

Abstract
Hippocampal network hyperexcitability is considered an early indicator of Alzheimer's disease (AD) memory impairment. Some AD mouse models exhibit similar network phenotypes. In this study we focused on dentate gyrus (DG) granule cell spontaneous and evoked properties in 9-month-old Tg2576 mice that model AD amyloidosis and cognitive deficits. Using whole-cell patch-clamp recordings, we found that Tg2576 DG granule cells exhibited spontaneous EPSCs that were higher in frequency but not amplitude compared with wild-type mice, suggesting hyperactivity of DG granule cells via a presynaptic mechanism. Further support of a presynaptic mechanism was revealed by increased I–O relationships and probability of release in Tg2576 DG granule cells.
Since we and others have shown that activation of the peroxisome proliferator-activated receptor gamma (PPARγ) axis improves hippocampal cognition in mouse models for AD as well as benefitting memory performance in some humans with early AD, we investigated how PPARγ agonism affected synaptic activity in Tg2576 DG. We found that PPARγ agonism normalized the I–O relationship of evoked EPSCs, frequency of spontaneous EPSCs, and probability of release that, in turn, correlated with selective expression of DG proteins essential for presynaptic SNARE function that are altered in patients with AD. These findings provide evidence that DG principal cells may contribute to early AD hippocampal network hyperexcitability via a presynaptic mechanism, and that hippocampal cognitive enhancement via PPARγ activation occurs through regulation of presynaptic vesicular proteins critical for proper glutamatergic neurotransmitter release, synaptic transmission, and short-term plasticity.
Keywords: Alzheimer's, animal model, mass spectrometry, patch clamp, SNARE proteins, synaptic plasticity
Introduction
Hippocampus-dependent learning and memory deficits are quintessential features of Alzheimer's disease (AD; Schiöth et al., 2012) that are evident in animal models of the disease including the Tg2576 mouse model of AD where overexpression of mutant amyloid precursor protein (APP) leads to β amyloid (Aβ) accumulation, synaptic damage, and hippocampus-dependent cognitive deficits (Dineley et al., 2002; Dineley et al., 2007). Tg2576 mice also exhibit a recently recognized feature of mild cognitive impairment (MCI) and early AD: dysregulated insulin signaling (Abramov and Duchen, 2010; Rodriguez-Rivera et al., 2011; Agrawal and Gomez-Pinilla, 2012; De Felice, 2013). Peripheral insulin resistance is correlated with cognitive deficits in human AD (Talbot et al., 2012), a phenotype reflected in the Tg2576 model that also manifests as dysregulated CNS insulin signaling (Rodriguez-Rivera et al., 2011; Denner et al., 2012).
A key mediator of insulin sensitivity is the nuclear transcription factor peroxisome proliferator-activated receptor gamma (PPARγ). Because of the emergent shared comorbidity of insulin resistance in AD and diabetes (Biessels et al., 2006), we and others have proposed that PPARγ is an opportunistic therapeutic target in MCI/AD patients with concomitant insulin dysregulation (Geldmacher et al., 2011; Sato et al., 2011). Indeed, PPARγ agonism with drugs such as rosiglitazone (RSG) improved cognition in some early AD patients and in several animal models of AD (Hamann et al., 2002; Pedersen et al., 2006; Hort et al., 2007; Hoefer et al., 2008; Escribano et al., 2010; Rodriguez-Rivera et al., 2011). Our previous findings showed that RSG enhanced hippocampus-dependent cognition in the Tg2576 mouse model through convergence of the PPARγ and ERK MAPK signaling pathways in the dentate gyrus (DG) (Denner et al., 2012). In the present study we investigated DG synaptic mechanisms in Tg2576 treated with or without the PPARγ agonist RSG to determine how these pathways that enhance cognition might also influence synaptic transmission.
Previous work suggested that mouse models for AD-like amyloidosis exhibit hippocampal network hyperexcitability similar to what is now considered an indicator of early AD memory impairment (Palop et al., 2007; Putcha et al., 2011; Bejanin et al., 2012). In the present study we investigated spontaneous and evoked EPSCs (sEPSCs; eEPSCs) in 9-month-old (9MO) Tg2576 DG, and whether these were affected by RSG. Because we found increased excitatory synaptic transmission in 9MO Tg2576 DG granule cells that reflected augmented probability of neurotransmitter release (Pr), which was normalized with RSG, we used mass spectrometry and bioinformatics approaches to focus on proteins important for synaptic vesicle function that were affected during cognitive enhancement following PPARγ activation. We found RSG induced expression of many proteins previously reported to be affected in the hippocampus of AD patients whose dysregulation correlated with poor cognitive performance and diminished cognitive reserve (Tannenberg et al., 2006; Honer et al., 2012). These findings indicate that DG principal cells may contribute to early AD hippocampal network hyperexcitability via presynaptic hyperactivity mechanisms that are normalized during cognitive enhancement following activation of PPARγ by presynaptic vesicular proteins critical for proper glutamatergic neurotransmitter release, synaptic transmission, and short-term plasticity.
Materials and Methods
Animals.
Animals were bred in the University of Texas Medical Branch (UTMB) animal care facility by mating heterozygous Tg2576 males with C57BL/6SJL (F1) females (The Jackson Laboratory). The UTMB operates in compliance with the United States Department of Agriculture Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and Institutional Animal Care and Use Committee-approved protocols. Mice were housed, n ≤ 5 per cage, with food and water ad libitum. All animal manipulations were conducted during the lights-on phase (0700–1900 h). Male and female 8MO Tg2576 and wild-type (WT) littermates were fed control or 30 mg/kg RSG diet (Bio-Serv) for 30 d, as previously described (Rodriguez-Rivera et al., 2011). Animals were killed by decapitation and the brain rapidly removed from the skull for hippocampus dissection.
Slice preparation.
Acute hippocampal slices were prepared from 9MO WT littermate control and Tg2576 mice treated or untreated with RSG. Animals were anesthetized with 2-2-2-tribromoethanol (Avertin; Sigma) then intracardiac perfusion was performed with sucrose-based artificial CSF (sucrose-based ACSF) consisting of the following (in mm): 56 NaCl, 100 Sucrose, 2.5 KCl, 20 glucose, 5 MgCl2, 1 CaCl2, 30 NaHCO3, and 1.25 NaH2PO4, osmolarity 300–310, pH 7.4. Brains were dissected and 250 μm horizontal hippocampal slices prepared with a Leica Vibratome 1200S (Leica) in iced sucrose-based ACSF continuously oxygenized and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2. Slices were then transferred to an incubation chamber with standard ACSF consisting of the following (in mm): 130 NaCl, 3.5 KCl, 10 glucose, 1.5 MgCl2, 1.4 CaCl2, 23 NaHCO3, and 1.25 NaH2PO4, osmolarity 300–310, oxygenated and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2 at 31°C. After 1–2 h of recovery, brain slices were placed in a submerged recording chamber on the stage of an upright microscope (Axioskop2 FS plus; Zeiss). Slices were continuously perfused at room temperature with standard ACSF (∼2 ml/min). All recordings were performed in the presence of 20 μm bicuculline to block GABAergic synaptic transmission.
Patch-clamp recording and data analysis.
Whole-cell patch-clamp recordings were obtained from visually identified DG granule cells using infrared DIC-IR optics. Recording pipettes (4–7 MΩ tip resistance) were fabricated from borosilicate glass (WPI) using a two-step vertical puller PP-83 (Narishige), and filled with intracellular solution containing the following (in mm): 120 CH3KO3S, 10 KCl, 10 HEPES, 10 glucose, 2 MgCl2, 0.5 EGTA, 2 MgATP, and 0.5 Na3GTP, osmolarity 280–290, pH 7.3, adjusted with KOH. Whole-cell somatic recordings were performed using an Axopatch 200A amplifier (Molecular Devices), low-pass filtered at 5 kHz, and sampled at 10–20 kHz using a Digidata 1200 analog-to-digital interface and pClamp7 acquisition software (Molecular Devices). After seal formation and membrane rupture, sEPSCs and eEPSCs (elicited either by paired pulse or 20 Hz stimulation) were recorded at a holding potential of −70 and −80 mV, respectively. sEPSCs and eEPSCs for each cell were analyzed using MiniAnalysis 6.0 (Synaptosoft) and Clampfit 9, respectively. All spontaneous events were automatically detected and visually inspected with the threshold amplitude for the detection of an event adjusted to 5 pA (≥2 SD above noise level).
eEPSCs were elicited by paired-pulse stimulation of variable current step intensities (25 μA increments) with 50 ms interpulse interval using an isolated pulse stimulator MODEL 2100 (A-M Systems) and a monopolar tungsten electrode (FHC) positioned in the middle third of the molecular layer to activate the medial perforate path. Minimal stimulus intensity was defined as the amount of current required to induce at least one synaptic response (eEPSC). I–O relationships (average of four EPSC traces at each given stimulus) were individually analyzed for each pulse from paired-pulse stimulation. Paired-pulse ratios (PPR) were calculated using paired-pulse stimulation at a fixed current (175 μA) with 10–15 traces to reduce variability of EPSCs. To estimate the size of the readily releasable pool (RRP) and the probability of release, a 1.6 s, 20 Hz stimulation was used to evoke a series of EPSCs and cumulative charge was calculated. Unpaired and paired Student's t tests were used to compare two sets of data. One-way ANOVA with either Bonferroni or Dunnett's post hoc analysis and Kruskal–Wallis one-way ANOVA with Dunn's post hoc were used for multiple comparisons.
Quantitative mass spectrometry.
Stable isotope labeling was used to quantify differential protein expression as previously described (Sadygov et al., 2010; Wu et al., 2013). Briefly, the DGs from 10 mice each of Tg2576 fed control or RSG diet were homogenized in TRIzol (Life Technologies) and the protein pellet resuspended in guanidine. Following reduction and alkylation, proteins were digested with trypsin and peptides desalted with SepPack C18 cartridges. Dried peptides were then treated with immobilized trypsin (Applied Biosystems) in normal water (H216O) or heavy water (H218O) for trypsin-mediated exchange of oxygen atoms from water onto the C terminus of peptides. Desalted peptides were then pooled to prepare a mixture of 16O-labeled peptides from control-fed mice and 18O-labeled peptides from RSG-fed mice. To reduce the sample complexity and increase the depth of analysis into the proteome, the peptide mixture was resolved into 60 fractions using strong cation exchange chromatography.
Two-dimensional liquid chromatography-tandem mass spectrometry.
Each SCX fraction was injected onto a C18 peptide trap (Agilent) and desalted, and eluted peptides were separated on a reversed phase nano-HPLC column with a linear gradient over 120 min at 200 nl/min. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments were performed with an LTQ linear ion trap MS (Thermo Finnigan) equipped with a nanospray source. The mass spectrometer was coupled on-line to a ProteomX nano-HPLC system (Thermo Finnigan). The mass spectrometer was operated in the data-dependent triple-play mode. In this mode, the three most intense ions in each MS survey scan were automatically selected for moderate resolution zoom scans, which were followed by MS/MS. Each of the peptide mixtures was repetitively analyzed by nano-HPLC-MS/MS three times. The acquired MS/MS spectra were searched with SEQUEST algorithm performed on the BioWorks 3.2 platform (Thermo Finnigan) using conservative filtering criteria of Sp ≥ 300, ΔCn ≥ 0.12, and Xcorr of 1.9, 2.0, and 3.0 for data from a singly, doubly, or triply charged precursor ions, respectively.
The zoom scan data were used to calculate the relative abundance ratios of 18O-labeled peptide/16O-unlabeled peptide pairs using MassXplorer (Sadygov et al., 2010). Peptides with charge > 3, false discovery rate > 3%, 18O/16O ratios < 0.1 or > 10, and reversed sequences were removed from further analysis. Calculated peptide ratios were log2 transformed and mean centered before statistical analysis. Significance was determined using the Wilcoxon rank-sum test with Benjamini–Hochberg false discovery rate correction for multiple testing comparisons as indicated (Benjamini and Hochberg, 1995).
Bioinformatics.
Data were analyzed through the use of the extensively curated Ingenuity Pathways Analysis (IPA; Ingenuity Systems) with a significance cutoff of p ≤ 0.05 and ≥20% change in protein expression. Network analysis generates a graphical representation of the molecular relationships between molecules. Molecules are represented as nodes, and the biological relationship between two nodes is represented as a line. All lines are supported by at least one reference from the literature, from a textbook, or from canonical information stored in the Ingenuity Knowledge Base. PPREs and CREs were from DECODE (http://www.sabiosciences.com) and the Montminy Lab CREB Target Gene Database at https://www.salk.edu/labs/pbl-m/contactus.php (Zhang et al., 2005).
Results
We previously demonstrated enhanced hippocampus-dependent cognitive function in 9MO Tg2576 AD mice following treatment with the PPARγ agonist RSG (Rodriguez-Rivera et al., 2011). RSG cognitive enhancement was mediated by CNS PPARγ and led to convergence of the hippocampal PPARγ and pERK pathways at the level of both the transcriptome and proteome (Denner et al., 2012). To investigate whether, along with compromised hippocampal cognitive function, Tg2576 also exhibit altered synaptic transmission and short-term plasticity, we performed whole-cell patch-clamp electrophysiology on DG granule cells in acute hippocampal slices prepared from 9MO WT and Tg2576 littermates treated with or without RSG for 1 month. To unmask Aβ-mediated impairment in synaptic transmission, all recordings were performed in 1.4 mm external Ca2+ to match physiological CSF calcium concentration in aging animals (Jones and Keep, 1988) and favor low release probability (Abramov and Duchen, 2010; Houeland et al., 2010). We found that sEPSCs in WT DG granule cells were rather infrequent and significantly fewer in number when compared with Tg2576 (Fig. 1A,B). RSG-treated Tg2576 did not differ from WT, suggesting that RSG treatment in vivo reversed the Tg2576 increased spontaneous synaptic activity. Amplitudes of sEPSCs were unaffected by genotype or treatment (Fig. 1A,C). Changes in sEPSCs frequency were further illustrated by cumulative probability plots showing that Tg2576 exhibit significantly higher sEPSC probability at short interevent intervals compared with either WT or Tg2576 treated with RSG (Fig. 1D), while WT and Tg2576 treated with RSG did not differ. Cumulative probability plots of sEPSC amplitudes did not differ among the groups (Fig. 1E). One-way ANOVA revealed there were no differences in sEPSC kinetics for rise time (Fig. 1F) or decay time (Fig. 1G). Altered sEPSC frequency, but not amplitude or kinetics, suggests that impaired hippocampal cognition in Tg2576 mice is accompanied by DG synaptic hyperactivity through a presynaptic mechanism that is normalized following activation of PPARγ.
Figure 1.
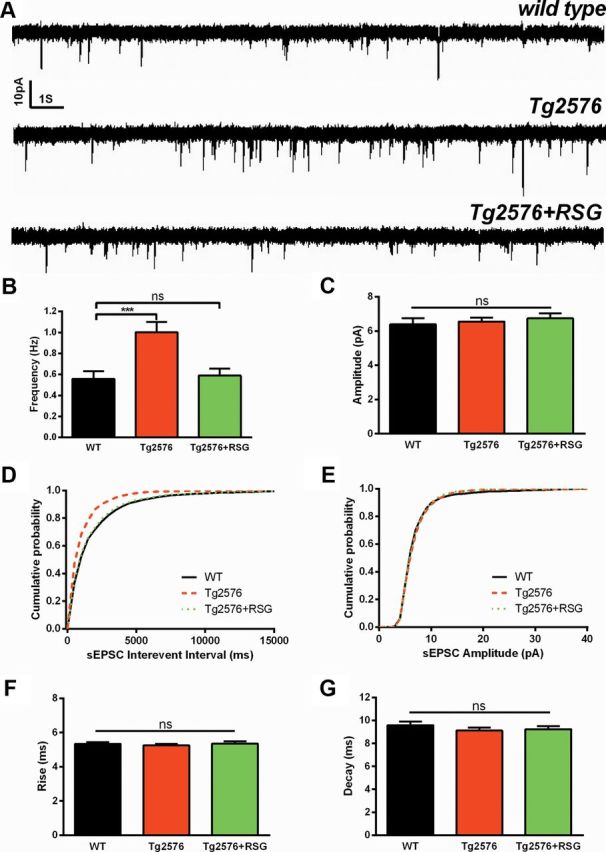
PPARγ agonism normalizes increased spontaneous synaptic activity in 9MO Tg2576 DG. A, Representative sEPSCs recorded from dentate granule cells in hippocampal slices from WT, Tg2576, and RSG-treated Tg2576. B, Frequency of sEPSCs was elevated in Tg2576 DG granule cells compared with WT (p < 0.001, one-way ANOVA with Bonferroni post hoc test), which did not differ from Tg2576 treated with RSG (p > 0.05, one-way ANOVA with Bonferroni post hoc test). C, There were no differences in sEPSCs amplitude among groups (p = 0.7, one-way ANOVA with Bonferroni post hoc test). D, The cumulative probability distribution of frequency was increased at short interevent intervals in Tg2576 compared with WT or to Tg2576 treated with RSG (p < 0.001; Kolmogorov–Smirnov test) while WT did not differ from Tg2576 treated with RSG (p = 0.36). E, Cumulative probability distribution of amplitude did not differ among groups (p > 0.1, Kolmogorov–Smirnov test). F, G, Kinetics of rise time (F(2,57) = 0.28, p = 0.76; F) and decay time (F(2,57) = 0.7, p = 0.5; G) did not differ among groups (p > 0.05, one-way ANOVA with Bonferroni post hoc test). Results represent mean ± SEM. Calibration: 10 pA, 1 s. n = 19 (WT), 19 (Tg2576), and 22 (Tg2576 + RSG). *p < 0.05, **p < 0.01, ***p < 0.001.
To further characterize synaptic transmission in Tg2576 and the beneficial activation of PPARγ, we examined short-term synaptic plasticity by stimulating the medial perforant path (mPP) and performing patch-clamp recordings from DG granule cells. Using variable current step intensities in a paired-pulse stimulation paradigm with 50 ms interpulse intervals, we analyzed the eEPSC I–O relationship (Fig. 2A1–A3). One-way ANOVA revealed that the responses to the first (Fig. 2B) and second (Fig. 2C) pulses at each input current intensity were significantly elevated in Tg2576 compared with WT, which did not differ from RSG-treated Tg2576. Analysis of I–O relationships separately for pulse1 and pulse2, showed that RSG reduced the amplitude of pulse2 EPSCs more than pulse1 EPSCs (Fig. 2D), suggesting that activation of PPARγ may have affected presynaptic neurotransmitter vesicle release machinery in Tg2576 DG (Fioravante and Regehr, 2011). Moreover, minimal stimulus intensity required to induce at least one synaptic response was significantly lower in Tg2576 mice compared with WT or RSG-treated Tg2576 (Fig. 2E), suggesting that afferent synaptic inputs from mPP to DG granule cells are hyperactive in Tg2576 mice. Finally, paired-pulse facilitation (PPF), estimated by analyzing PPR of eEPSCs, was significantly reduced in Tg2576 compared with WT, which can be associated with increased Pr; RSG-treated Tg2576 did not differ from WT (Fig. 2F). Together, these results are consistent with the interpretation that Tg2576 exhibit aberrant synaptic transmission and short-term plasticity mediated by a presynaptic mechanism.
Figure 2.
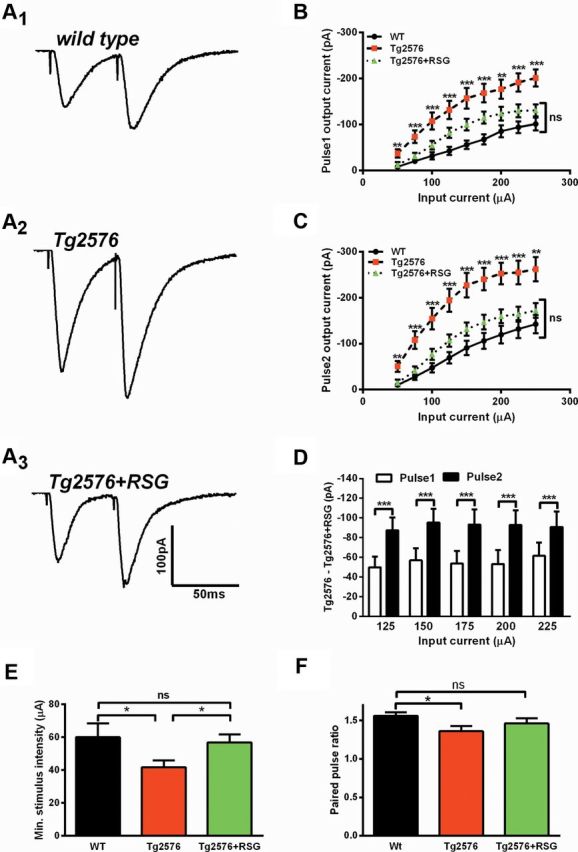
PPARγ agonism normalizes short-term plasticity and elevated probability of neurotransmitter release in 9MO Tg2576 DG. A, Representative traces of dentate granule cell eEPSCs using a paired-pulse stimulation paradigm by stimulating the mPP in hippocampal slices from WT (A1), Tg2576 (A2), and RSG-treated Tg2576 (A3). B, C, Synaptic I–O curves representing pulse1 (B) and pulse2 (C) show that eEPSCs were elevated in Tg2576 compared with WT (p < 0.01 or p < 0.001, one-way ANOVA with Bonferroni post hoc test), which did not differ from Tg2576 treated with RSG (p > 0.05, one-way ANOVA with Bonferroni post hoc test). D, RSG reduced the response to pulse2 (filled bars) more than pulse1 (open bars). For each input current, the output current from RSG-treated Tg2576 was subtracted from Tg2576 (p < 0.001, paired Student's t test). E, Minimal stimulus intensity was lower for Tg2576 than WT or RSG-treated Tg2576 (p < 0.05 unpaired Student's t test). WT did not differ from RSG-treated Tg2576 (p = 0.37, unpaired Student's t test). F, The ratio of pulse2 to pulse1 was reduced in Tg2576 compared with WT (p < 0.05, Kruskal–Wallis test with Dunn's post hoc test), which did not differ from RSG-treated Tg2576 (p > 0.05, Kruskal–Wallis test with Dunn's post hoc test). Results represent mean ± SEM, n = 10 (WT), 9 (Tg2576), and 11 (Tg2576 + RSG) for B–E and n = 15 (WT), 12 (Tg2576), and 13 (Tg2576 + RSG) for F. Calibration; 100 pA, 50 ms; *p < 0.05, **p < 0.01, ***p < 0.001.
Aberrant presynaptic hyperactivity can manifest via changes in the Pr or the RRP. While we found changes in Tg2576 PPF, which is frequently used to estimate Pr, this is an imprecise method due to the influence of postsynaptic components to PPR (Wang and Kelly, 1997). Therefore, to provide a more accurate assessment of Pr and, thereby, short-term plasticity, a series of evoked EPSCs elicited by high-frequency stimulation of 20 Hz and duration of 1.6 s (Fig. 3A) was used to calculate cumulative charge transfer (Fig. 3B). The size of the RRP was calculated by fitting the last 11 stimuli and back-extrapolating to the y-intercept. As shown in Figure 3C, the size of the RRP did not differ among the groups. The Pr was then calculated using the magnitude of the response to the first pulse divided by the size of the RRP. As shown in Figure 3D, one-way ANOVA revealed that Pr was significantly higher in Tg2576 compared with WT, which did not differ from Tg2576 treated with RSG, further supporting the interpretation that Tg2576 may have dysregulated neurotransmitter vesicle release machinery and PPARγ agonism rectifies this condition to restore synaptic transmission and short-term plasticity to WT conditions. Finally, to estimate the influence of RSG treatment on WT littermate controls we compared WT on control or RSG diet. We did not find any significant changes in (1) frequency of sEPSC, (2) I–O relationships of eEPSC, (3) RRP size, or (4) Pr (Table 1). These results provide support that PPARγ agonism specifically modulates synaptic dysfunction in the cognitively impaired Tg2576 and does not affect normal synaptic transmission and short-term synaptic plasticity.
Figure 3.

PPARγ agonism normalizes Pr but not the size of the RRP in 9MO Tg2576 DG. A, Representative traces recorded from dentate granule cell evoked responses to mPP high-frequency stimulation (20 Hz, 1.6 s). B, eEPSC cumulative charge transfer did not differ among groups. C, The RRP did not differ among groups (F(2,23) = 1.8, p = 0.18, one-way ANOVA with Dunnett's post hoc comparison). D, The Pr was higher in Tg2576 compared with WT, which did not differ from Tg2576 treated with RSG (p > 0.05, one-way ANOVA with Dunnett's post hoc comparison). Results are mean ± SEM; n = 11 (WT), 7 (Tg2576), and 8 (Tg2576 + RSG). Calibration: 25 pA, 250 ms; *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1.
Effect of RSG on synaptic properties in WT mice
| WT | WT + RSG | p | |
|---|---|---|---|
| Frequency of sEPSC (Hz) | 0.56 ± 0.07 (n = 19) | 0.51 ± 0.06 (n = 23) | 0.63 |
| Amplitude of sEPSC (pA) | −6.34 ± 0.35 (n = 19) | −6.39 ± 0.24 (n = 23) | 0.98 |
| Injected current: μA | |||
| Pulse1 125 μA | −42.51 ± 8.27 (n = 10) | −51.61 ± 9.94 (n = 9) | 0.49 |
| eEPSC 175 μA | −67.51 ± 10.99 (n = 10) | −73.50 ± 12.14 (n = 9) | 0.72 |
| Amplitude (pA) 225 μA | −94.19 ± 12.48 (n = 10) | −84.28 ± 12.95 (n = 9) | 0.59 |
| Injected current: μA | |||
| Pulse 2 125 μA | −69.30 ± 12.27 (n = 10) | −74.71 ± 12.27 (n = 9) | 0.76 |
| eEPSC 175 μA | −106.16 ± 16.96 (n = 10) | −102.85 ± 16.22 (n = 9) | 0.89 |
| Amplitude (pA) 225 μA | −131.95 ± 19.06 (n = 10) | −119.99 ± 16.15 (n = 9) | 0.64 |
| RRP size (nC) | −9.40 ± 0.70 (n = 10) | −7.64 ± 0.88 (n = 5) | 0.15 |
| Release probability (Pr%) | 15.46 ± 2.07 (n = 10) | 17.34 ± 2.06 (n = 5) | 0.53 |
Mean and SEM are listed as well as p value resulting from Student's t test.
To elucidate proteins regulated by RSG during normalization of synaptic function and hippocampal memory, we performed quantitative MS on the DG of Tg2576 mice treated with or without RSG for 1 month. Benjamini–Hochberg analysis of protein expression revealed that RSG significantly increased (p < 0.05) expression of many proteins seminal to SNARE regulation of vesicle dynamics including vesicle-associated membrane protein 2 (VAMP2/synaptobrevin) and complexin-2 (CPLX2), while SNAP-25 and syntaxin-1A were unchanged (Table 2). Proteins that in turn regulate SNAREs were also induced including syntaxin binding protein-5/tomosyn, phosphofurin acidic cluster sorting protein 1 (PACS1), puromycin-sensitive aminopeptidase (PSA), piccolo, and vesicle-fusing ATPase. The specificity of induction was illustrated by the lack of statistically significant changes in many highly related proteins involved in vesicle fusion dynamics such as complexin-1 (CPLX1), VAMP1, and several isoforms of SNAPs, syntaxins, and their cognate binding proteins. Bioinformatics studies using IPA revealed extensive inter-relationships between these RSG-regulated proteins and their cognate pathways (Fig. 4). Many of these proteins are transcribed from genes containing PPREs and CREs in their proximal promoters (Table 2), further supporting our previous observation of the critical convergence of the PPARγ and ERK signaling pathways in RSG-mediated hippocampal cognitive enhancement. Thus, these findings indicate that hippocampal information processing occurs through multiple levels of pathways and networks to achieve cognitive enhancement through proteins involved in the highly regulated process of vesicular fusion underlying synaptic transmission.
Table 2.
SNARE Core and SNARE-associated proteins regulated by RSG
| Category | UniProt # | Ratio* | UniProt Name | Name | PPRE | CRE |
|---|---|---|---|---|---|---|
| SNARE core | ||||||
| VAMP | P63044 | 1.41 | VAMP2 | Vesicle-associated membrane protein 2 | ✓ | ✓ |
| SNAP | P60879 | 1 | SNP25 | Synaptosomal-associated protein 25 | ||
| STX | O35526 | 1 | STX1A | Syntaxin-1A | ✓ | ✓ |
| SNARE-associated protein families | ||||||
| Complexin | P84086 | 1.45 | CPLX2 | Complexin-2 | ✓ | ✓ |
| P63040 | 1 | CPLX1 | Complexin-1 | ✓ | ✓ | |
| VAMP | Q62442 | 1 | VAMP1 | Vesicle-associated membrane protein 1 | ✓ | ✓ |
| Q9WV55 | 1 | VAPA | Vesicle-associated membrane protein-associated protein A | ✓ | ||
| Q9QY76 | 1 | VAPB | Vesicle-associated membrane protein-associated protein B | ✓ | ||
| SNAP | O09044 | 1 | SNP23 | Synaptosomal-associated protein 23 | ✓ | |
| Q8R570 | 1 | SNP47 | Synaptosomal-associated protein 47 | |||
| STX | Q8K400 | 1.30 | STXB5 | Syntaxin-binding protein 5 (tomosyn-1) | ✓ | |
| P61264 | 1 | STX1B | Syntaxin-1B | ✓ | ||
| Q60770 | 1 | STXB3 | Syntaxin-binding protein 3 | ✓ | ||
| O08599 | 1 | STXB1 | Syntaxin-binding protein 1 | ✓ | ||
| SYT | P46096 | 1 | SYT1 | Synaptotagmin-1 | ||
| Q9R0N7 | 1 | SYT7 | Synaptotagmin-7 | ✓ | ✓ | |
| RIMS | Q99NE5 | 1 | RIMS1 | Regulating synaptic membrane exocytosis protein 1 | ✓ | |
| Q9EQZ7 | 1 | RIMS2 | Regulating synaptic membrane exocytosis protein 2 | ✓ | ||
| NSF | P46460 | 1.10 | NSF | Vesicle-fusing ATPase | ✓ | |
| Q9DB05 | 1 | SNAA | α-Soluble NSF attachment protein | ✓ | ✓ | |
| P28663 | 1 | SNAB | β-Soluble NSF attachment protein | |||
| Q9CWZ7 | 1 | SNAG | γ-Soluble NSF attachment protein | ✓ | ||
| Others | Q11011 | 1.13 | PSA | Puromycin-sensitive aminopeptidase | ✓ | |
| Q8K212 | 1.19 | PACS1 | Phosphofurin acidic cluster sorting protein 1 | ✓ | ||
| Q9QYX7 | 1.12 | PCLO | Protein piccolo | ✓ | ✓ |
*Quantitative MS of protein expression in Tg2576 with RSG diet over Tg2576 with control diet. Also denoted is the presence of PPREs and/or CREs in the target gene promoters.
Figure 4.

DG network of SNARE-regulatory proteins modulated by RSG treatment of Tg2576. Proteins identified via quantitative MS were subjected to IPA to reveal the extensive inter-relationships between the indicated proteins. The highest scoring network is shown (score = 64). Green, induced by RSG; white, no change; STXBP5, syntaxin-binding protein 5; NSF, vesicle-fusing ATPase; PCLO, protein piccolo; RIMS1 and RIMS2, regulating synaptic membrane exocytosis proteins 1 and 2; SNAP-25, synaptosomal-associated protein 25; STX1A, syntaxin-1A; SYT1, synatpotagmin-1; VAPA and VAPB, vesicle-associated membrane protein-associated proteins A and B.
Discussion
In some humans with early AD and mouse models for AD-like amyloidosis, impaired memory can be improved with PPARγ activation (Watson et al., 2005; Pedersen et al., 2006; Risner et al., 2006; Hort et al., 2007; Hoefer et al., 2008; Escribano et al., 2010; Rodriguez-Rivera et al., 2011). In the present study, we investigated Tg2576 synaptic transmission in the DG, the hippocampal gateway for the formation of new memories, which is particularly susceptible to disruption in early AD (Goodrich-Hunsaker et al., 2008). We found that spontaneous and mPP-evoked excitatory synaptic transmission in Tg2576 DG granule cells is hyperactive due to presynaptic mechanisms that were normalized following PPARγ activation with RSG. A role for presynaptic function in RSG-dependent cognitive enhancement was further corroborated by quantitative MS-based proteomics, which revealed that PPARγ activation induced expression of proteins critical to presynaptic SNARE complex regulation. Many of these proteins are coded for by genes with promoter regulatory elements that are responsive to PPARγ (PPREs) and ERK MAPK (CREs), further supporting previous findings that the PPARγ axis converges upon pathways that control synaptic transmission and hippocampal memory formation (Denner et al., 2012).
Our findings of altered synaptic transmission in DG of Tg2576 mice provide additional evidence for the well established role of the DG in AD pathology and is consistent with numerous reports linking Aβ pathology to synaptic dysfunction (Kamenetz et al., 2003; Rowan et al., 2005; Hermann et al., 2009; Abramov and Duchen, 2010; Cuevas et al., 2011; Chakroborty et al., 2012; Park et al., 2013; Ripoli et al., 2013). Elevated synaptic transmission observed in 9MO Tg2576 neurons is consistent with previous reports describing Aβ-induced hyperactivity of hippocampal neural networks (Palop et al., 2007; Busche et al., 2012). We found a significant increase in the frequency of sEPSCs and in Tg2576 neurons, suggesting such responses derive from presynaptic mechanisms. In accordance with increased frequency of sEPSCs, we found that paired-pulse stimulation of mPP displayed lower stimulus intensity and higher amplitude of I–O relationships in dentate granule cells of Tg2576 providing additional evidence in support of elevated synaptic activity at mPP-DG granule cells synapses. Enhanced sEPSC activity in Tg2576 DG granule cells due to presynaptic mechanisms raised the question of whether this also led to aberrant DG plasticity, specifically short-term presynaptic plasticity. When we examined mPP inputs to DG granule cells using paired-pulse stimulation to assess PPF, we found PPR was decreased in Tg2576 and normalized to WT with RSG. Further, using high-frequency stimulation to estimate RRP size and Pr, we concluded that increased Pr, but not the RRP, underlies the observed synaptic dysfunction. Together, our studies provide strong evidence for a hyperactive DG circuit during early stage AD pathology that can be attributed to presynaptic and short-term plasticity deficits.
Emerging evidence indicates links between impaired brain insulin signaling, cognition, and AD (Bosco et al., 2011; Rodriguez-Rivera et al., 2011; Schiöth et al., 2012; Talbot et al., 2012). Because insulin sensitizers improve insulin resistance in diabetes, they have also been tested for cognitive improvement in AD. Although previous large-scale clinical trials for dementia due to AD failed to show efficacy of insulin sensitizer therapy in AD, evolving consensus deduces their ineffectiveness was due to testing in late stage disease, similar to the failure of many other AD drug candidates (Becker and Greig, 2013). This evolving perspective derives, in part, from recent clinical trials on patients with MCI where insulin sensitizers as well as intranasal insulin provided significant cognitive benefit (Stockhorst et al., 2004; Watson et al., 2005; Risner et al., 2006; Sato et al., 2011; Craft et al., 2012).
In mouse models of AD-like amyloidosis, PPARγ agonists ameliorate Aβ-related pathology and restore ERK MAPK signaling pathways underlying hippocampus-dependent cognition (Pedersen et al., 2006; Escribano et al., 2009, 2010; Rodriguez-Rivera et al., 2011; Denner et al., 2012; Mandrekar-Colucci et al., 2012). The present studies showed rescue of hippocampal presynaptic transmission in Tg2576 mice and provide a novel functional mechanism that adds to previously identified effects of PPARγ agonism on Aβ-mediated synaptic plasticity deficits (Costello et al., 2005). We found that 1 month of treatment with the PPARγ agonist RSG restored the following synaptic dysfunction in Tg2576 DG granule cells: sEPSCs frequency, eEPSC I–O relationships, and Pr without affecting these functions in WT. Interestingly, studying I–O relationships with paired-pulse stimulation revealed that activation of PPARγ led to greater normalization of pulse2 compared with pulse1, pointing for a role of PPARγ on vesicle release machinery. Together these findings suggest that impaired hippocampus-dependent cognitive function in 9MO Tg2576 is founded, at least in part, on loss of fidelity in entorhinal cortex communication to the DG, which is a likely contributing factor to memory deficits in early AD.
MS and bioinformatics analyses identified candidate proteins involved in restoring presynaptic function in DG and hippocampal cognition. This revealed several proteins important for synaptic transmission, particularly those important to SNARE complex integrity and function (Lao et al., 2000; Schoch et al., 2001; Sakisaka et al., 2008; Maximov et al., 2009; Jorquera et al., 2012; Cao et al., 2013). Extensive literature confirms that altered expression of these proteins occurs in several neurological disorders including AD (Sze et al., 1997; Eastwood and Harrison, 2001; Freeman and Morton, 2004). Furthermore, many of the genes coding for these proteins contain promoter regulatory elements that are responsive to both PPARγ (PPREs) and ERK MAPK (CREs), further supporting the finding that PPARγ activation leads to convergence of these two pathways to achieve hippocampal memory enhancement (Denner et al., 2012). A potential common integrator is CRE binding protein, a nuclear histone acetylase that mediates actions of ERK through CREB and directly regulates PPARγ to enhance hippocampal cognition in AD mouse models (Guzowski and McGaugh, 1997; Caccamo et al., 2010; Denner et al., 2012).
The majority of the vesicle-related proteins detected were unaffected by RSG, indicating that PPARγ-mediated cognitive enhancement is defined by a specific genomic/transcriptomic/proteomic program. For example, of the SNARE core proteins, only VAMP2/synaptobrevin was upregulated in Tg2576 DG by RSG while SNAP25 and syntaxin-1A were unchanged. Decreased VAMP2/synaptobrevin correlates with cognitive decline in AD patients (Tannenberg et al., 2006; Honer et al., 2012), and interaction of VAMP2/synaptobrevin with synaptophysin is modulated by Aβ (Russell et al., 2012). Similarly, CPLX2 and syntaxin-binding protein 5 were induced by PPARγ activation while related isoforms and genes were unaffected. This indicates that altered expression of a select subgroup of proteins occurred during RSG-mediated cognitive enhancement.
Bioinformatics analysis revealed a highly interconnected network of DG synaptic vesicle proteins regulated by activation of PPARγ and convergence with the ERK MAPK pathway (Fig. 4). VAMP2 exemplifies this interconnectivity both as a PPRE- and CRE-containing gene as well being designated as a central interaction node connecting several SNARE and SNARE regulatory proteins including: (1) PACS1, which plays a critical role in the balance of amyloidogenic and non-amyloidogenic processing of APP to regulate levels of Aβ (Schmidt et al., 2007); (2) syntaxin binding protein 5 (tomosyn-1; STXBP5), which regulates SNARE complex formation, hippocampal synaptic transmission, and spatial memory in the Morris water maze (Barak et al., 2013); (3) PSA, which is enriched in synapses and important for synaptic transmission (Hui et al., 1998); and (4) CPLX2.
Since complexins are integral to SNARE regulation of vesicle priming, fusion, and subsequent synchronous neurotransmitter release (Huntwork and Littleton, 2007; Jorquera et al., 2012; Kaeser-Woo et al., 2012; Lin et al., 2013), our finding of increased CPLX2 in Tg2576 DG with PPARγ agonism-mediated cognitive enhancement suggests that this protein may occupy a significant role in hippocampal synaptic function during early AD. The decreased CPLX2 expression in Tg2576 DG would be predicted to result in increased spontaneous release and reduced synaptic dynamic range (Wragg et al., 2013), thus providing a potential mechanism for the Tg2576 synaptic phenotype. Since VAMP2 is also reduced in AD hippocampus where levels correlate with progressive accumulation of Aβ oligomers (Pham et al., 2010), coordinate induction of VAMP2 and CPLX2 by RSG further supports that the memory-enhancing effects of PPARγ agonism impinge upon synaptic vesicle dynamics.
In summary, hippocampal network hyperexcitability is considered an early indicator of AD memory impairment resulting from altered medial temporal lobe function where early hyperactivity is followed by failure of hippocampal activation as dementia develops (Putcha et al., 2011; Bejanin et al., 2012). Neuronal circuits vulnerable to AD are also affected in mouse models for AD-like amyloidosis and have shown aberrant hippocampal network hyperactivity (Palop et al., 2007; Verret et al., 2012). In a complementary fashion, we provide evidence that DG principal cells may contribute to hippocampal hyperexcitability via a presynaptic mechanism involving elevated glutamate release probability through deficits in SNARE and complexin proteins that are coded for by PPRE- and/or CRE-containing genes. These findings provide a putative mechanism for cognitive enhancement by RSG activation of PPARγ to ameliorate Tg2576 dysfunctional glutamatergic synaptic transmission, short-term plasticity, and synaptic vesicle regulatory proteins.
Footnotes
This work was supported by the National Institutes of Health under Grant AG031859 to K.T.D. and L.D. Additional funding was provided by the American Health Assistance Foundation and The Sealy Foundation for Biomedical Research to K.T.D., by the Emmett and Miriam McCoy Foundation to L.D., by a kind gift from J. and W. Mohn to K.T.D. and L.D., and the Cullen Trust for Health care to the Mitchell Center. Expert technical assistance was provided by Wei Song. Expert animal husbandry was provided by Dr. Caterina M. Hernandez.
The authors declare no competing financial interests.
References
- Abramov AY, Duchen MR. Impaired mitochondrial bioenergetics determines glutamate-induced delayed calcium deregulation in neurons. Biochim Biophys Acta. 2010;1800:297–304. doi: 10.1016/j.bbagen.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Agrawal R, Gomez-Pinilla F. ‘Metabolic syndrome’ in the brain: deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J Physiol. 2012;590:2485–2499. doi: 10.1113/jphysiol.2012.230078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak B, Okun E, Ben-Simon Y, Lavi A, Shapira R, Madar R, Wang Y, Norman E, Sheinin A, Pita MA, Yizhar O, Mughal MR, Stuenkel E, van Praag H, Mattson MP, Ashery U. Neuron-specific expression of tomosyn1 in the mouse hippocampal dentate gyrus impairs spatial learning and memory. Neuromolecular Med. 2013;15:351–363. doi: 10.1007/s12017-013-8223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RE, Greig NH. Fire in the ashes: can failed Alzheimer's disease drugs succeed with second chances? Alzheimers Dement. 2013;9:50–57. doi: 10.1016/j.jalz.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejanin A, Viard A, Chételat G, Clarys D, Bernard F, Pélerin A, de La Sayette V, Eustache F, Desgranges B. When higher activations reflect lower deactivations: a PET study in Alzheimer's disease during encoding and retrieval in episodic memory. Front Hum Neurosci. 2012;6:107. doi: 10.3389/fnhum.2012.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med. 2011;15:1807–1821. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Yang X, Südhof TC. Complexin activates exocytosis of distinct secretory vesicles controlled by different synaptotagmins. J Neurosci. 2013;33:1714–1727. doi: 10.1523/JNEUROSCI.4087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer's disease mice. J Neurosci. 2012;32:8341–8353. doi: 10.1523/JNEUROSCI.0936-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, O'Leary DM, Herron CE. Agonists of peroxisome proliferator-activated receptor-gamma attenuate the Abeta-mediated impairment of LTP in the hippocampus in vitro. Neuropharmacology. 2005;49:359–366. doi: 10.1016/j.neuropharm.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas ME, Haensgen H, Sepúlveda FJ, Zegers G, Roa J, Opazo C, Aguayo LG. Soluble Abeta(1–40) peptide increases excitatory neurotransmission and induces epileptiform activity in hippocampal neurons. J Alzheimers Dis. 2011;23:673–687. doi: 10.3233/JAD-2011-091717. [DOI] [PubMed] [Google Scholar]
- De Felice FG. Alzheimer's disease and insulin resistance: translating basic science into clinical applications. J Clin Invest. 2013;123:531–539. doi: 10.1172/JCI64595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon BA, Wood TG, Dineley KT. Cognitive enhancement with rosiglitazone links the hippocampal PPARgamma and ERK MAPK signaling pathways. J Neurosci. 2012;32:16725–16735a. doi: 10.1523/JNEUROSCI.2153-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ. Synaptic pathology in the anterior cingulate cortex in schizophrenia and mood disorders. a review and a Western blot study of synaptophysin, GAP-43 and the complexins. Brain Res Bull. 2001;55:569–578. doi: 10.1016/S0361-9230(01)00530-5. [DOI] [PubMed] [Google Scholar]
- Escribano L, Simón AM, Pérez-Mediavilla A, Salazar-Colocho P, Del Río J, Frechilla D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochem Biophys Res Commun. 2009;379:406–410. doi: 10.1016/j.bbrc.2008.12.071. [DOI] [PubMed] [Google Scholar]
- Escribano L, Simón AM, Gimeno E, Cuadrado-Tejedor M, López de Maturana R, García-Osta A, Ricobaraza A, Pérez-Mediavilla A, Del Río J, Frechilla D. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman W, Morton AJ. Regional and progressive changes in brain expression of complexin II in a mouse transgenic for the Huntington's disease mutation. Brain Res Bull. 2004;63:45–55. doi: 10.1016/j.brainresbull.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol. 2011;68:45–50. doi: 10.1001/archneurol.2010.229. [DOI] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Hunsaker MR, Kesner RP. The interactions and dissociations of the dorsal hippocampus subregions: how the dentate gyrus, CA3, and CA1 process spatial information. Behav Neurosci. 2008;122:16–26. doi: 10.1037/0735-7044.122.1.16. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci U S A. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S, Monarch ES, Goldstein FC. Impaired fear conditioning in Alzheimer's disease. Neuropsychologia. 2002;40:1187–1195. doi: 10.1016/S0028-3932(01)00223-8. [DOI] [PubMed] [Google Scholar]
- Hermann D, Both M, Ebert U, Gross G, Schoemaker H, Draguhn A, Wicke K, Nimmrich V. Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein-overexpressing mice without altering behaviorally-correlated sharp wave-ripple complexes. Neuroscience. 2009;162:1081–1090. doi: 10.1016/j.neuroscience.2009.05.044. [DOI] [PubMed] [Google Scholar]
- Hoefer M, Allison SC, Schauer GF, Neuhaus JM, Hall J, Dang JN, Weiner MW, Miller BL, Rosen HJ. Fear conditioning in frontotemporal lobar degeneration and Alzheimer's disease. Brain. 2008;131:1646–1657. doi: 10.1093/brain/awn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honer WG, Barr AM, Sawada K, Thornton AE, Morris MC, Leurgans SE, Schneider JA, Bennett DA. Cognitive reserve, presynaptic proteins and dementia in the elderly. Transl Psychiatry. 2012;2:e114. doi: 10.1038/tp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hort J, Laczó J, Vyhnalek M, Bojar M, Bures J, Vlcek K. Spatial navigation deficit in amnestic mild cognitive impairment. Proc Natl Acad Sci U S A. 2007;104:4042–4047. doi: 10.1073/pnas.0611314104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houeland G, Romani A, Marchetti C, Amato G, Capsoni S, Cattaneo A, Marie H. Transgenic mice with chronic NGF deprivation and Alzheimer's disease-like pathology display hippocampal region-specific impairments in short- and long-term plasticities. J Neurosci. 2010;30:13089–13094. doi: 10.1523/JNEUROSCI.0457-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KS, Saito M, Hui M. A novel neuron-specific aminopeptidase in rat brain synaptosomes. Its identification, purification, and characterization. J Biol Chem. 1998;273:31053–31060. doi: 10.1074/jbc.273.47.31053. [DOI] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Jones HC, Keep RF. Brain interstitial fluid calcium concentration during development in the rat: control levels and changes in acute plasma hypercalcaemia. Physiol Bohemoslov. 1988;37:213–216. [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci. 2012;32:18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Südhof TC. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci. 2012;32:2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Lao G, Scheuss V, Gerwin CM, Su Q, Mochida S, Rettig J, Sheng ZH. Syntaphilin: a syntaxin-1 clamp that controls SNARE assembly. Neuron. 2000;25:191–201. doi: 10.1016/S0896-6273(00)80882-X. [DOI] [PubMed] [Google Scholar]
- Lin MY, Rohan JG, Cai H, Reim K, Ko CP, Chow RH. Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol. 2013;591:2463–2473. doi: 10.1113/jphysiol.2012.244517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Jang M, Chang S. Deleterious effects of soluble amyloid-beta oligomers on multiple steps of synaptic vesicle trafficking. Neurobiol Dis. 2013;55:129–139. doi: 10.1016/j.nbd.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, Yates JR, Glabe C, Masliah E. Progressive accumulation of amyloid-beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha D, Brickhouse M, O'Keefe K, Sullivan C, Rentz D, Marshall G, Dickerson B, Sperling R. Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non-demented elderly adults. J Neurosci. 2011;31:17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli C, Piacentini R, Riccardi E, Leone L, Li Puma DD, Bitan G, Grassi C. Effects of different amyloid beta-protein analogues on synaptic function. Neurobiol Aging. 2013;34:1032–1044. doi: 10.1016/j.neurobiolaging.2012.06.027. [DOI] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rivera J, Denner L, Dineley KT. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav Brain Res. 2011;216:255–261. doi: 10.1016/j.bbr.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Anwyl R. Synaptic plasticity disruption by amyloid beta protein: modulation by potential Alzheimer's disease modifying therapies. Biochem Soc Trans. 2005;33:563–567. doi: 10.1042/BST0330563. [DOI] [PubMed] [Google Scholar]
- Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P. Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One. 2012;7:e43201. doi: 10.1371/journal.pone.0043201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadygov RG, Zhao Y, Haidacher SJ, Starkey JM, Tilton RG, Denner L. Using power spectrum analysis to evaluate (18)O-water labeling data acquired from low resolution mass spectrometers. J Proteome Res. 2010;9:4306–4312. doi: 10.1021/pr100642q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka T, Yamamoto Y, Mochida S, Nakamura M, Nishikawa K, Ishizaki H, Okamoto-Tanaka M, Miyoshi J, Fujiyoshi Y, Manabe T, Takai Y. Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J Cell Biol. 2008;183:323–337. doi: 10.1083/jcb.200805150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Schiöth HB, Craft S, Brooks SJ, Frey WH, 2nd, Benedict C. Brain insulin signaling and Alzheimer's disease: current evidence and future directions. Mol Neurobiol. 2012;46:4–10. doi: 10.1007/s12035-011-8229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, Willnow TE. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem. 2007;282:32956–32964. doi: 10.1074/jbc.M705073200. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Stockhorst U, de Fries D, Steingrueber HJ, Scherbaum WA. Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol Behav. 2004;83:47–54. doi: 10.1016/S0031-9384(04)00348-8. [DOI] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR. Selective loss of synaptic proteins in Alzheimer's disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49:631–639. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Postsynaptic calcineurin activity downregulates synaptic transmission by weakening intracellular Ca2+ signaling mechanisms in hippocampal CA1 neurons. J Neurosci. 1997;17:4600–4611. doi: 10.1523/JNEUROSCI.17-12-04600.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–958. doi: 10.1097/00019442-200511000-00005. [DOI] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS. Synaptic vesicles position complexin to block spontaneous fusion. Neuron. 2013;77:323–334. doi: 10.1016/j.neuron.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Zhao Y, Haidacher SJ, Wang E, Parsley MO, Gao J, Sadygov RG, Starkey JM, Luxon BA, Spratt H, Dewitt DS, Prough DS, Denner L. Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J Neurotrauma. 2013;30:775–788. doi: 10.1089/neu.2012.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]