDS-PAGE and a representative blot from 2 independent studies. These findings indicate a time-dependent effect of glucose on LCMT1. D, INS-1 832/13 cells were incubated with basal glucose (2.5mM), HG (10mM), 3MG (10mM), or mannitol (10mM) for 24 hours. Relative abundance of LCMT1 is quantitated by Western blotting. A representative blot from 3 independent experiments is shown here.
HG-induced activation of PP2A may not be mediated via ER stress
Several lines of evidence implicate that HG effects on the islet β-cell involve alterations in intracellular metabolism, including induction of ER stress (27, 28). Therefore, we next asked the question whether glucose-induced augmentation of PP2Ac CML and catalytic activity are due to increased ER stress caused by HG conditions. To address this, we quantitated the effects of HG conditions on the induction of CHOP protein expression, which is a known marker for ER stress (28, 29). As a positive control, we studied the effects of TG, a known inducer of ER stress. Data in Figure 7 demonstrate a significant increase in CHOP expression in INS-1 832/13 cells exposed to HG (Figure 7A). As expected, we noticed a significant increase in CHOP expression in these cells after exposure to TG (Figure 7B). However, unlike HG, TG failed to increase the expression of LCMT1 and the CML of PP2Ac (Figure 7C) and PP2A activity (Figure 7D). Together, our findings suggest that ER stress-derived signals may not be contributing to the increased CML of PP2Ac and associated activation of PP2A under HG exposure conditions.
Figure 7.

Unlike HG, ER stress conditions fail to increase LCMT1 expression and PP2A activity in INS-1 832/13 cells. INS-1 832/13 cells were treated with LG, HG for 48 hours (A), or TG (0.25μM) for 6 hours (B). CHOP expression was determined in lysates by the Western blotting method. A representative blot from 3 independent studies is shown. Relative abundance of LCMT1 and methylated and total PP2Ac in control and TG-treated cell lysates is shown in C. Data of PP2A activity measurements in control and TG-treated cell lysates are shown in D. Data represented are from 3 independent determinations. NS, not significant.
HG increases the expression of B55α in INS-1 832/13 cells via an ER stress-independent mechanism
It is widely felt that the regulatory B subunits dictate the subcellular targeting and substrate specificity of the PP2A. However, recent evidence implicates additional regulatory roles for these subunits in the function of PP2A. For example, recent studies by Yan et al (30) have demonstrated involvement of a B55α-containing PP2A holoenzyme in the dephosphorylation of Forkhead box protein O1 (FOXO1) in islet β-cells under conditions of H2O2-induced oxidative stress. These investigators have also reported increased expression of B55α subunits in islets derived from db/db mouse (30). The aforementioned observations have prompted us to quantify expression of B55α subunits in INS-1 832/13 cells exposed to HG conditions. Data shown in Figure 8 demonstrate a significant increase in the expression of B55α (∼1.5-fold) (Figure 8, A and B) subunits under these conditions. Further, data in Figure 8C indicated that B55α expression in these cells remained unchanged under conditions of forced induction of ER stress using TG. Together, these data are suggestive of differential effects of HG on PP2A activation, including expression and function of several regulatory proteins involved in this signaling cascade (see Discussion). Based on our observations, we propose a model that links activation of PP2A to loss in GSIS seen in HG exposure conditions (see below).
Figure 8.

HG exposure, but not TG treatment conditions, increases the expression of B55α subunit of PP2A in INS-1 832/13 cells. INS-1 832/13 cells were treated with LG or HG for 48 hours or TG (0.25μM) for 6 hours. Expression levels of B55α was determined by Western blotting followed by densitometry. Representative blot indicating increased expression of B55α under HG is shown in A. Densitometric quantitation of B55α from 3 independent experiments is provided in B. These data are expressed as mean ± SEM. *, P < .05 vs LG. A representative blot to demonstrate lack of effects of TG treatment conditions on the expression of B55α is provided in C.
Discussion
The overall objective of the current study was to examine functional regulation of PP2A in normal rat islets and INS-1 832/13 cells under the duress of HG conditions. Salient findings of this study are: 1) PP2A is activated in normal rat islets and insulin-secreting INS-1 832/13 cells after exposure to HG; 2) HG, but not 3MG or mannitol, significantly stimulated the CML of PP2Ac; 3) siRNA-PP2Ac or siRNA-LCMT1 significantly attenuated glucose-induced activation of PP2A; and 4) HG increased the expression of LCMT1 and B55α subunit of PP2A. Based on these findings, we conclude that HG conditions regulate PP2A activity at multiple levels, including posttranslational CML of PP2Ac.
It is well established that the C-terminal tail of PP2Ac (TPDYFL 304–309) undergoes a variety of posttranslational modifications, including methylation and phosphorylation (4, 7–10, 22, 23). Thr-304 and Tyr-307 undergo phosphorylation, whereas the C-terminal Leu-309 undergoes methylation (4, 9). Unlike the methylation of prenylated cysteine (eg, small G proteins and nuclear lamins), which is mediated by the prenylcysteine methyltransferase (20, 31), the methylation of Leu-309 of PP2Ac is catalyzed by LCMT1 (3, 15). Published evidence implicates functional activation of PP2A after Leu-309 methylation, presumably via increased assembly of the PP2A heterotrimeric holoenzyme. For example, Favre et al (7) have reported that the CML of PP2Ac increases PP2A activity, thereby promoting dephosphorylation of a phosphopeptide and phosphorylase a. Along these lines, we provided additional evidence to indicate that the CML of PP2Ac leads to functional activation of PP2A in pancreatic β-cells (3). Bryant et al (22) suggested that the CML of PP2Ac is critical for binding of regulatory B subunit. Evans and Hemmings (23) further suggested the requisite nature for Leu-309 in binding of regulatory B subunit (PR55) in yeast.
Therefore, it is likely that the ultimate status of PP2A catalytic activation depends on several regulatory events, which include, reversible methylation and phosphorylation. Our current findings demonstrate regulation of Leu-309 methylation under HG conditions. We observed that the CML of PP2Ac and PP2A activity are significantly increased within 12 hours under HG treatment conditions. Existing evidence in other cell types provide evidence in support of activation of PPs (eg, PP2A) in both in vitro and in in vivo models of glucotoxicity, lipotoxicity, and diabetes (21, 32–36). For example, studies by Du et al (21) recently demonstrated novel regulatory roles for activated PP2A in bovine aortic endothelial cells under the duress of HG conditions. They also observed activation of PP2A in retina and aorta isolated from diabetic rats compared with the cognate preparations from the control animals. Rastogi et al (33) reported an increase in PP activity in the hearts of streptozotocin (STZ)-induced diabetic rats 1 week after STZ injection with persistence lasting until 8 weeks and attributed this activity to an increase in the protein levels of both PP1 and PP2A. Li et al (37) noticed a significant reduction in 5′ adenosine monophosphate-activated protein kinase phosphorylation due to activation of PP2A in STZ-induced diabetic mice.
It is noteworthy that emerging evidence suggests key alterations in the expression and activation of PP2A in human diabetes. For example, Højlund et al (36) have examined regulatory effects of insulin on PP2A expression in muscle cells from type diabetes mellitus (DM) humans and their age-matched control subjects. Their findings suggested a significant reduction in the expression of PP2Acα after insulin treatment in skeletal muscle from control subjects; these data correlated well with insulin-mediated glucose disposal, glucose oxidation, and decrease in lipid oxidation. Interestingly, however, insulin-mediated effects on these metabolic indices were not seen in muscle preparations from type 2DM subjects. These investigators postulated that impaired down-regulation of PP2Acα expression by insulin might serve as a biomarker for insulin resistance and could contribute to the onset of type 2DM. Along these lines, we demonstrated in the current study that incubation of islets from normal human donors with HG markedly stimulated the CML of PP2Ac.
On a mechanistic note, our data rule out the involvement of ER stress in glucose-induced effects on PP2A. These conclusions were drawn based on our findings that although TG and HG induced CHOP expression in INS-1 832/13 cells, HG, but not TG, elicited effects on the expression of LCMT1 and B55α, PP2Ac methylation, and PP2A activity. One potential caveat here is that mechanisms underlying TG-induced ER stress may be different from the ER stress induced by misfolded proteins under the duress of HG, lipids, and nitric oxide (38–41). Additional studies are needed to further validate this model. However, it is possible that increase in oxidative stress under glucotoxic conditions promotes downstream signaling pathways to regulate PP2A function, including the expression of B55α subunit (Figure 8). Indeed, recent observations from our laboratory have demonstrated significant increase in phagocyte-like NADPH oxidase-derived reactive oxygen species generation in β-cell models of gluco-, lipotoxicity, exposure to proinflammatory cytokines, and diabetes (42–46). Our current findings gain additional support from recent studies by Yan et al (30), who demonstrated that a B55α-containing PP2A holoenzyme is involved in the dephosphorylation of FOXO1 in clonal β-[INS-1 and βTC-3] cells exposed to H2O2 (30). Interestingly, they demonstrated a significant increase in the expression of B55α and nuclear translocation of FOXO1 in H2O2-treated cells as well as in islets derived from the db/db mice, a model for type 2 diabetes. Based on these findings, they concluded that PP2A is a key regulator of FOXO1 activity in vivo. Increased dephosphorylation and nuclear translocation of FOXO1 in db/db islets support our current observations of increased PP2A activity under HG conditions. Given our current findings indicating a significant increase in the expression of B55α under glucotoxic conditions, it remains to be seen whether HG-induced PP2A activation leads to dephosphorylation and nuclear association of FOXO1.
It is noteworthy that increase in LCMT1 expression and the CML of PP2Ac were demonstrable in INS-1 832/13 cells incubated in the presence of 10mM glucose for 24 hours. These data are compatible with observations of Olson et al (12), who demonstrated significant inhibition of insulin mRNA levels in INS-1 cells by 8mM glucose within 24 hours of exposure. Interestingly, these investigators reported reversal of inhibitory effects of glucose after incubation with 4mM glucose. In this context, at least based on our recent findings, we speculate that the effects of glucose on LCMT1/PP2Ac/PP2A signaling axis under these conditions may not be reversible. Along these lines, we have recently reported significant activation of caspase-3 and subsequent degradation of nuclear lamin-B in INS-1 832/13 cells and normal rodent islets within 12 hours of exposure to HG; these findings are indicative of irreversible structural damage at the levels of mitochondria and nucleus (47). However, additional studies are needed to conclusively determine whether glucose-induced effects on LCMT1 expression, the CML of PP2Ac, and the catalytic activation of PP2A are reversible.
In the context of glucose-induced activation of PPs, Castermans et al (48) recently demonstrated posttranslational activation of PP2A and PP1 in yeast by glucose. Using a variety of complementary experimental conditions, they noted rapid CML of PP2Ac after exposure to glucose. Interestingly, however, based on additional deletion analyses, they concluded that the CML of PP2Ac is necessary, but not sufficient, for full glucose-induced activation of PP2A, thus raising the possibility that additional regulatory mechanisms are needed for PP2A activation under these conditions. In further support of this hypothesis, these researchers presented evidence to suggest that glucose-induced PP2A activation also involves the cAMP-protein kinase A signaling cascade. Due to the scope of the current studies, we have not looked into potential regulation by phosphorylation in the cascade of events leading to glucose-mediated activation of PP2A in pancreatic β-cells. As reviewed by us recently (4), the PP2A family of enzymes are subject to multifactorial regulation and participate in a variety of islet β-cell under acute or chronic exposure to glucose.
Lastly, it is likely that in addition to increasing the expression of LCMT1 and CML of PP2Ac, HG conditions might exert direct effects of the de-esterification of the methylated PP2Ac. In this context, we published the first evidence to suggest that PP2Ac undergoes methylation-demethylation cycles in clonal β-cells, normal rodent, and human islets (3). We have characterized a demethylating (esterase) activity, and demonstrated its role in GSIS. Using ebelactone, a selective inhibitor of PP2Ac esterase, we have demonstrated significant inhibition of GSIS and KCl-induced insulin secretion from normal rat islets, suggesting a critical requirement for PP2Ac methylation-demethylation in insulin secretion under acute conditions. Furthermore, the effects of ebelactone were rapidly reversible, because removal of this inhibitor from the medium reversed its inhibitory effects on insulin secretion (3). Therefore, it is likely that glucotoxic conditions promote inactivation of PP2Ac methyl-esterase, thereby retaining the PP2A in its methylated active state. Future studies will determine potential temporal relationships between posttranslational methylation-demethylation and phosphorylation-dephosphorylation of PP2Ac to further evaluate their roles in the activation of PP2A under glucotoxic conditions.
Based on our current findings, we propose that exposure of pancreatic β-cells to HG leads to increased expression of LCMT1 and the resultant increase in the CML of PP2Ac. Several previous have demonstrated that the CML of PP2Ac leads to increased affinity/association between the structural, regulatory, and catalytic subunits (ie, increased assembly of PP2A holoenzyme), culminating in the functional activation of PP2A (4, 6, 32). In addition to the CML of PP2Ac, our current observations revealed that HG induces increased expression of B55α, a regulatory subunit of PP2A, which has been implicated in the activation of a PP2A holoenzyme involved in the dephosphorylation and inactivation of key proteins (FOXO1) (30) necessary for islet function. Studies are in progress to determine the identity of potential substrate proteins (eg, FOXO1, Akt, and Bcl2) for the activated PP2A, dephosphorylation of which could contribute to metabolic dysregulation, loss in GSIS and eventual demise of the β-cell.
Acknowledgments
We thank Mr Giridhar Jangati and Dr Chandrashekara Kyathanahalli for excellent technical assistance in these studies. We also thank Professor Chris Newgard for providing INS-1 832/13 cells.
This work was supported in part by the Department of Veterans Affairs, Office of Research and Development (Biomedical Laboratory Research and Development) Merit Review Award 1BX000469 (to A.K.) and by National Institutes of Health Grants RO1 DK74921 (to A.K.), DK69455 (to S.R.), and GM051366 (to B.E.W.). A.K. is also the recipient of the Department of Veterans Affairs Senior Research Career Scientist Award 13S-RCS-006. A.M. was supported by a postdoctoral fellowship from Wayne State University. B.M. was supported by the T32 Training Program in Endocrine and Diabetes Research Fellowship DK080657.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CHOP
- C/EBP homology protein
- CML
- carboxymethylation
- DM
- diabetes mellitus
- ER
- endoplasmic reticulum
- FOXO1
- Forkhead box protein O1
- GSIS
- glucose-induced insulin secretion
- HG
- hyperglycemia
- LCMT1
- leucine carboxymethyl transferase 1
- LG
- low glucose
- 3MG
- 3-O-methyl glucose
- PP
- protein phosphatase
- PP2Ac
- catalytic subunit of PP2A
- siRNA
- small interfering RNA
- STZ
- streptozotocin
- TG
- thapsigargin.
References
- 1. Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1185–1248 [DOI] [PubMed] [Google Scholar]
- 2. Jones PM, Persaud SJ. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic β-cells. Endocr Rev. 1998;19:429–461 [DOI] [PubMed] [Google Scholar]
- 3. Kowluru A, Seavey SE, Rabaglia ME, Nesher R, Metz SA. Carboxylmethylation of the catalytic subunit of protein phosphatase 2A in insulin-secreting cells: evidence for functional consequences on enzyme activity and insulin secretion. Endocrinology. 1996;137:2315–2323 [DOI] [PubMed] [Google Scholar]
- 4. Kowluru A. Novel regulatory roles for protein phosphatase-2A in the islet β cell. Biochem Pharmacol. 2005;69:1681–1691 [DOI] [PubMed] [Google Scholar]
- 5. Leulliot N, Quevillon-Cheruel S, Sorel I, et al. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J Biol Chem. 2004;279:8351–8358 [DOI] [PubMed] [Google Scholar]
- 6. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Favre B, Zolnierowicz S, Turowski P, Hemmings BA. The catalytic subunit of protein phosphatase 2A is carboxyl-methylated in vivo. J Biol Chem. 1994;269:16311–16317 [PubMed] [Google Scholar]
- 8. Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2013;280:644–661 [DOI] [PubMed] [Google Scholar]
- 9. Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264 [DOI] [PubMed] [Google Scholar]
- 10. Begum N, Ragolia L. cAMP counter-regulates insulin-mediated protein phosphatase-2A inactivation in rat skeletal muscle cells. J Biol Chem. 1996;271:31166–31171 [DOI] [PubMed] [Google Scholar]
- 11. Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev. 2008;29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olson LK, Qian J, Poitout V. Glucose rapidly and reversibly decreases INS-1 cell gene transcription via decrements in STF-1 and C1 activator transcription factor activity. Mol Endocrinol. 1998;12:207–219 [DOI] [PubMed] [Google Scholar]
- 13. Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442 [DOI] [PubMed] [Google Scholar]
- 14. Fonseca SG, Urano F, Burcin M, Gromada J. Stress hyperactivation in the β-cell. Islets. 2010;2:1–9 [DOI] [PubMed] [Google Scholar]
- 15. Longin S, Zwaenepoel K, Martens E, et al. Spatial control of protein phosphatase 2A (de)methylation. Exp Cell Res. 2008;314:68–81 [DOI] [PubMed] [Google Scholar]
- 16. Kowluru A, Veluthakal R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes. 2005;54:3523–3529 [DOI] [PubMed] [Google Scholar]
- 17. Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic β-cells. Diabetes. 2010;59:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kowluru A, Metz SA. Stimulation by PGE2 of a high-affinity GTPase in the secretory granules of normal rat and human pancreatic islets. Biochem J. 1994;297:399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kowluru A, Rabaglia ME, Muse KE, Metz SA. Subcellular localization and kinetic characterization of guanine nucleotide binding proteins in normal rat and human pancreatic islets and transformed β-cells. Biochim Biophys Acta. 1994;1222:348–359 [DOI] [PubMed] [Google Scholar]
- 20. Li G, Kowluru A, Metz SA. Characterization of prenylcysteine methyltransferase in insulin-secreting cells. Biochem J. 1996;316:345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du Y, Kowluru A, Kern TS. PP2A contributes to endothelial death in high glucose: inhibition by benfotiamine. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1610–R1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bryant JC, Westphal RS, Wadzinski BE. Methylated C-terminal leucine residue of PP2A catalytic subunit is important for binding of regulatory Bα subunit. Biochem J. 1999;339:241–246 [PMC free article] [PubMed] [Google Scholar]
- 23. Evans DR, Hemmings BA. Mutation of the C-terminal leucine residue of PP2Ac inhibits PR55/B subunit binding and confers supersensitivity to microtubule destabilization in saccharomyces cerevisiae. Mol Gen Genet. 2000;264:425–432 [DOI] [PubMed] [Google Scholar]
- 24. Reddy AB, Ramana KV, Srivastava S, Bhatnagar A, Srivastava SK. Aldose reductase regulates high glucose-induced ectodomain shedding of tumor necrosis factor (TNF)-α via protein kinase c-δ and TNF-α converting enzyme in vascular smooth muscle cells. Endocrinology. 2009;150:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kowluru A, Seavey SE, Li G, et al. Glucose- and GTP-dependent stimulation of the carboxyl methylation of Cdc42 in rodent and human pancreatic islets and pure β cell. J Clin Invest. 1996;98:540–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jangati GR, Veluthakal R, Kowluru A. siRNA-mediated depletion of endogenous protein phosphatase 2Acα markedly attenuates ceramide-activated protein phosphatase activity in insulin-secreting INS-832/13 cells. Biochem Biophys Res Commun. 2006;348:649–652 [DOI] [PubMed] [Google Scholar]
- 27. Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61 [DOI] [PubMed] [Google Scholar]
- 30. Yan L, Guo S, Brault M, et al. The B55α-containing PP2A holoenzyme dephosphorylates FOXO1 in islet β-cells under oxidative stress. Biochem J. 2012;444:239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kowluru A. Small G proteins in islet β-cell function. Endocr Rev. 2010;31:52–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol. 2012;84:591–597 [DOI] [PubMed] [Google Scholar]
- 33. Rastogi S, Sentex E, Elimban V, Dhalla NS, Netticadan T. Elevated levels of protein phosphatase 1 and phosphatase 2A may contribute to cardiac dysfunction in diabetes. Biochem Biophys Acta. 2003;1638:273–277 [DOI] [PubMed] [Google Scholar]
- 34. Galbo T, Olsen GS, Quistorff B, Nishimura E. Free fatty acid-induced PP2A hyperactivity selectively impairs hepatic insulin action on glucose metabolism. PLoS One. 2011;6:e27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788 [DOI] [PubMed] [Google Scholar]
- 36. Højlund K, Poulsen M, Staehr P, Brusgaard K, Beck-Nielsen H. Effect of insulin on protein phosphatase 2A expression in muscle in type 2 diabetes. Eur J Clin Invest. 2002;32:918–923 [DOI] [PubMed] [Google Scholar]
- 37. Li Q, Li J, Ren J. UCF-101 mitigates streptozotocin-induced cardiomyocyte dysfunction: role of AMPK. Am J Physiol Endocrinol Metab. 2009;297:E965–E973 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Intern Med. 2003;42:7–14 [DOI] [PubMed] [Google Scholar]
- 39. Meares GP, Hughes KJ, Naatz A, et al. IRE1-dependent activation of AMPK in response to nitric oxide. Mol Cell Biol. 2011;31:4286–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baldwin AC, Green CD, Olson LK, Moxley MA, Corbett JA. A role for aberrant protein palmitoylation in FFA-induced ER stress and β-cell death. Am J Physiol Endocrinol Metab. 2012;302:E1390–E1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou ZQ, Li HL, Gao L, Pan L, Zhao JJ, Li GW. Involvement of chronic stresses in rat islets and INS-1 cell glucotoxicity induced by intermittent high glucose. Mol Cell Endocrinol. 2008;291:71–78 [DOI] [PubMed] [Google Scholar]
- 42. Syed I, Jayaram B, Subasinghe W, Kowluru A. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and loss of mitochondrial membrane potential in pancreatic β-cells. Biochem Pharmacol. 2010;80:874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol. 2011;300:R12–R20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Syed I, Kyathanahalli CN, Jayaram B, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60:2843–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mohammed AM, Syeda K, Hadden T, Kowluru A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic β-cells: attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol. 2013;85:109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Syeda K, Mohammed AM, Arora DK, Kowluru A. Glucotoxic conditions induce endoplasmic stress to cause caspase 3 mediated lamin B degradation in pancreatic β-cells: protection by nifedipine. Biochem Pharmacol. 2013;86:1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Castermans D, Somers I, Kriel J, et al. Glucose-induced posttranslational activation of protein phosphatases PP2A and PP1 in yeast. Cell Res. 2012;22:1058–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]