

Abstract
The ventral pallidum (VP) is a target of dense nucleus accumbens projections. Many of these projections coexpress GABA and the neuropeptide enkephalin, a δ and μ opioid receptor (MOR) ligand. Of these two, the MOR in the VP is known to be involved in reward-related behaviors, such as hedonic responses to palatable food, alcohol intake, and reinstatement of cocaine seeking. Stimulating MORs in the VP decreases extracellular GABA, indicating that the effects of MORs in the VP on cocaine seeking are via modulating GABA neurotransmission. Here, we use whole-cell patch-clamp on a rat model of withdrawal from cocaine self-administration to test the hypothesis that MORs presynaptically regulate GABA transmission in the VP and that cocaine withdrawal changes the interaction between MORs and GABA. We found that in cocaine-extinguished rats pharmacological activation of MORs no longer presynaptically inhibited GABA release, whereas blocking the MORs disinhibited GABA release. Moreover, MOR-dependent long-term depression of GABA neurotransmission in the VP was lost in cocaine-extinguished rats. Last, GABA neurotransmission was found to be tonically suppressed in cocaine-extinguished rats. These substantial synaptic changes indicated that cocaine was increasing tone on MOR receptors. Accordingly, increasing endogenous tone by blocking the enzymatic degradation of enkephalin inhibited GABA neurotransmission in yoked saline rats but not in cocaine-extinguished rats. In conclusion, our results indicate that following withdrawal from cocaine self-administration enkephalin levels in the VP are elevated and the opioid modulation of GABA neurotransmission is impaired. This may contribute to the difficulties withdrawn addicts experience when trying to resist relapse.
Keywords: addiction, cocaine, electrophysiology, enkephalin, μ opioid receptor, ventral pallidum
Introduction
The ventral pallidum (VP) is a nucleus in the basal forebrain that contains largely GABAergic projection neurons and receives substantial GABAergic input from the nucleus accumbens. As part of the basal ganglia, the VP is important for reward-related behavior. For example, rats have been shown to electrically self-stimulate in the VP (Panagis et al., 1995), and inhibition of the VP either by lesions (Hubner and Koob, 1990; Hiroi and White, 1993) or by GABA agonists (McFarland and Kalivas, 2001) decreases drug-seeking behavior. In addition, in vivo electrophysiology studies have shown that VP neurons change their firing patterns during cocaine self-administration (Root et al., 2010, 2012, 2013).
A characteristic of the VP is the abundance of enkephalin (Zahm et al., 1985; Tripathi et al., 2010), a μ and δ opioid receptor ligand, and the expression of opioid receptors (Mansour et al., 1988) and mRNA (Mansour et al., 1994). Although all three classes of opioid receptors are found in the VP, behavioral data favor the μ opioid receptor (MOR) as the most relevant for the reinforcing properties of drugs, such as heroin (Olive and Maidment, 1998), cocaine (Skoubis and Maidment, 2003; Tang et al., 2005), and ethanol (Kemppainen et al., 2012) as well as of natural rewards (Smith and Berridge, 2005; Tindell et al., 2006; Smith et al., 2009). Enkephalin in the VP arises from the nucleus accumbens where it is coexpressed with GABA and D2 dopamine receptors (Zahm et al., 1985). The colocalization of enkephalin with nonopioid neurotransmitters has been observed in other brain regions and enkephalin is suggested to have an inhibitory effect on the release of the coexpressed neurotransmitter (Spanagel et al., 1990; Finnegan et al., 2006). Thus, the effects of activating MORs in the VP on reward-related behavior might be mediated by inhibiting GABA neurotransmission. Indeed, activation of MORs in the VP reduces extracellular GABA levels in the VP (Kalivas et al., 2001), and reduces the inhibitory effect of nucleus accumbens projections on VP neurons (Napier and Mitrovic, 1999). This is of behavioral relevance because GABA levels in the VP have been shown to change during cocaine self-administration (Wydra et al., 2013), as well as during reinstatement of cocaine seeking in cocaine-extinguished rats (Tang et al., 2005). In the latter case, the decrease in GABA levels and reinstatement of cocaine seeking were abolished by intra-VP microinjection of a MOR antagonist.
Although the above evidence describes an intimate relationship between activation of pallidal MORs and addictive behavior, it is not known how self-administration of drugs changes the capacity of MORs to modulate GABAergic transmission at accumbens-VP synapses. Here, we whole-cell patch-clamped VP neurons to characterize MOR regulation of GABA neurotransmission, and determine whether and how the modulation of GABA neurotransmission by MORs in the VP is altered after extinction from cocaine self-administration.
Materials and Methods
Animal housing and surgery.
All experiments were conducted in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals, and the Institutional Animal Care and Use Committee at the Medical University of South Carolina approved all procedures. Male Sprague-Dawley rats (250 g; Charles River Laboratories) were single housed under controlled temperature and humidity with a 12 h light/dark cycle. All behavioral training occurred during the dark cycle. Rats were acclimated in the vivarium for 1 week before surgeries, and fed and watered ad libitum until 2 d before behavioral training, during which food was restricted to 20 g/d. Rats were anesthetized with ketamine HCl (87.5 mg/kg Ketaset, Fort Dodge Animal Health) and xylazine (5 mg/kg Rompum, Bayer), and implanted with intravenous catheters. Intravenous catheters were flushed daily with cefazolin (0.2 ml of 0.1 g/ml) and heparin (0.2 ml of 100 IU) to prevent infection and maintain catheter patency, and rats recovered for a week before behavioral training.
Self-administration and extinction procedures.
Rats were trained to self-administer cocaine (NIH) in operant chambers with two retractable levers. The self-administration regimen consisted of 12 d of self-administration or until reaching the criterion of 10 or more infusions of cocaine per session during 10 consecutive sessions (average duration of the self-administration regimen was 11.0 ± 0.3 d for yoked saline rats and 11.1 ± 0.2 d for rats self-administering cocaine; minimum self-administration period was 10 d; all rats completed self-administration). Daily sessions lasted 2 h, with an active lever press resulting in 0.2 mg in 0.05 ml cocaine infusion (dissolved in sterile 0.9% saline) >3 s, whereas inactive lever presses were of no consequence. Average self-administered cocaine dose on last day of self-administration was 18.0 ± 1.2 mg/kg in a 2 h session. Yoked saline controls received a noncontingent infusion of saline in parallel with cocaine rats receiving a self-administered infusion of cocaine. A 5 s cue tone (2900 Hz) and light were presented with each cocaine infusion. Cocaine infusions were also followed by a 20 s timeout during which active lever presses had no consequence. After reaching the self-administration criterion rats began extinction training, in which pressing either lever had no consequence. Extinction training lasted at least 10 d and was continued until rats reached the extinction criterion of <25 active lever presses for three consecutive sessions (average duration of extinction training was 18.7 ± 1.8 d in the yoked saline group and 16.4 ± 0.9 d in the cocaine group; the statistically insignificant difference between the groups stems from the fact that only one rat was used per day for slice electrophysiology).
Slice preparation.
Sagittal slices were prepared as described previously (Kupchik and Kalivas, 2013) from yoked saline or cocaine-extinguished rats 24 h after the last extinction training session. Rats were anesthetized with ketamine (100 mg/kg) and decapitated. The brain was removed and sagittal VP brain slices (220 μm; VT1200S Leica vibratome) were collected into a vial containing aCSF (in mm: 126 NaCl, 1.4 NaH2PO4, 25 NaHCO3, 11 glucose, 1.2 MgCl2, 2.4 CaCl2, 2.5 KCl, 2.0 NaPyruvate, 0.4 ascorbic acid, bubbled with 95% O2 and 5% CO2) and a mixture of 5 mm kynurenic acid and 50 μm d-(-)-2-amino-5-phosphonopentanoic acid (D-AP5). Slices were stored at room temperature until used for recording.
In vitro whole-cell recording.
All recordings were collected at 32°C (TC-344B, Warner Instrument). Neurons were visualized with an Olympus BX51WI microscope. Excitatory synaptic transmission was blocked with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm). Multiclamp 700B (Molecular Devices) was used to record IPSCs under −80 mV voltage-clamp whole-cell configuration. Glass microelectrodes (1–2 MΩ) were filled with internal solution (in mm: 68 potassium chloride, 65 d-gluconic acid potassium salt, 7.5 HEPES potassium, 1 EGTA, 1.25 MgCl2, 10 NaCl, 2.0 MgATP, and 0.4 NaGTP, pH 7.2–7.3, 275 mOsm). We targeted only “classic” pallidal GABAergic cells which were identified by their soma size and membrane potential and avoided the newly discovered accumbens-like rostral VP neurons (Kupchik and Kalivas, 2013) and cholinergic cells (Bengtson and Osborne, 2000). All patched cells were located in the VP within the following approximate boundaries (coordinates relative to bregma): 1.9–2.9 medial-lateral, 0 to −1 rostral-caudal, 7.5–8.5 dorsal-ventral (Paxinos and Watson, 2006). Data were acquired at 10 kHz, and filtered at 2 kHz using AxoGraph X software (AxoGraph Scientific). To evoke IPSCs, a bipolar stimulating electrode was placed ∼200–300 μm rostral of the recorded cell to maximize chances of stimulating accumbens afferents. Stimulating amplitude was set to evoke an IPSC that was 200–600 pA, which was usually 30–70% of maximal IPSCs. In Figures 7, 8 we used a high-frequency stimulation (HFS) protocol (Nugent et al., 2007) to induce synaptic plasticity. The HFS protocol consisted of two trains separated by 20 s, each delivering 100 stimulations at 100 Hz. During the HFS protocol, membrane potential was allowed to change freely (i.e., current-clamp configuration). Series resistance (Rs) measured with a 5 mV depolarizing step (10 ms) given with each stimulus and holding current were always monitored online. Recordings with unstable Rs, or when Rs exceeded 20 MΩ were aborted.
Figure 7.
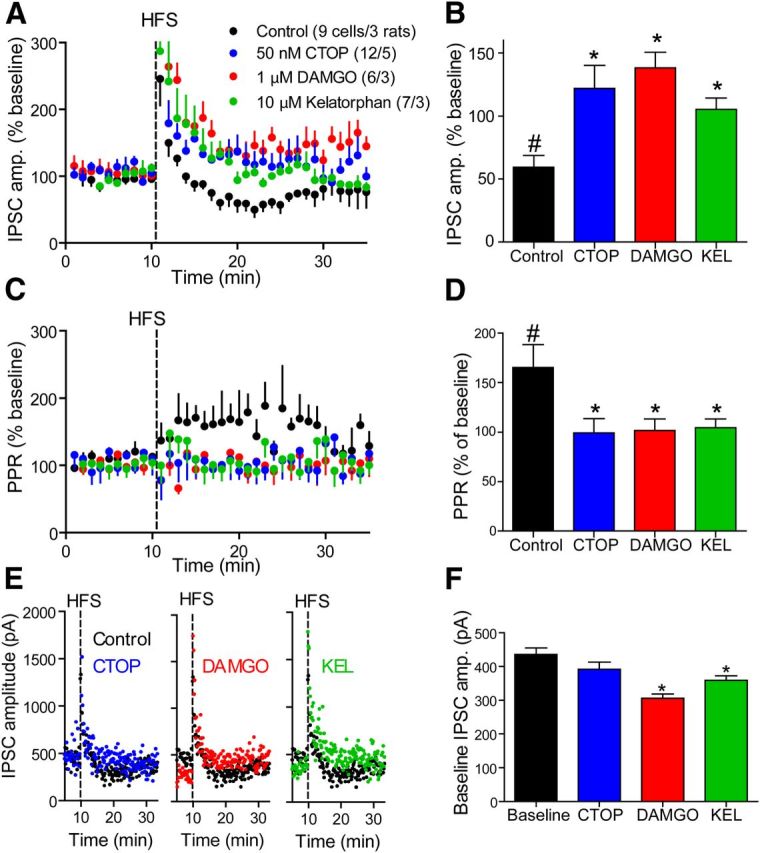
Loss of LTDGABA under tonic activation or inhibition of μ-opioid receptors. A, Time course of GABA eIPSCs after HFS (vertical dashed line) in control (black) and in the presence of either 50 nm CTOP (blue), 1 μm DAMGO (red), or 10 μm kelatorphan (green). Compounds were present throughout the experiment and were never washed out. The HFS generated a LTDGABA that was abolished in the presence of either compound. Number of cells in legend is the same for B–D, F. B, Average eIPSC amplitude taken from 18 to 24 min in A. In control conditions the eIPSC is inhibited to 62.1 ± 12.5% of baseline and is significantly different from in the presence of CTOP (121.9 ± 18.1% of baseline), DAMGO (138.3 ± 12.3% of baseline) and kelatorphan (105.4 ± 8.9% of baseline). C, Time course of the PPR in the same experiment shown in A. The PPR was elevated only in the control conditions and the elevation was synchronous with the LTDGABA shown in A. D, Average PPR during 18–24 min from experiment depicted in A. PPR was elevated in control conditions to 165.4 ± 23.2%. In all other conditions, PPR remained unchanged. E, Representative raw experiments with each compound compared with control condition. Note the difference in baseline between control condition and in the presence of DAMGO or kelatorphan. F, Average raw baseline eIPSC amplitudes in each condition. In the presence of DAMGO or kelatorphan the eIPSC was under tonic depression. KEL, Kelatorphan. #p < 0.05 compared with 100% using a one-sample t test. *p < 0.05 compared with control condition (C, D) or baseline (H) using one-way ANOVA and Dunnett's multiple-comparison post hoc analysis. Data presented as mean ± SEM.
Figure 8.

LTDGABA in the VP is lost after extinction of cocaine self-administration. A, Time course of GABA eIPSCs after HFS (vertical dashed line) in yoked saline (filled) and cocaine-extinguished (open) rats. Yoked saline data are the same as presented in Figure 6A. Number of cells and rats in legend is the same for B–D. B, Average eIPSC amplitude taken from 18 to 24 min in A. In yoked saline rats the eIPSC was inhibited to 62.1 ± 12.5% of baseline, whereas in cocaine-extinguished rats there was no change (112.8 ± 12% of baseline). C, Time course of the PPR in the same experiment shown in A. The PPR was elevated only in yoked saline rats and the elevation was synchronous with the LTDGABA shown in A. D, Average PPR during 18–24 min from experiment depicted in A. PPR was elevated in yoked saline rats to 165.4 ± 23.18 but remained unchanged in cocaine-extinguished rats. Coc, Cocaine-extinguished. #p < 0.05 compared with 100% using a one-sample t test. *p < 0.05 compared with control condition using one-way ANOVA and Dunnett's multiple-comparison post hoc analysis. Data presented as mean ± SEM.
Materials.
All salts were purchased from Sigma-Aldrich. D-AP5, CNQX, and kynurenic acid were purchased from Abcam. KC-2-009 was synthesized as described previously (Cheng et al., 2011) and provided by K.C.R, kelatorphan was provided by B.P.R.
Statistics.
All statistical analysis was performed in GraphPad Prism 5. Statistical tests are indicated in the figure legends.
Results
Cocaine self-administration
Rats self-administered cocaine for a minimum of 10 d until they reached a stable high level of active lever presses (Fig. 1A) and drug infusions (Fig. 1B). Active lever pressing was extinguished for at least 10 d, and rats were then killed for the various experimental procedures described below.
Figure 1.
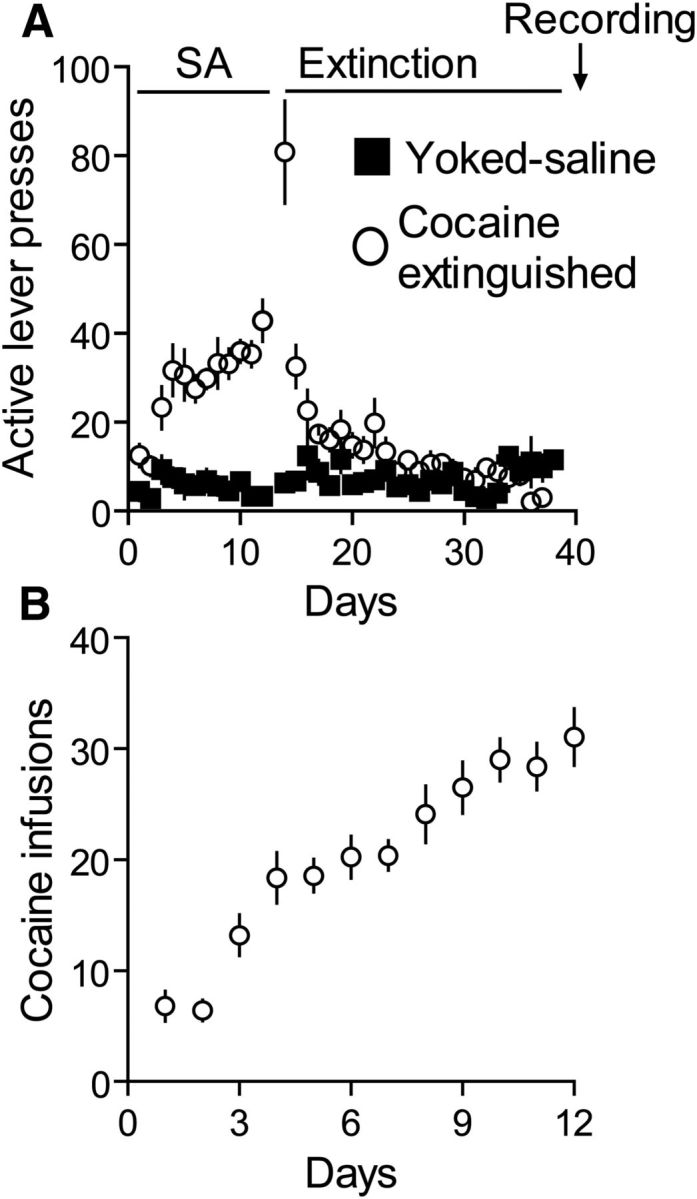
Cocaine self-administration and extinction of cocaine-seeking behavior. A, Active lever presses of rats receiving either saline (■) or cocaine (○) during self-administration training and extinction of cocaine-seeking behavior. Electrophysiological recordings were performed 24 h after the last day of extinction training. B, Number of cocaine infusions self-administered by rats during the self-administration training.
MORs fail to modulate GABA transmission in the VP of cocaine-extinguished rats
Sagittal slices of the VP were prepared from rats extinguished from cocaine self-administration or yoked saline rats. IPSCs were recorded from VP GABAergic neurons using the whole-cell patch-clamp technique. Evoked IPSCs (eIPSCs) were induced by a bipolar stimulating electrode placed 200–300 μm rostral to the recording pipette to increase probability of stimulating nucleus accumbens afferents (Fig. 2A). of [D-Ala2, N-MePhe4, Gly(ol)5]-enkephalin (DAMGO), a MOR selective agonist, were washed on VP slices and eIPSCs were examined in voltage-clamp configuration (Fig. 2B,C). DAMGO dose-dependently inhibited eIPSCs in yoked saline rats by ∼50% at the highest concentration (1 μm). However, application of DAMGO on slices of cocaine-extinguished rats failed to inhibit the eIPSCs. Examination of the spontaneous IPSCs (sIPSCs; Fig. 2D–F) or the miniature IPSCs (mIPSCs; Fig. 2G,H) in cocaine-extinguished rats revealed that the average amplitude and frequency of sIPSCs or mIPSCs were not changed by DAMGO. In contrast, yoked saline animals showed a DAMGO-induced reduction in the frequency but not amplitude of both sIPSCs and mIPSCs, indicating that the decrease in eIPSCs in saline rats was a presynaptic event that had been abolished in cocaine-extinguished rats.
Figure 2.

DAMGO, a μ-opioid receptor agonist, inhibits GABA release in yoked saline but not cocaine-extinguished rats. A, Recordings were performed on sagittal slices. Patched cells were located under the anterior commissure (AC) and the stimulating electrode was positioned 200–300 μm rostral to the recorded cell. Core, Core subcompartment of nucleus accumbens; IPAC, interstitial nucleus of the posterior part of the anterior commissure; Shell, shell subcompartment of nucleus accumbens. B, DAMGO dose-dependently inhibited evoked GABA IPSCs in yoked saline rats but not in rats that underwent cocaine self-administration and extinction [two-way ANOVA; F(1,80) = 8.94, p < 0.05 for the main effect of group (saline vs cocaine); F(3,80) = 3.92, p < 0.05 for interaction group × DAMGO]. Number of replicates is presented as number of cells/number of rats for each data point. Number of cells and rats is the same for D, E. C, Representative traces of eIPSCs with increasing doses of DAMGO (0, 10, 100, 1000 nm) in yoked saline and cocaine-extinguished rats. D, sIPSC amplitude was unchanged after treatment with DAMGO in yoked saline and cocaine-extinguished rats. E, sIPSC frequency was reduced by DAMGO in yoked saline but not cocaine-extinguished rats (two-way ANOVA; F(1,88) = 11.36, p < 0.05 for the main effect of group; F(3,88) = 3.78, p < 0.05 for the interaction group × DAMGO). F, Representative traces of sIPSCs in yoked saline (left) and cocaine-extinguished (right) rats with increasing DAMGO concentrations. Note the decrease in sIPSC frequency in yoked saline rats. G, mIPSC amplitude was unchanged by DAMGO in both yoked saline and cocaine-extinguished rats. H, mIPSC frequency was reduced by DAMGO in yoked saline but not cocaine-extinguished rats (two-way ANOVA; F(1,41) = 9.69, p < 0.05 for main effect of group; F(2,41) = 3.65, p < 0.05 for interaction group × DAMGO). Number of cells as in G. I, DAMGO hyperpolarized VP neurons in yoked saline rats but not in rats that underwent cocaine self-administration and extinction (two-way ANOVA; F(1,61) = 4.04, p < 0.05 for the main effect of group). J, DAMGO application dose-dependently decreased action potential (AP) frequency in yoked saline rats (44.12 ± 10.10% of baseline with 1000 nm DAMGO; one-way ANOVA; F(3,38) = 6.44, p < 0.05). Number of cells is presented above the number of rats (in parentheses). *p < 0.05 comparing the two groups in a given DAMGO concentration using two-way ANOVA and Bonferroni post hoc analysis. #p < 0.05 compared with baseline using one-way ANOVA and Bonferroni post hoc analysis. Data presented as mean ± SEM.
In current-clamp experiments, similar to the voltage-clamp experiments, DAMGO was found to hyperpolarize the membrane potential of VP neurons in yoked saline, but not cocaine-extinguished rats (Fig. 2I). The DAMGO-induced hyperpolarization in yoked saline rats resulted in a reduction in action potential firing frequency of VP neurons (Fig. 2J). Although these postsynaptic effects of DAMGO may be of importance, our experiments show that in the voltage-clamp configuration postsynaptic activation of the MORs does not affect the amplitude of the GABAergic IPSC. Therefore, we did not further pursue the postsynaptic effects of opioids in the VP.
Blocking MORs enhances GABA transmission more efficiently in cocaine-extinguished rats
We next examined the effect of D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) and D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), two selective MOR antagonists, on IPSCs in the VP. Surprisingly, in contrast to the effect of DAMGO, both antagonists were highly effective in cocaine-extinguished rats but not in yoked saline rats (Fig. 3A,D,F). A CTOP concentration as low as 5 nm significantly increased eIPSC amplitude in cocaine-extinguished rats by ∼45%, whereas only the highest CTOP concentration used (500 nm) elicited a nonsignificant trend toward an increase in the eIPSCs. Examination of the sIPSCs shows, akin to the effect of DAMGO, that the effects of CTOP and CTAP were likely of presynaptic origin, as both groups of rats differed in the sIPSC frequency (Fig. 3C,E,F) but not amplitude (Fig. 3B,F). These results suggest that the MORs in the VP are tonically occupied in cocaine-extinguished rats.
Figure 3.
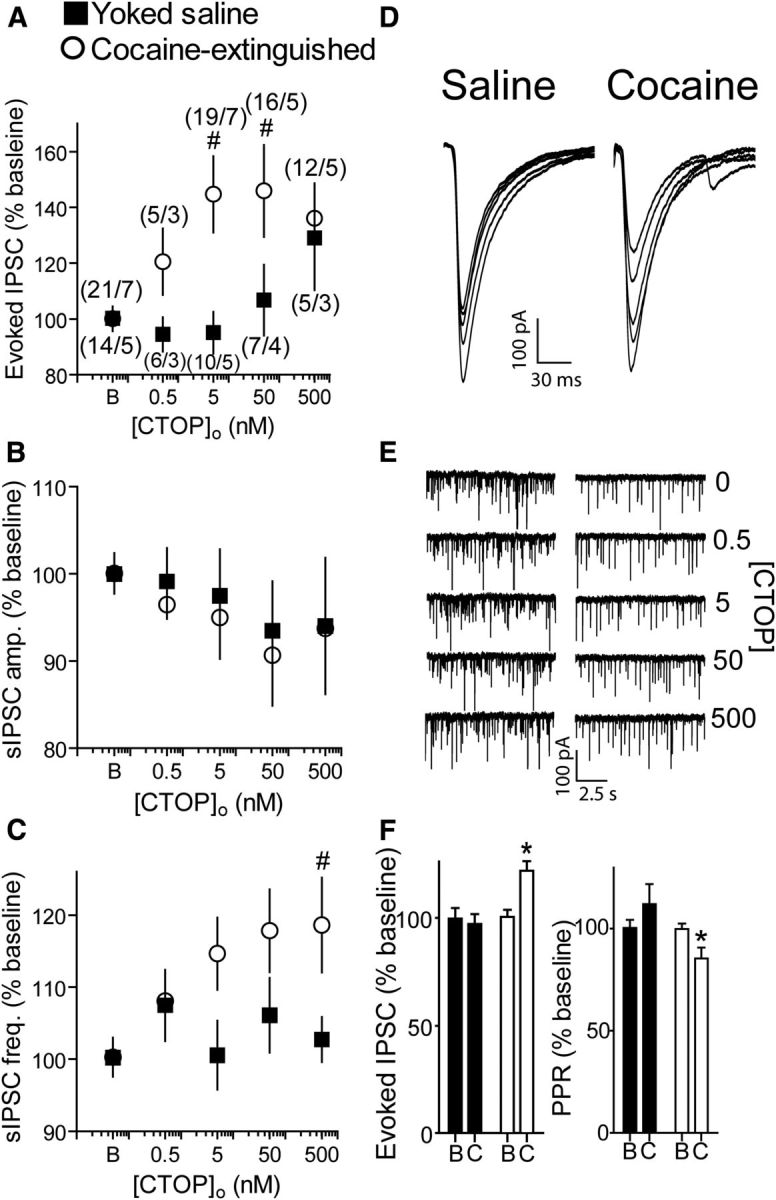
CTOP, a μ-opioid receptor antagonist, potentiated GABA release in cocaine-extinguished but not yoked saline rats. A, CTOP dose-dependently potentiated GABA eIPSCs after extinction of cocaine self-administration but not in yoked saline rats (two-way ANOVA; F(1,104) = 7.44, p < 0.05 for main effect of group). Number of replicates is presented as number of cells/number of rats for each data point. Number of cells and rats is the same for B, C. B, CTOP did not alter sIPSC amplitude in either group. C, CTOP increased sIPSC frequency in cocaine-extinguished rats but not in yoked saline rats (two-way ANOVA; F(1,106) = 5.79, p < 0.05 for main effect of group). D, Representative traces of eIPSCs with increasing doses of CTOP (0, 0.5, 5, 50, 500 nm) in yoked-saline and cocaine-extinguished rats. E, Representative traces of sIPSCs in yoked-saline (left) and cocaine-extinguished (right) rats with increasing CTOP concentrations. Note the increase in sIPSC frequency in cocaine-extinguished rats. F, CTAP, a second μ-opioid receptor antagonist, potentiated eIPSCs in cocaine-extinguished rats but not in yoked saline rats (left, two-way ANOVA; F(1,29) = 8.27, p < 0.05 for main effect of group; F(1,29) = 7.31 for interaction effect group × treatment). This was accompanied by a change in PPR in cocaine-extinguished rats but not in yoked saline rats (two-way ANOVA; F(1,27) = 5.65, p < 0.05 for main effect of group; F(1,27) = 5.34, p < 0.05 for interaction effect group × treatment). Data collected from three yoked saline rats (six cells) and four cocaine-extinguished rats (11 cells). B, Baseline; C, 50 nm CTOP. *p < 0.05 compared with CTAP in yoked saline using two-way ANOVA and Bonferroni post hoc analysis. #p < 0.05 compared with baseline using one-way ANOVA and Bonferroni post hoc analysis. Data presented as mean ± SEM.
As another measure of tonic MOR occupancy we applied the MOR inverse agonist (+)-3-((1S,5R)-2-((Z)-3-Phenylallyl)-2-azabicyclo[3.3.1]nonan-5-yl)phenol hydrochloride (KC-2-009; Cheng et al., 2011) on VP slices of yoked saline and cocaine-extinguished rats. In accordance with the effects of CTOP and CTAP, KC-2-009 increased eIPSC amplitude (by ∼30%) and caused a corresponding decrease in the paired-pulse ratio (PPR) in cocaine-extinguished but not yoked saline subjects (Fig. 4). Note that although KC-2-009 has been reported in the literature to affect constitutive activity in the MORs (Sadée et al., 2005; Meye et al., 2012), the effects of the antagonists on GABA neurotransmission indicate that in our experiments KC-2-009 is most likely antagonizing activity induced by binding of an endogenous ligand.
Figure 4.
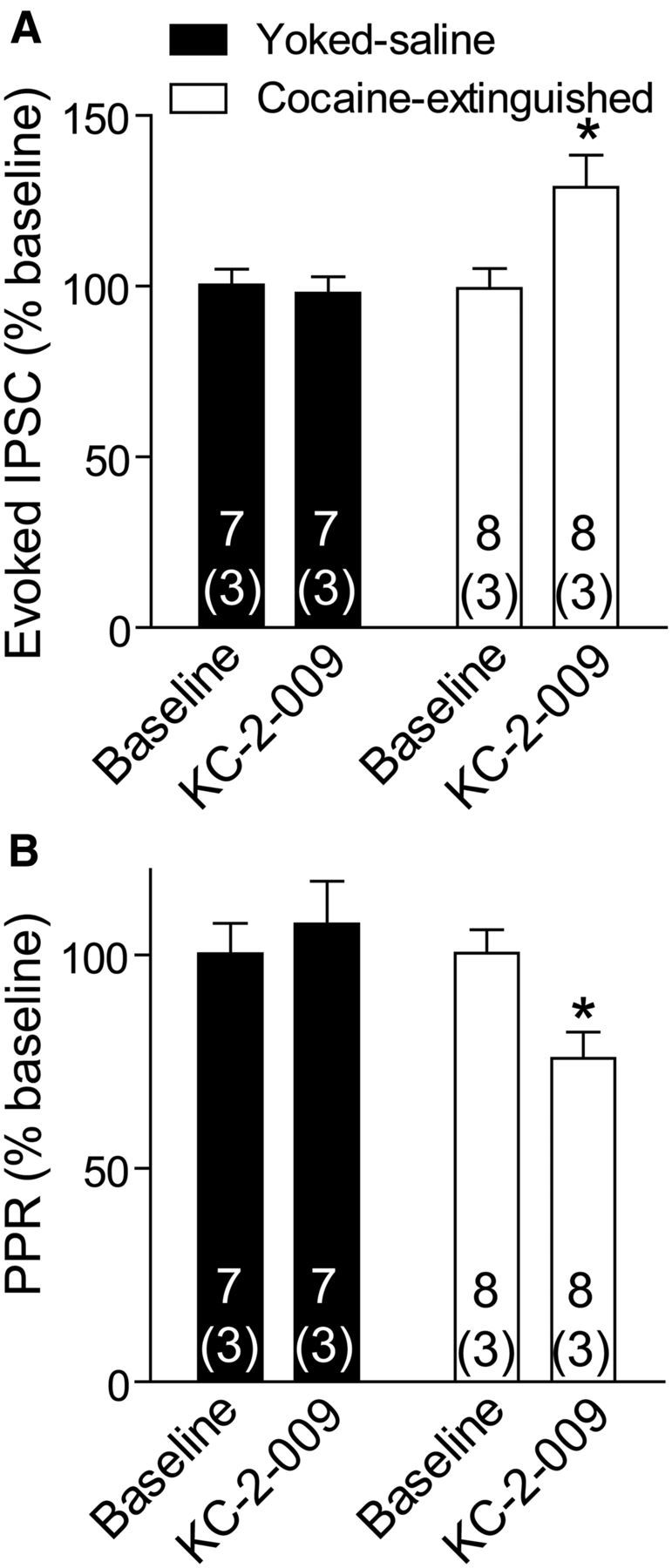
KC-2-009, an inverse agonist of the μ-opioid receptor, potentiated GABA release in cocaine-extinguished rats but not in yoked saline rats. A, 1 μm KC-2-009 increased the amplitude of GABA eIPSCs after extinction of cocaine self-administration (two-way ANOVA; F(1,26) = 4.59, p < 0.05 for main effect of group; F(1,26) = 7.48, p < 0.05 for interaction group × treatment). Number of cells presented above number of rats (in parentheses). B, KC-2-009 altered the PPR only in cocaine-extinguished rats (two-way ANOVA; F(1,18) = 4.43, p < 0.05 for main effect of group; F(1,18) = 4.51, p < 0.05 for interaction group × treatment). *p < 0.05 comparing the two groups at 1 μm KC-2-009 using two-way ANOVA and Bonferroni post hoc analysis. Data presented as mean ± SEM.
Blocking enkephalin catabolism does not affect GABA transmission in cocaine-extinguished rats
The increased basal activity of VP MORs suggests that there may be increased tone by endogenous enkephalin in cocaine-extinguished rats. This might be caused by reduced degradation of released enkephalin (Daugé et al., 1996). To check for this possibility, we washed the slices with kelatorphan, a dual inhibitor of the two enzymes that scavenge enkephalin, enkephalinase, and aminopeptidase N (Fournie-Zaluski et al., 1984; Roques et al., 2012). Akin to the addition of an exogenous MOR agonist in Figure 2B, a concentration of kelatorphan known to increase enkephalin levels in brain slices (Bourgoin et al., 1986), was able to inhibit eIPSCs in yoked saline rats but not in cocaine-extinguished rats (Fig. 5). Moreover, the effect of kelatorphan was counteracted by CTOP, indicating that the reduced eIPSC amplitude was mediated by MORs. In contrast to the results shown so far, we could not detect an effect of kelatorphan on sIPSC frequency or amplitude or on PPR (data not shown). Therefore, we cannot conclude that the effect of kelatorphan was either presynaptic or postsynaptic. Nonetheless, the dependence on MOR and similarity to the effects of DAMGO imply that kelatorphan is acting to increase levels of endogenous enkephalin. Possibly, inhibition of the two peptidases by kelatorphan affects the sIPSC in a more complicated manner due to changes in levels of peptides other than enkephalin that are known to also be degraded by enkephalinase and aminopeptidase (for review, see Roques et al., 2012). For example, a dual inhibitor of enkephalin degradation was able to reduce stress-induced behavior even in preproenkephalin knock-out mice (Noble et al., 2008).
Figure 5.

Kelatorphan, a dual inhibitor of enkephalin-degrading enzymes, inhibited GABA neurotransmission in yoked saline rats but not in cocaine-extinguished rats. GABA eIPSCs were inhibited by 10 μm kelatorphan in yoked saline rats but not in cocaine-extinguished rats (two-way ANOVA on yoked saline and cocaine-extinguished groups; F(1,52) = 6.14, p < 0.05 for main effect of group; F(1,52) = 4.11, p < 0.05 for interaction group × treatment). The inhibitory effect of kelatorphan was blocked by 500 nm CTOP. No changes had been observed in spontaneous activity or PPR (data not shown). Number of cells presented above number of rats (in parentheses). B, Baseline; K, 10 μm kelatorphan; K+C, 10 μm kelatorphan + 500 nm CTOP. *p < 0.05 compared with the respective bar in the cocaine-extinguished group using two-way ANOVA and Bonferroni post hoc analysis. #p < 0.05 compared with baseline using one-way ANOVA and Bonferroni post hoc analysis. Data presented as mean ± SEM.
GABA release in the VP is tonically depressed in cocaine-extinguished rats
The above results indicate that in cocaine-extinguished rats there is tone on presynaptic MORs in the VP imposed by high levels of enkephalin. This suggests, but does not prove, that GABA neurotransmission in the VP of cocaine-extinguished rats was inhibited. If this were true then, on average, the same amplitude of stimulation should result in larger amplitude eIPSCs in yoked saline compared with cocaine-extinguished rats. To examine this possibility, we calculated the ratio between the eIPSC amplitude and the stimulation amplitude (eIPSC/stimulation) in every cell and compared the ratios between the yoked saline and cocaine-extinguished rats (higher ratio indicating more readily evoked GABA release). Figure 6A shows that the eIPSC/stimulation was higher in yoked saline rats than in cocaine-extinguished rats by ∼40%. This supports the notion that GABA neurotransmission is tonically inhibited after extinguishing cocaine self-administration.
Figure 6.
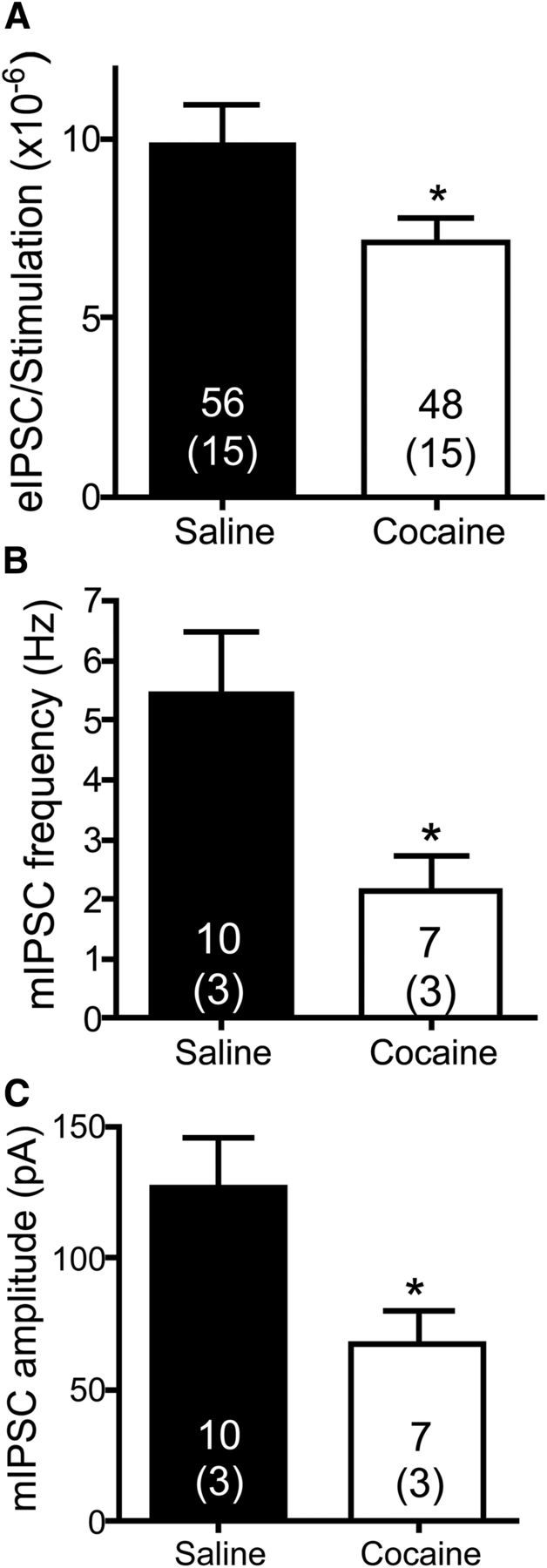
GABA neurotransmission in the VP is depressed in cocaine-extinguished rats. A, GABA IPSC amplitudes (pA) were divided by the stimulation amplitude (μA) used to generate them. The eIPSC/stimulation ratio was higher in yoked saline rats [9.87 ± 1.1 (×10−6)] than in cocaine-extinguished rats [7.10 ± 0.69 (×10−6)], indicating that for a given stimulation amplitude the resultant eIPSC was lower in cocaine-extinguished rats (unpaired two-tailed t test, t(102) = 2.059, p = 0.042). Number of cells presented above number of rats (in parentheses). B, The frequency of mIPSCs was significantly lower in cocaine-extinguished rats (2.15 ± 0.6 Hz) compared with yoked saline rats (5.43 ± 1 Hz), indicating a tonic presynaptic inhibition of GABA neurotransmission (unpaired two tailed t test, t(15) = 2.97, p = 0.010). C, The amplitude of mIPSCs was significantly lower in cocaine-extinguished rats (67.8 ± 12.2 pA) compared with yoked saline rats (127.0 ± 18.8 pA), indicating postsynaptic inhibition of GABAergic receptors (unpaired two-tailed t test, t(15) = 2.77, p = 0.014). *p < 0.05 compared with yoked saline.
Examination of sIPSCs and mIPSCs in the VP of yoked saline and cocaine-extinguished rats provides a similar conclusion. Although the reduction in sIPSC amplitude and frequency after extinction of cocaine seeking did not reach significance (data not shown), both amplitude and frequency of the mIPSCs were significantly reduced in cocaine-extinguished rats compared with yoked saline rats (Fig. 6B,C), indicating both presynaptic and postsynaptic mechanisms. The difference between the sIPSCs and mIPSCs implies that the decreased GABA neurotransmission in cocaine-extinguished rats is caused by lower probability of vesicle release at the presynaptic terminal rather than by lower excitability of GABAergic neurons. Importantly, whereas the presynaptic inhibition of GABA neurotransmission is alleviated by application of MOR antagonists (Fig. 3), the latter did not have a postsynaptic effect, suggesting that the decrease in mIPSC amplitude found in cocaine-extinguished rats may not be mediated by MORs.
High-frequency stimulation induces LTDGABA through activation of MORs
The inhibitory state of GABA neurotransmission in the VP of cocaine-extinguished rats may result in an altered ability to undergo synaptic plasticity. MOR-dependent long-term plasticity in GABAergic synapses was shown previously in several regions (Nugent et al., 2007; Dacher and Nugent, 2011; Wamsteeker Cusulin et al., 2013) and may also occur in the VP. Moreover, changes in basal MOR activation may affect this long-term plasticity. To examine this possibility we applied a HFS protocol similar to that used by Nugent et al. (2007), to VP slices of yoked saline rats and recorded GABA eIPSCs. Figure 7A shows that in control animals the HFS caused an immediate and transient increase in the eIPSC amplitude which declined within 3 min and was then followed by a long-term depression in GABAergic synapses (LTDGABA) that reached a maximum ∼50% reduction in baseline amplitude by 10 min and endured for up to 25 min (Fig. 7B). The observed LTDGABA was accompanied by an increase in the PPR that was time-locked to the inhibitory effect on the eIPSCs (Fig. 7C,D). This indicates the likely presynaptic origin of the LTDGABA.
To test whether the LTDGABA was mediated by MORs, we applied the HFS protocol in the presence of 50 nm CTOP. In the presence of CTOP, the LTDGABA was lost (Fig. 7A), indicating a role for MORs in mediating the LTDGABA. Interestingly, applying the HFS protocol in the presence of the MOR agonist DAMGO or the inhibitor of enkephalin degradation kelatorphan also resulted in a loss of the LTDGABA akin to that seen after CTOP (Fig. 7A). When the average change in eIPSC during the peak of inhibition in control condition (8–14 min after the HFS) was compared, all three treatments were significantly elevated compared with control (Fig. 7B). Although the blockade of LTD by CTOP implicates mediation by MOR stimulation, it seems paradoxical that direct (DAMGO) or indirect (kelatorphan) activation of MORs would also block LTD. However, as was shown in Figure 2B, stimulating MORs reduces sIPSC amplitude, and Figure 7E,F shows that having either DAMGO or kelatorphan in the bath during the HFS experiments significantly reduced the baseline sIPSC amplitude. Thus, the blockade of LTD is likely a masking event where the neurons were already in an LTD-like state from MOR stimulation by DAMGO or endogenous enkephalin (i.e., after kelatorphan).
LTDGABA is abolished in the VP of cocaine-extinguished rats
The results in Figures 2–5 suggest that in cocaine-extinguished rats the MORs are under a tonic activation by endogenous enkephalin, and Figure 7 shows that tonic activation of MORs in the VP following bath application of DAMGO or kelatorphan leads to a loss of LTDGABA. Therefore, we predicted that cocaine-extinguished rats would show a loss of LTDGABA. Figure 8A,C shows that the HFS protocol in VP slices from cocaine-extinguished rats did not induce LTDGABA. Similarly, HFS did not alter PPR in cocaine-extinguished animals, supporting a presynaptic action (Fig. 8B,D).
Discussion
The important role for the VP in addictive behavior has gained increasing interest in recent years (Kemppainen et al., 2012; Mahler and Aston-Jones, 2012; Hjelmstad et al., 2013; Stefanik et al., 2013). In particular, cocaine-induced adaptations in MOR signaling in the VP have emerged as possible candidates for the development and expression of addictive behavior (Olive and Maidment, 1998; Skoubis and Maidment, 2003; Tang et al., 2005; Kemppainen et al., 2012). Our findings mechanistically amplify this body of work by showing profound adaptations in MOR regulation of presynaptic GABA transmission in rats extinguished from cocaine self-administration. We demonstrate that compared with yoked saline rats, GABA neurotransmission in the VP of cocaine-extinguished rats is depressed by tonic activation of presynaptic MORs. As a result, the presynaptic inhibition of GABAergic eIPSCs in VP neurons by the MOR agonist DAMGO seen in yoked saline rats is markedly attenuated in cocaine-extinguished animals. This attenuation was confirmed to result from increased endogenous tone on MORs that masked the inhibitory effect of additional MOR stimulation either by a MOR agonist DAMGO or by preventing the degradation of endogenous MOR ligands with kelatorphan. The masking produced by increased MOR tone was revealed most clearly by the fact that selective MOR antagonists increased eIPSC amplitude only in cocaine-extinguished subjects. Similarly, the capacity to induce LTDGABA in VP neurons was found to depend on MOR stimulation and was masked in cocaine-extinguished animals or by DAMGO pretreatment in control rats.
Increased endogenous tone on presynaptic MORs after extinction from cocaine self-administration
The predominant endogenous opioid in the VP is enkephalin (Skoubis et al., 2005). Enkephalin arises almost entirely in GABAergic afferents from the nucleus accumbens and is not synthesized by VP neurons (Zahm et al., 1985). Previous in vitro studies indicate that one role for enkephalin coexpression with GABA in striatal medium spiny neurons is for enkephalin to presynaptically inhibit the release of GABA (Maneuf et al., 1994; Stanford and Cooper, 1999; Miura et al., 2007, 2008). Also, in vivo iontophoresis of a MOR agonist presynaptically reduces the GABAergic inhibition of VP neuron firing (Johnson and Napier, 1997), and microdialysis studies reveal MOR agonist-mediated reductions in extracellular GABA content in the VP (Tang et al., 2005). Our data demonstrate that MORs presynaptically regulate GABA release, and show that endogenous tone on MORs was elevated after extinction from cocaine self-administration. Given the dense enkephalinergic innervation of the VP, it is likely that colocalized enkephalin/GABA afferents are the source of increased endogenous tone in cocaine-extinguished subjects. Increases in enkephalinergic tone on MORs can have various effects, including MOR endocytosis (Sternini et al., 2000; Williams et al., 2013) and affinity changes (Birdsong et al., 2013). However, reductions in MOR signaling are not the likely mechanism underpinning the lack of response to MOR agonist in cocaine-extinguished animals because an inverse MOR agonist had full efficacy in the VP of cocaine-extinguished rats, and a MOR antagonist markedly potentiated eIPSCs only in cocaine subjects. These findings clearly reveal substantial tonic MOR activation in cocaine-extinguished animals. The presence of MOR tone in VP neurons after extinction from cocaine was also manifested in GABAergic synaptic plasticity, where HFS produced MOR-dependent LTDGABA that was absent in cocaine-extinguished rats as well as when MORs were either blocked or tonically activated.
MORs versus other types of opioid receptors
Whereas the VP expresses δ (DOR) and κ (KOR), as well as μ opioid receptors (Mansour et al., 1988), the MORs are considered most important for addictive behavior (Johnson and Napier, 2000; Skoubis and Maidment, 2003; Tang et al., 2005). Supporting this notion, elevating endogenous enkephalin levels with the enkephalinase inhibitor kelatorphan reduced GABA eIPSC amplitude and was completely reversed by CTOP. This indicates that kelatorphan-mediated increases in extracellular enkephalin are not activating DORs or KORs to modulate GABA neurotransmission in the VP. A distinction between MORs and DORs in the VP is also supported by higher levels of MORs compared with DORs in the more dorsal globus pallidus (Waksman et al., 1986). Moreover, a double-labeling study showing that whereas MORs are found both presynaptically on terminals of nucleus accumbens afferents and postsynaptically on VP neurons, DORs are found only postsynaptically, mostly on neurons that project back to the nucleus accumbens (Olive et al., 1997). Given this preferential postsynaptic localization, it seems unlikely that DORs significantly affect the presynaptic modulation of GABA neurotransmission that was abolished in the VP of cocaine-extinguished rats.
A role for tonic MOR stimulation in the VP on cocaine addiction
The projection from the accumbens to VP has long been known to regulate a variety of reward-seeking behaviors (Mogenson and Yang, 1991), including reinstated cocaine-seeking (McFarland and Kalivas, 2001; Stefanik et al., 2013), and MOR signaling in the VP is essential in this regard. Thus, injection of MOR antagonists into the VP prevents reinstatement of cocaine-seeking (Tang et al., 2005) and cocaine-induced conditioned-place preference (Skoubis and Maidment, 2003). Similarly, microinjection of MOR agonists into the VP greatly affects reward-related behavior (Smith and Berridge, 2005; Tindell et al., 2006; Kemppainen et al., 2012), and potentiates cocaine reinstatement (Tang et al., 2005). Interestingly, whereas microinjection of morphine induces reinstatement of drug-seeking behavior through a MOR-dependent mechanism (Tang et al., 2005), application of DAMGO in cocaine-extinguished rats did not affect GABA neurotransmission. This seeming contradiction may stem from the use of different MOR agonists [DAMGO here and morphine by Tang et al. (2005); MORs have been suggested to respond differently to different types of agonists; Piñeyro and Archer-Lahlou, 2007], differences between in vivo and in vitro examinations, and last, the morphine-induced reinstatement of cocaine-seeking may be mediated by an effect of MORs on the release of neurotransmitters other than GABA.
Implicating enkephalin rather than endorphin in the modulation of the VP via MORs, preproenkephalin knock-out mice are unable to express the conditioned place aversion induced by microinjecting naloxone into the VP (Skoubis et al., 2005). Our findings show that there is enduring tone on MORs in the VP following extinction of cocaine self-administration. This indicates that in addition to changes in the acute regulation of drug- and reward-seeking behaviors, regulation of basal enkephalin levels in the VP may undergo long-lasting adaptations in response to repeated use of addictive drugs. Consistent with this possibility, microdialysis experiments in the VP reveal long-lasting increases in the basal levels of enkephalin following chronic systemic noncontingent injection of opiates (Olive et al., 1995; Olive and Maidment, 1998). Also, enkephalin mRNA in accumbens cells is elevated during (Hurd et al., 1992), and after extinction from cocaine self-administration (Crespo et al., 2001). A role for the loss of MOR-dependent LTDGABA in VP neurons of cocaine-extinguished subjects in reinstated cocaine seeking is especially provocative. Synaptic plasticity is required for the learning of new behaviors (Kauer and Malenka, 2007), and the inability of inhibitory GABAergic synapses in the VP to undergo LTDGABA may impair the ability to modify cocaine-seeking and contribute to the relapse vulnerability that characterizes addiction.
Summary and future directions
Our findings demonstrate that MORs presynaptically regulate GABA transmission in the VP, and in doing so can promote synaptic plasticity by inducing LTDGABA. We also demonstrated tonic MOR presynaptic regulation of GABA transmission in cocaine-extinguished animals and a corresponding masking of LTDGABA. This work is consistent with previous studies showing an important role for enkephalin transmission in the accumbens to VP projection in regulating reward seeking behaviors (Smith and Berridge, 2005; Tindell et al., 2006; Kemppainen et al., 2012), including reinstated cocaine seeking (Tang et al., 2005). Enkephalin in the VP is released at synapses arising from a subset of accumbens medium spiny neurons that also express D2 dopamine receptors, dynorphin, and neurotensin. Thus, the release of enkephalin and subsequent presynaptic regulation of GABA release may be accompanied by neurotensin and dynorphin release. Although we definitively show a cocaine-induced adaptation in MOR signaling in the VP that has an obligatory role in reinstated cocaine seeking (Tang et al., 2005), there is also microdialysis evidence that VP neurotensin promotes GABA release and reinstated cocaine seeking, although the involvement of neurotensin was modulatory and not obligatory in cocaine reinstatement (Torregrossa and Kalivas, 2008). In addition, D1-expressing accumbens GABAergic neurons that project to the VP coexpress dynorphin, substance P, and cocaine and amphetamine-regulated transcript peptide (Zahm and Heimer, 1990; Zhou et al., 2003); all of which have been shown to modulate cocaine-related behaviors including locomotion, reward, and/or reinstatement (Shippenberg et al., 2007; Commons, 2010; Hubert et al., 2010). Given that MORs may also be localized to these D1-expressing GABAergic terminals, and that enkephalin released from the D2-expressing GABAergic input neurons may stimulate these receptors (especially following kelatorphan), a complete understanding of the role played by the VP in reinstated cocaine seeking will require evaluation of interactions between enkephalin, GABA and the various other colocalized neuropeptides.
Footnotes
This work was supported by Grants DA015369, DA003906, and DA012513 from the National Institutes of Health and by the Neuroscience Institute Fellowship for International Trainees, Medical University of South Carolina. A portion of this work was supported by the Intramural Research Programs of the National Institute on Drug Abuse and National Institute on Alcohol and Alcoholism.
The authors declare no competing financial interests.
References
- Bengtson CP, Osborne PB. Electrophysiological properties of cholinergic and noncholinergic neurons in the ventral pallidal region of the nucleus basalis in rat brain slices. J Neurophysiol. 2000;83:2649–2660. doi: 10.1152/jn.2000.83.5.2649. [DOI] [PubMed] [Google Scholar]
- Birdsong WT, Arttamangkul S, Clark MJ, Cheng K, Rice KC, Traynor JR, Williams JT. Increased agonist affinity at the μ-opioid receptor induced by prolonged agonist exposure. J Neurosci. 2013;33:4118–4127. doi: 10.1523/JNEUROSCI.4187-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgoin S, Le Bars D, Artaud F, Clot AM, Bouboutou R, Fournie-Zaluski MC, Roques BP, Hamon M, Cesselin F. Effects of kelatorphan and other peptidase inhibitors on the in vitro and in vivo release of methionine-enkephalin-like material from the rat spinal cord. J Pharmacol Exp Ther. 1986;238:360–366. [PubMed] [Google Scholar]
- Cheng K, Lee YS, Rothman RB, Dersch CM, Bittman RW, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 41. Unusual inverse μ-agonists and potent μ-opioid antagonists by modification of the N-substituent in enantiomeric 5-(3-hydroxyphenyl)morphans. J Med Chem. 2011;54:957–969. doi: 10.1021/jm1011676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons KG. Neuronal pathways linking substance P to drug addiction and stress. Brain Res. 2010;1314:175–182. doi: 10.1016/j.brainres.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JA, Manzanares J, Oliva JM, Corchero J, Palomo T, Ambrosio E. Extinction of cocaine self-administration produces a differential time-related regulation of proenkephalin gene expression in rat brain. Neuropsychopharmacology. 2001;25:185–194. doi: 10.1016/S0893-133X(01)00221-4. [DOI] [PubMed] [Google Scholar]
- Dacher M, Nugent FS. Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology. 2011;61:1166–1171. doi: 10.1016/j.neuropharm.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Daugé V, Mauborgne A, Cesselin F, Fournié-Zaluski MC, Roques BP. The dual peptidase inhibitor RB101 induces a long-lasting increase in the extracellular level of met-enkephalin-like material in the nucleus accumbens of freely moving rats. J Neurochem. 1996;67:1301–1308. doi: 10.1046/j.1471-4159.1996.67031301.x. [DOI] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, Pan HL. μ Opioid receptor activation inhibits GABAergic inputs to basolateral amygdala neurons through Kv1.1/1.2 channels. J Neurophysiol. 2006;95:2032–2041. doi: 10.1152/jn.01004.2005. [DOI] [PubMed] [Google Scholar]
- Fournie-Zaluski MC, Chaillet P, Bouboutou R, Coulaud A, Cherot P, Waksman G, Costentin J, Roques BP. Analgesic effects of kelatorphan, a new highly potent inhibitor of multiple enkephalin degrading enzymes. Eur J Pharmacol. 1984;102:525–528. doi: 10.1016/0014-2999(84)90575-2. [DOI] [PubMed] [Google Scholar]
- Hiroi N, White NM. The ventral pallidum area is involved in the acquisition but not expression of the amphetamine conditioned place preference. Neurosci Lett. 1993;156:9–12. doi: 10.1016/0304-3940(93)90426-L. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Xia Y, Margolis EB, Fields HL. Opioid modulation of ventral pallidal afferents to ventral tegmental area neurons. J Neurosci. 2013;33:6454–6459. doi: 10.1523/JNEUROSCI.0178-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert GW, Manvich DF, Kuhar MJ. Cocaine and amphetamine-regulated transcript-containing neurons in the nucleus accumbens project to the ventral pallidum in the rat and may inhibit cocaine-induced locomotion. Neuroscience. 2010;165:179–187. doi: 10.1016/j.neuroscience.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CB, Koob GF. The ventral pallidum plays a role in mediating cocaine and heroin self-administration in the rat. Brain Res. 1990;508:20–29. doi: 10.1016/0006-8993(90)91112-T. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Brown EE, Finlay JM, Fibiger HC, Gerfen CR. Cocaine self-administration differentially alters mRNA expression of striatal peptides. Brain Res Mol Brain Res. 1992;13:165–170. doi: 10.1016/0169-328X(92)90058-J. [DOI] [PubMed] [Google Scholar]
- Johnson PI, Napier TC. GABA- and glutamate-evoked responses in the rat ventral pallidum are modulated by dopamine. Eur J Neurosci. 1997;9:1397–1406. doi: 10.1111/j.1460-9568.1997.tb01494.x. [DOI] [PubMed] [Google Scholar]
- Johnson PI, Napier TC. Ventral pallidal injections of a mu antagonist block the development of behavioral sensitization to systemic morphine. Synapse. 2000;38:61–70. doi: 10.1002/1098-2396(200010)38:1<61::AID-SYN7>3.0.CO%3B2-6. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Jackson D, Romanidies A, Wyndham L, Duffy P. Involvement of pallidothalamic circuitry in working memory. Neuroscience. 2001;104:129–136. doi: 10.1016/S0306-4522(01)00054-9. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kemppainen H, Raivio N, Suo-Yrjo V, Kiianmaa K. Opioidergic modulation of ethanol self-administration in the ventral pallidum. Alcohol Clin Exp Res. 2012;36:286–293. doi: 10.1111/j.1530-0277.2011.01611.x. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Kalivas PW. The rostral subcommissural ventral pallidum is a mix of ventral pallidal neurons and neurons from adjacent areas: an electrophysiological study. Brain Struct Funct. 2013;218:1487–1500. doi: 10.1007/s00429-012-0471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Aston-Jones GS. Fos activation of selective afferents to ventral tegmental area during cue-induced reinstatement of cocaine seeking in rats. J Neurosci. 2012;32:13309–13326. doi: 10.1523/JNEUROSCI.2277-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneuf YP, Mitchell IJ, Crossman AR, Brotchie JM. On the role of enkephalin cotransmission in the GABAergic striatal efferents to the globus pallidus. Exp Neurol. 1994;125:65–71. doi: 10.1006/exnr.1994.1007. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, Watson SJ. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438. doi: 10.1002/cne.903500307. [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meye FJ, van Zessen R, Smidt MP, Adan RA, Ramakers GM. Morphine withdrawal enhances constitutive mu-opioid receptor activity in the ventral tegmental area. J Neurosci. 2012;32:16120–16128. doi: 10.1523/JNEUROSCI.1572-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Saino-Saito S, Masuda M, Kobayashi K, Aosaki T. Compartment-specific modulation of GABAergic synaptic transmission by mu-opioid receptor in the mouse striatum with green fluorescent protein-expressing dopamine islands. J Neurosci. 2007;27:9721–9728. doi: 10.1523/JNEUROSCI.2993-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Masuda M, Aosaki T. Roles of micro-opioid receptors in GABAergic synaptic transmission in the striosome and matrix compartments of the striatum. Mol Neurobiol. 2008;37:104–115. doi: 10.1007/s12035-008-8023-2. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Yang CR. The contribution of basal forebrain to limbic-motor integration and the mediation of motivation to action. Adv Exp Med Biol. 1991;295:267–290. doi: 10.1007/978-1-4757-0145-6_14. [DOI] [PubMed] [Google Scholar]
- Napier TC, Mitrovic I. Opioid modulation of ventral pallidal inputs. Ann N Y Acad Sci. 1999;877:176–201. doi: 10.1111/j.1749-6632.1999.tb09268.x. [DOI] [PubMed] [Google Scholar]
- Noble F, Benturquia N, Bilkei-Gorzo A, Zimmer A, Roques BP. Use of preproenkephalin knock-out mice and selective inhibitors of enkephalinases to investigate the role of enkephalins in various behaviours. Psychopharmacology (Berl) 2008;196:327–335. doi: 10.1007/s00213-007-0964-z. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Olive MF, Maidment NT. Repeated heroin administration increases extracellular opioid peptide-like immunoreactivity in the globus pallidus/ventral pallidum of freely moving rats. Psychopharmacology (Berl) 1998;139:251–254. doi: 10.1007/s002130050712. [DOI] [PubMed] [Google Scholar]
- Olive MF, Bertolucci M, Evans CJ, Maidment NT. Microdialysis reveals a morphine-induced increase in pallidal opioid peptide release. Neuroreport. 1995;6:1093–1096. doi: 10.1097/00001756-199505300-00005. [DOI] [PubMed] [Google Scholar]
- Olive MF, Anton B, Micevych P, Evans CJ, Maidment NT. Presynaptic versus postsynaptic localization of mu and delta opioid receptors in dorsal and ventral striatopallidal pathways. J Neurosci. 1997;17:7471–7479. doi: 10.1523/JNEUROSCI.17-19-07471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagis G, Miliaressis E, Anagnostakis Y, Spyraki C. Ventral pallidum self-stimulation: a moveable electrode mapping study. Behav Brain Res. 1995;68:165–172. doi: 10.1016/0166-4328(94)00169-G. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. San Diego: Academic; 2006. [DOI] [PubMed] [Google Scholar]
- Piñeyro G, Archer-Lahlou E. Ligand-specific receptor states: implications for opiate receptor signalling and regulation. Cell Signal. 2007;19:8–19. doi: 10.1016/j.cellsig.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Root DH, Fabbricatore AT, Ma S, Barker DJ, West MO. Rapid phasic activity of ventral pallidal neurons during cocaine self-administration. Synapse. 2010;64:704–713. doi: 10.1002/syn.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Fabbricatore AT, Pawlak AP, Barker DJ, Ma S, West MO. Slow phasic and tonic activity of ventral pallidal neurons during cocaine self-administration. Synapse. 2012;66:106–127. doi: 10.1002/syn.20990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Ma S, Barker DJ, Megehee L, Striano BM, Ralston CM, Fabbricatore AT, West MO. Differential roles of ventral pallidum subregions during cocaine self-administration behaviors. J Comp Neurol. 2013;521:558–588. doi: 10.1002/cne.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques BP, Fournié-Zaluski MC, Wurm M. Inhibiting the breakdown of endogenous opioids and cannabinoids to alleviate pain. Nat Rev Drug Discov. 2012;11:292–310. doi: 10.1038/nrd3673. [DOI] [PubMed] [Google Scholar]
- Sadée W, Wang D, Bilsky EJ. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 2005;76:1427–1437. doi: 10.1016/j.lfs.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoubis PD, Maidment NT. Blockade of ventral pallidal opioid receptors induces a conditioned place aversion and attenuates acquisition of cocaine place preference in the rat. Neuroscience. 2003;119:241–249. doi: 10.1016/S0306-4522(03)00121-0. [DOI] [PubMed] [Google Scholar]
- Skoubis PD, Lam HA, Shoblock J, Narayanan S, Maidment NT. Endogenous enkephalins, not endorphins, modulate basal hedonic state in mice. Eur J Neurosci. 2005;21:1379–1384. doi: 10.1111/j.1460-9568.2005.03956.x. [DOI] [PubMed] [Google Scholar]
- Smith KS, Berridge KC. The ventral pallidum and hedonic reward: neurochemical maps of sucrose “liking” and food intake. J Neurosci. 2005;25:8637–8649. doi: 10.1523/JNEUROSCI.1902-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, Berridge KC. Ventral pallidum roles in reward and motivation. Behav Brain Res. 2009;196:155–167. doi: 10.1016/j.bbr.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- Stanford IM, Cooper AJ. Presynaptic mu and delta opioid receptor modulation of GABAA IPSCs in the rat globus pallidus in vitro. J Neurosci. 1999;19:4796–4803. doi: 10.1523/JNEUROSCI.19-12-04796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, Brown RM, Kalivas PW. Optogenetic evidence that pallidal projections, not nigral projections, from the nucleus accumbens core are necessary for reinstating cocaine seeking. J Neurosci. 2013;33:13654–13662. doi: 10.1523/JNEUROSCI.1570-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternini C, Brecha NC, Minnis J, D'Agostino G, Balestra B, Fiori E, Tonini M. Role of agonist-dependent receptor internalization in the regulation of mu opioid receptors. Neuroscience. 2000;98:233–241. doi: 10.1016/S0306-4522(00)00118-4. [DOI] [PubMed] [Google Scholar]
- Tang XC, McFarland K, Cagle S, Kalivas PW. Cocaine-induced reinstatement requires endogenous stimulation of mu-opioid receptors in the ventral pallidum. J Neurosci. 2005;25:4512–4520. doi: 10.1523/JNEUROSCI.0685-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tindell AJ, Smith KS, Peciña S, Berridge KC, Aldridge JW. Ventral pallidum firing codes hedonic reward: when a bad taste turns good. J Neurophysiol. 2006;96:2399–2409. doi: 10.1152/jn.00576.2006. [DOI] [PubMed] [Google Scholar]
- Torregrossa MM, Kalivas PW. Neurotensin in the ventral pallidum increases extracellular gamma-aminobutyric acid and differentially affects cue- and cocaine-primed reinstatement. J Pharmacol Exp Ther. 2008;325:556–566. doi: 10.1124/jpet.107.130310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Prensa L, Cebrián C, Mengual E. Axonal branching patterns of nucleus accumbens neurons in the rat. J Comp Neurol. 2010;518:4649–4673. doi: 10.1002/cne.22484. [DOI] [PubMed] [Google Scholar]
- Waksman G, Hamel E, Fournié-Zaluski MC, Roques BP. Autoradiographic comparison of the distribution of the neutral endopeptidase “enkephalinase” and of mu and delta opioid receptors in rat brain. Proc Natl Acad Sci U S A. 1986;83:1523–1527. doi: 10.1073/pnas.83.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker Cusulin JI, Füzesi T, Inoue W, Bains JS. Glucocorticoid feedback uncovers retrograde opioid signaling at hypothalamic synapses. Nat Neurosci. 2013;16:596–604. doi: 10.1038/nn.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wydra K, Golembiowska K, Zaniewska M, Kamińska K, Ferraro L, Fuxe K, Filip M. Accumbal and pallidal dopamine, glutamate and GABA overflow during cocaine self-administration and its extinction in rats. Addict Biol. 2013;18:307–324. doi: 10.1111/adb.12031. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Heimer L. Two transpallidal pathways originating in the rat nucleus accumbens. J Comp Neurol. 1990;302:437–446. doi: 10.1002/cne.903020302. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Zaborszky L, Alones VE, Heimer L. Evidence for the coexistence of glutamate decarboxylase and Met-enkephalin immunoreactivities in axon terminals of rat ventral pallidum. Brain Res. 1985;325:317–321. doi: 10.1016/0006-8993(85)90331-2. [DOI] [PubMed] [Google Scholar]
- Zhou L, Furuta T, Kaneko T. Chemical organization of projection neurons in the rat accumbens nucleus and olfactory tubercle. Neuroscience. 2003;120:783–798. doi: 10.1016/S0306-4522(03)00326-9. [DOI] [PubMed] [Google Scholar]