

Abstract
The ability of axons to regrow after injury is determined by the complex interplay of intrinsic growth programs and external cues. In Caenorhabditis elegans mechanosensory neuron, axons exhibit robust regenerative regrowth following laser axotomy. By surveying conserved metabolic signaling pathways, we have identified the ribosomal S6 kinase RSKS-1 as a new cell-autonomous inhibitor of axon regeneration. RSKS-1 is not required for axonal development but inhibits axon regrowth after injury in multiple neuron types. Loss of function in rsks-1 results in more rapid growth cone formation after injury and accelerates subsequent axon extension. The enhanced regrowth of rsks-1 mutants is partly dependent on the DLK-1 MAPK cascade. An essential output of RSKS-1 in axon regrowth is the metabolic sensor AMP kinase, AAK-2. We further show that the antidiabetic drug phenformin, which activates AMP kinase, can promote axon regrowth. Our data reveal a new function for an S6 kinase acting through an AMP kinase in regenerative growth of injured axons.
Keywords: AMP kinase, Caenorhabditis elegans, laser axotomy, ribosomal S6 kinase, DLK kinase
Introduction
Axons in the adult nervous system have limited regenerative ability after injury compared with embryonic stages (Schwab and Bartholdi, 1996). In the mammalian CNS, this phenomenon has long been attributed to an inhibitory extrinsic environment composed of CNS myelin-associated proteins and chondroitin sulfate proteoglycans (Yiu and He, 2006). However, a diminished intrinsic regenerative capacity is widely documented in many CNS neurons, including cerebellar, cortical, and retinal cells (Goldberg et al., 2002; Liu et al., 2011). Numerous cell-autonomous factors, such as cAMP and CREB, can stimulate neurite outgrowth on inhibitory substrates (Cai et al., 2001; Gao et al., 2004). More recently, a decrease in mammalian target of rapamycin (mTOR) signaling has been associated with the decline of intrinsic regrowth ability. Deletion of the gene for phosphatase and tensin homolog, PTEN, a negative regulator of mTOR, promotes axon regrowth after injury to the optic nerve or spinal cord, even in an inhibitory environment (Park et al., 2008; Christie et al., 2010). Moreover, neurons derived from embryonic neural cells undergo extensive axon growth in the injured adult spinal cord (Lu et al., 2012). Overexpression of a constitutively active (CA) version of the transcriptional factor KLF7 also enhances axon regeneration in the adult spinal cord (Blackmore et al., 2012). These studies have led to the emerging view that manipulation of the intrinsic growth state of neurons can improve axon regeneration despite the inhibitory environment of the mammalian CNS. Nonetheless, our understanding of the intrinsic signaling pathways operating in the adult nervous system to promote regenerative ability remains limited.
Caenorhabditis elegans is a tractable experimental model with which to dissect the mechanisms of axon regeneration (Chen and Chisholm, 2011). Single axons can be severed using laser axotomy, and their regrowth can be analyzed quantitatively and unambiguously in vivo. We screened mutants in a set of 25 genes affecting mitochondrial function, protein translation, the insulin/IGF-1 signaling pathway, and cellular stress responses to identify intrinsic regulators of axon regrowth after injury (Chen et al., 2011). We find that axon regrowth is significantly enhanced in loss-of-function mutants of rsks-1, which encodes the C. elegans ortholog of p70 ribosomal S6 kinase (p70S6K). RSKS-1 likely acts in parallel to the DLK-1 MAPK cascade, which is critical for the initiation of axon regeneration (Hammarlund et al., 2009; Yan et al., 2009). RSKS-1 acts cell autonomously to restrain axon regrowth through the AMP kinase AAK-2. We further show that the antidiabetic drug phenformin can enhance axon regrowth, likely in an AAK-2/AMPK-dependent manner. Our results uncover a previously unknown function for p70S6K, and suggest that regenerative axon regrowth can be enhanced by stimulation of the AMPK pathway.
Materials and Methods
Genetics.
C. elegans was grown on nematode growth medium agar plates at 20°C. For drug experiments, metformin (PHR1084, Sigma) or phenformin (P7045, Sigma) was added to the agar to a final concentration of 50 or 4.5 mm, respectively. L4 hermaphrodites were put on drug plates and their L4 progeny were used for axotomy. AICAR (5-aminoimidazole-4-carboxamide ribonucleoside; A9978, Sigma) was dissolved in DMSO before dilution in M9. We incubated worms in drug solution (containing Escherichia coli OP50) for 3 h before axotomy and recovered in drug solution for 24 h after axotomy. Control animals were incubated in M9-containing DMSO solvent to the equivalent concentration.
Touch neurons were visualized using either muIs32[Pmec-7-GFP] or zdIs5[Pmec-4-GFP], as described previously (Wu et al., 2007). Alleles used are as follows: aak-1(tm1944), aak-2(ok524, rr48), dlk-1(ju476, tm4024), hif-1(ia4), rskn-1(tm4834), and rsks-1(sv31, tm1714, ok1255). Deletion mutations were outcrossed to N2 wild type at least twice before analysis; primer sequences for genotyping are available on request.
Molecular biology and transgenes.
rsks-1 expression transgenes were generated from the rsks-1 cDNA yk290d1 (a gift from Yuji Kohara, National Institute of Genetics, Mishima, Japan). The cDNA was PCR amplified using primers 5′-atggctgacgtgttcgagtt (YJ9494) and 5′-tcagaaaaagtggaagaaca (YJ9495), and was subcloned into pCR8 (Invitrogen) to generate a Gateway entry clone. The rsks-1 entry clone was recombined with an appropriate destination vector to generate final clones pCZGY1870 (Pmec-4-RSKS-1) and pCZGY1872 (Pmec-4-GFP::RSKS-1). To generate CA RSKS-1, we mutated residues involved in S6K activation (Pullen and Thomas, 1997) by substituting T404 (T389 in p70S6K) to glutamate or S439 (S411 in p70S6K) to aspartate. Kinase-dead (KD) RSKS-1 was made by mutating K115 to glutamine. These constructs were used to generate pCZGY2249 (Pmec-4-RSKS-1T404E), pCZGY2250 (Pmec-4-RSKS-1S439D) and pCZGY2248 (Pmec-4-RSKS-1K115Q).
aak-2 genomic DNA was amplified from fosmid WRM067cF08 using primers 5′-tgggattccgtcaaagaaggacatg (YJ9492) and 5′-ctgaaaatgaaagcggcact (YJ9493). The resulting fragment contained 3.0 kb sequences upstream of and 241 bp downstream of the aak-2 coding sequence. aak-2 cDNA was amplified using RT-PCR from wild-type mRNAs using primers 5′-atgttttctcatcaagatcgaga (YJ9781) and 5′-ctgaaaatgaaagcggcact (YJ9493), and was subcloned to generate pCZGY2244 (Pmec-4-AAK-2).
We used standard procedures to generate transgenic animals (Mello et al., 1991). Plasmid DNAs were used at 25–50 ng/μl, a linear PCR fragment of aak-2 genomic DNA was used at 15 ng/μl, and coinjection marker Pttx-3-RFP or Pttx-3-GFP was used at 50 ng/μl. For each construct, at least two extrachromosomal transgenic lines were analyzed.
Prgef-1-DLK-1L(juSi50) was inserted at the ttTi5606 site, as described previously (Yan and Jin, 2012). Primers F (YJ9683; 5′gtcctccgacttctctacag) with R (YJ9684; 5′gccattcaagttcggagatag) and F (YJ9685; 5′ gagattcttgaagacgacgag) with R (YJ9686; 5′ tcttgataaggagttccacg) were used to distinguish the juSi50 insertion from the endogenous dlk-1 locus.
Axon regeneration.
Touch neuron axotomy and measurement of axon regeneration were essentially as described previously (Wu et al., 2007). New regrowing processes were considered neurites if they were ≥5 μm in length. Regenerating axons fused with distal process were excluded from measurement. To compare data from distinct genetic backgrounds or collected on different days, total axon regrowth was normalized to a wild-type control dataset from the same day. All statistical analyses used GraphPad Prism. The distribution of the total regrowth length of axons in wild-type cells and controls passed tests of normality. For comparisons of two groups, we used a two-tailed Student's t test or Fisher's exact test for proportion; for comparison of multiple groups, we used one-way ANOVA followed by Bonferroni's correction or Dunnett's post hoc test.
Quantitation of GFP::RSKS-1.
z-stacks of Pmec-4-GFP::RSKS-1(juEx4484) fluorescence in posterior lateral microtubule (PLM) neurons were taken using the spinning disk confocal microscope using constant imaging parameters. Maximum intensity projections were made, and fluorescence was quantitated in a 1 × 10 μm line scan ROI close to the axotomy site using MetaMorph.
Results
Ribosomal S6 kinase rsks-1 is a cell-autonomous inhibitor of axon regrowth
The axon outgrowth of mechanosensory neurons is normal in rsks-1 loss of function mutants. To examine the role of RSKS-1 in axon regeneration, we performed laser axotomy on PLM axons in L4 larvae, and imaged regrowth 24 h later (see Materials and Methods). Three genetic null mutations of rsks-1 caused significantly increased PLM axon regrowth (Fig. 1A,B; Table 1). Animals mutant for the related p90 S6K homolog rskn-1 showed normal regrowth (Table 1; see Fig. 3B). We also examined axon regeneration of anterior lateral microtubule (ALM) neurons, and observed significantly increased regrowth in rsks-1(0) mutants (Fig. 1C). PLM axons do not regrow when cut distally from their synaptic branch, but show significantly enhanced regrowth if the synaptic branch is also cut (Wu et al., 2007). Loss of RSKS-1 strongly enhanced distal PLM regrowth only when the synaptic branch was also cut (Table 1), suggesting that, although RSKS-1 is not the synaptic branch inhibitory signal, it functions in a related growth-inhibitory process.
Figure 1.

Ribosomal S6 kinase RSKS-1 inhibits axon regrowth in C. elegans. A, Representative images of PLM axons 24 h postaxotomy in animals, with genotypes indicated. GFP::RSKS-1 fully rescues the rsks-1(0) increased regrowth. Stars, Location of axotomy; arrows, tip of regrowing axon. B, PLM regrowth at 24 h postaxotomy in rsks-1(0) mutants, normalized to the appropriate control axotomized on the same day. Touch neuron-specific expression of rsks-1 cDNA or of GFP::RSKS-1 rescues rsks-1(0)-enhanced regrowth. Constitutively active forms of RSKS-1, T404E, and S439D inhibit regrowth when expressed in wild-type background, while the expression of a KD RSKS-1 fails to rescue rsks-1(0). C, ALM regrowth at 24 h postaxotomy in rsks-1(0) mutants shows increased regrowth. D, GFP::RSKS-1 is diffusely localized in the soma and axon of PLM, and is concentrated in the synaptic branch (arrow) in the ventral nerve cord. E, Examples of growth cones in control and rsks-1(0) mutants at 6 h after axotomy. F, rsks-1(0) mutants display increased axon regrowth at 24 and 48 h postaxotomy. G, The growth rate is higher in rsks-1(0) mutants at 0–6, 10–24, and 24–48 h. ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001. ns, Not significant. Scale bars: A, D, E, 20 μm.
Table 1.
PLM and ALM axon regrowth length measurement in selected genotypes
| Strain | Genotype | PLM regrowth (μm)a | ALM regrowth (μm)b |
|---|---|---|---|
| CZ10175 | Pmec-4-GFP(zdIs5) | 117.1 ± 6.8 | 90.23 ± 5.1; 204.4 ± 18.3 |
| CZ10969 | Pmec-7-GFP(muIs32) | 107.9 ± 8.5 | ND |
| CZ17112 | zdIs5;rsks-1(sv31) | 153.3 ± 9.9 | 158.8 ± 10.6 |
| CZ15318 | zdIs5;rsks-1(tm1714) | ND | 145.7 ± 14.9 |
| CZ15317 | muIs32;rsks-1(tm1714) | 135.6 ± 5.2 | ND |
| CZ14511 | zdIs5;rsks-1(ok1255) | 157.1 ± 8.2 | 149.6 ± 8.6 |
| CZ11327 | zdIs5;dlk-1(ju476) | 15.9 ± 2.4 | 68.2 ± 5.8; 111.9 ± 7.8 |
| CZ18141 | zdIs5;dlk-1(ju476);rsks-1(sv31) | 17.9 ± 2.3 | 87.2 ± 8.5; 181.8 ± 14.0 |
| CZ16351 | zdIs5;dlk-1(tm4024) | 33.4 ± 2.8 | 86.6 ± 10.5 |
| CZ19019 | zdIs5;dlk-1(tm4024);rsks-1(sv31) | 45.6 ± 6.5 | 129.4 ± 8.4 |
| CZ15571 | zdIs5;Prgef-1-DLK-1L(juSi50) | 173.5 ± 9.0 | 122.0 ± 7.5 |
| CZ17120 | zdIs5;Prgef-1-DLK-1L(juSi50);rsks-1(sv31) | 154.5 ± 9.4 | 190.6 ± 8.3 |
| CZ16434 | zdIs5;aak-2(ok524) | 57.7 ± 7.0 | 36.6 ± 7.7 |
| CZ19720 | zdIs5;aak-2(ok524); dlk-1(tm4024) | ND | 50.8 ± 7.7 |
| CZ14507 | zdIs5;aak-2(rr48) | 75.2 ± 8.0 | ND |
| CZ17121 | zdIs5;rsks-1(sv31);aak-2(ok524) | 83.5 ± 8.0 | ND |
| CZ17111 | rskn-1(tm4834);muIs32 | 136.7 ± 7.4 | ND |
| CZ11879 | zdIs5;hif-1(ia4) | 108.4 ± 7.3 | ND |
| CZ18142 | zdIs5;rsks-1(sv31);hif-1(ia4) | 156.2 ± 7.1 | ND |
| CZ19017 | zdIs5;aak-1(tm1944) | 130.6 ± 9.4 | ND |
| CZ19016 | zdIs5;aak-1(tm1944);rsks-1(sv31) | 152.2 ± 10.1 | ND |
| CZ15745 | zdIs5;Pmec-4-RSKS-1(juEx4395) | 110.7 ± 8.2 | ND |
| CZ17167 | zdIs5;rsks-1(sv31);Pmec-4-RSKS-1(juEx4395) | 111.0 ± 14.1 | ND |
| CZ17809 | zdIs5;rsks-1(sv31);Pmec-4-GFP::RSKS-1(juEx4484) | 97.4 ± 8.8 | ND |
| CZ17814 | zdIs5;Pmec-4-RSKS-1(T404E) (juEx5315) | 81.4 ± 8.2 | ND |
| CZ17812 | zdIs5;Pmec-4-RSKS-1(S439D) (juEx5313) | 51.4 ± 9.2 | ND |
| CZ18135 | zdIs5;Pmec-4-RSKS-1(K115Q) (juEx5420) | 165.0 ± 11.0 | ND |
| CZ18139 | zdIs5;rsks-1(sv31);Pmec-4-RSKS-1(K115Q) (juEx5420) | 174.1 ± 11.4 | ND |
| CZ19022 | zdIs5;juSi50;Pmec-4-RSKS-1(T404E) (juEx5316) | 107.9 ± 10.7 | ND |
| CZ16432 | zdIs5;aak-2(ok524);Paak-2-AAK-2(juEx4742) | 168.1 ± 6.9 | ND |
| CZ17106 | zdIs5;Paak-2-AAK-2(juEx4742) | 155.4 ± 11.8 | ND |
| CZ17800 | zdIs5;aak-2(ok524);Pmec-4-AAK-2(juEx5309) | 135.9 ± 9.4 | ND |
| CZ17118 | zdIs5;rsks-1(sv31);Paak-2-AAK-2(juEx4742) | 163.6 ± 10.9 | ND |
| CZ10175 | zdIs5 PLM distal | −1.8 ± 1.2 | ND |
| CZ17112 | zdIs5;rsks-1(sv31) PLM distal | 0.7 ± 1.0 | ND |
| CZ10175 | zdIs5 PLM distal + branch | 6.0 ± 4.3 | ND |
| CZ17112 | zdIs5;rsks-1(sv31) PLM distal + branch | 39.6 ± 4.4 | ND |
aValues are given at 24 h.
bRoman type indicates values at 24 h; bold type indicates values at 48 h.
Figure 3.

AAK-2 acts downstream of RSKS-1 in axon regrowth. A, Representative images of PLM axons 24 h after axotomy in wild-type, aak-2(0) single mutants, rsks-1(0); aak-2(0) double mutants, and wild-type animals grown on 4.5 mm phenformin. Scale bar, 20 μm. B, The enhanced PLM regrowth is abolished in rsks-1(0); aak-2(0) double mutants. Cell-autonomous expression of aak-2 cDNA rescues the low regrowth of aak-2(0). C, Normalized PLM regrowth 24 h after axotomy in wild-type, aak-2(0), and rsks-1(0) animals grown and recovered on plates containing 4.5 mm phenformin. PLM regrowth of wild-type animals was significantly enhanced when grown on 4.5 mm phenformin plates. ANOVA (B), t test (C): *p < 0.05; **p < 0.01; ***p < 0.001. ns, Not significant.
To address whether RSKS-1 functions cell autonomously in regrowth, we expressed an rsks-1 cDNA specifically in touch neurons. Expression of this cDNA did not affect regrowth in the wild-type background and fully rescued enhanced regrowth in rsks-1(0) mutants (Fig. 1B). Functional GFP::RSKS-1 was diffusely localized throughout the PLM axon, with notable concentration in the synaptic branch (Fig. 1D); the level and localization of GFP::RSKS-1 was not appreciably altered after axotomy (Fig. 2E,F). We next addressed whether the ability of RSKS-1 to inhibit regrowth depends on its kinase activity. We generated KD RSKS-1 by mutating the ATP binding site (von Manteuffel et al., 1997). Expression of RSKS-1 (KD) failed to rescue the enhanced regrowth of rsks-1(0) mutants, and did not affect regrowth in an rsks-1(+) background (Fig. 1B). Conversely, we generated two CA forms of RSKS-1 by making phosphomimetic changes T404E or S439D, predicted to cause S6K activation. When expressed in wild-type animals, both RSKS-1 (CA) transgenes significantly inhibited regrowth (Fig. 1B). Together, these data indicate that RSKS-1 can act cell autonomously to inhibit axon regrowth through its kinase activity.
Figure 2.
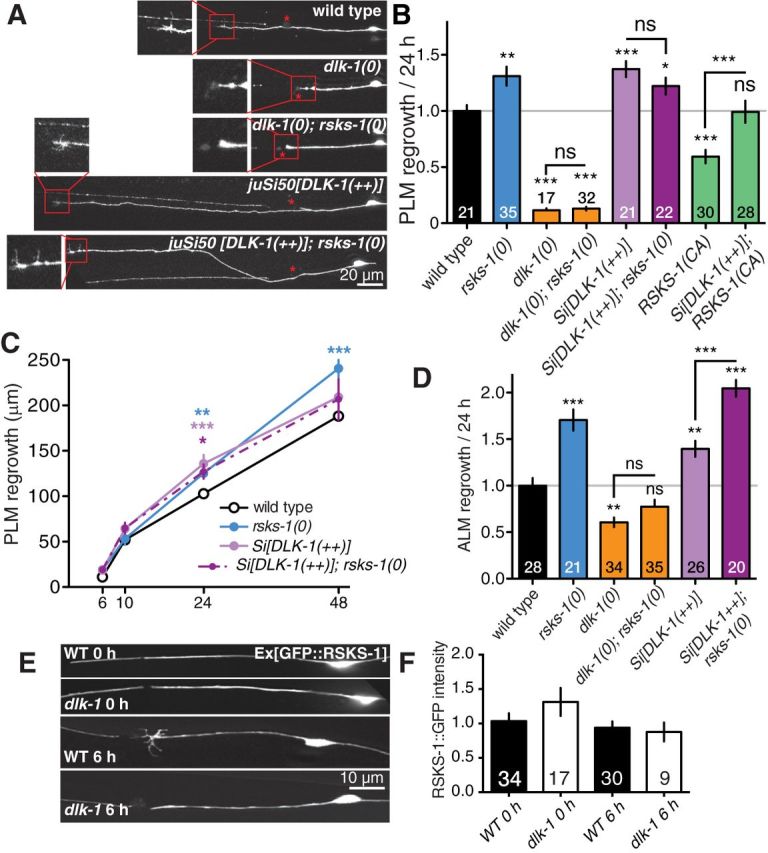
RSKS-1 may act upstream or in parallel with the DLK-1 pathway in axon regrowth. A, Representative images of PLM axons 24 h postaxotomy in genotypes as indicated. Scale bar, 20 μm. Red stars, Site of axotomy; red boxes, regrowing axon tip at higher magnification. B, Normalized PLM regrowth 24 h postaxotomy. C, PLM regrowth in rsks-1(0); Si[DLK-1(++)] double mutants postaxotomy is similar to each single mutant. D, Normalized ALM regrowth 24 h postaxotomy shows increased regrowth in rsks-1(0); Si[DLK-1(++)] double mutants. E, F, Pmec-4-GFP::RSKS-1 fluorescence in PLM in wild-type and in dlk-1(0), before and 6 h after axotomy, does not show significant difference. ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001. ns, Not significant.
We wanted to determine when in axon regrowth RSKS-1 has its inhibitory effect. In wild-type animals, ∼30% of injured PLM axons display growth cones at around 6 h postaxotomy, while ∼90% of injured PLM axons in rsks-1(0) mutants developed growth cones (Fig. 1E). We analyzed regrowth at multiple time points after axotomy (Fig. 1F). rsks-1(0) mutants displayed slight increased regrowth at 6 h postaxotomy, although the increase did not become statistically significant until 24 h. By pooling regrowth measurements at different time points, we found that compared with wild type, the rate of axon extension initially increased, then decreased from 6 to 10 h, and later increased between 10 and 48 h postaxotomy (Fig. 1G). These results suggest that RSKS-1 may play distinct roles in inhibiting growth cone formation early after injury and also later in inhibiting axon extension.
Regrowth in rsks-1 mutants is partly dependent on DLK-1
As the DLK-1 MAPKKK pathway is a key intrinsic determinant of axon regrowth in several neuron types (Hammarlund et al., 2009; Yan et al., 2009), we next addressed the relationship between RSKS-1 and DLK-1 in double mutants. dlk-1(0)-null mutants blocked PLM regrowth, with no visible growth cone formed after axotomy. rsks-1(0) did not significantly suppress dlk-1(0) phenotypes in PLM (Fig. 2A,B; Table 1). Animals expressing the DLK-1 long isoform under the control of the pan-neuronal rgef-1 promoter (dlk-1(++), genotype juSi50) display normal PLM development and enhanced PLM axon regrowth (Yan and Jin, 2012). PLM axon regrowth in dlk-1(++);rsks-1(0) double mutants was comparable to either single mutant over the course of 48 h postaxotomy (Fig. 2A–C). In addition, overexpression of DLK-1 was able to partly overcome the regrowth inhibition caused by rsks-1 (CA) (Fig. 2B).
As dlk-1(0) is completely epistatic to rsks-1(0) in PLM regrowth, RSKS-1 may act upstream of DLK-1 in a negative regulatory pathway. However, the strong dependence of PLM axon regrowth on the DLK-1 pathway precludes a precise mechanistic interpretation of these epistasis results. To attempt to resolve whether RSKS-1 might act independently of DLK-1, we turned to ALM neurons. ALM and PLM neurons are known to differ in their capacity to regenerate axons (Pinan-Lucarre et al., 2012). dlk-1(0) reduced but did not abolish ALM axon regrowth (Fig. 2D; Table 1). rsks-1(0) dramatically increased ALM regrowth. dlk-1(0);rsks-1(0) double mutants showed noticeable, although not statistically significant, increased ALM regrowth relative to dlk-1(0) in 24 h postaxotomy (Fig. 2D); this enhanced regrowth was significant over 48 h postaxotomy (p < 0.001, Student's t test; Table 1). rsks-1(0) also significantly suppressed the partial ALM regrowth defect of dlk-1(0) (Table 1). This result shows that loss of rsks-1 can partly bypass the requirement for DLK-1 in ALM regrowth, likely through enhancing axon extension. Notably, in dlk-1(0) single mutants and in dlk-1(0);rsks-1(0) double-mutant ALM regrew without forming morphologically distinct growth cones (data not shown). In addition, ALM axon regrowth was further increased in dlk-1(++);rsks-1(0) double mutants compared with either rsks-1(0) or dlk-1(++) mutants (Fig. 2D). Pmec-4-GFP::RSKS-1 fluorescence levels were not altered in dlk-1(0) mutants, and did not significantly change after axotomy (Fig. 2E,F). In summary, these results are generally consistent with the model that RSKS-1 can negatively regulate the axonal growth state independently of DLK-1, although some functions of RSKS-1 may depend on DLK-1 in specific neurons.
Increased regrowth in rsks-1 mutants requires AMP kinase AAK-2
Several genes act downstream of RSKS-1 in distinct contexts including pha-4/FOXA, hif-1/HIF1, and aak-2/AMPK (Sheaffer et al., 2008; Chen et al., 2009; Selman et al., 2009). pha-4 is essential for embryonic development, complicating the analysis of its role in adult axon regeneration. hif-1 mutants displayed normal regrowth and did not further enhance rsks-1 mutants (Fig. 3B; Table 1). AAK-1 and AAK-2 are the two C. elegans catalytic subunits of the cellular energy sensor AMPK (Apfeld et al., 2004). AAK-2 phosphorylation is increased in rsks-1 mutants, and the loss of aak-2 abolishes the longer lifespan of rsks-1 mutants (Selman et al., 2009). To address whether AAK-2 is activated in rsks-1(0) mutants, we took advantage of an AAK-2 activity reporter based on the nuclear translocation of the CREB-regulated transcriptional coactivator (CRTC-1) in intestinal cells (Mair et al., 2011). Activation of AAK-2 results in phosphorylation and nuclear export of CRTC-1. We found that CRTC-1::RFP was constitutively excluded from the nuclei of intestinal cells in rsks-1(0), indicating that AAK-2 is activated in these animals (data not shown). We therefore tested whether AAK-2 acts downstream of RSKS-1 in axon regeneration. We found that aak-2(0) mutants showed decreased PLM regrowth, while aak-1(0) mutants showed regrowth similar to that in the wild type (Fig. 3A,B). The expression of aak-2 driven by its own promoter or a touch neuron promoter not only rescued the reduced regrowth of aak-2(0), but also resulted in increased regrowth compared with wild type. Moreover, aak-2(0), but not aak-1(0), abolished the enhanced regrowth of rsks-1(0) mutants (Fig. 3B; Table 1). Transgenic overexpression of aak-2(+) also rescued the aak-2(0) regrowth defect to a level comparable to that of rsks-1(0) single mutant. AAK-2 overexpression in wild-type animals increased PLM regrowth, and this was not further enhanced in an rsks-1(0) background (Fig. 3B). aak-2(0); dlk-1(0) double mutants displayed reduced ALM regrowth compared with dlk-1(0) single mutants, although this was not statistically significant (Table 1). AAK-2 may therefore act partly in parallel with DLK-1. These results support the conclusion that AAK-2 acts downstream of RSKS-1 in PLM regrowth and suggest that increased AMPK activity enhances regenerative capacity.
Pharmacological activation of the AMPK pathway enhances axon regrowth
Several antihyperglycemic agents widely used to treat type 2 diabetes activate the AMPK pathway, such as the AMPK-activating agent AICAR and the biguanide drugs metformin and phenformin (Hawley et al., 2002). In C. elegans, metformin and phenformin treatment increases lifespan via AMPK, and phenformin has been shown to activate AAK-2 (Onken and Driscoll, 2010; Cabreiro et al., 2013). To test whether pharmacological activation of AAK-2 might enhance axon regrowth, we cultured animals in AICAR, metformin, or phenformin, using concentrations compatible with animal health and increased longevity (Onken and Driscoll, 2010; Cabreiro et al., 2013). Among the three drugs, phenformin treatment significantly enhanced PLM axon regrowth in wild-type animals, but not in aak-2 mutants (Fig. 3A,C). Phenformin treatment did not further enhance the axon regrowth of rsks-1(0) animals (Fig. 3C). These results suggest that pharmacological activation of AMPK can promote axon regrowth after injury.
Discussion
The ability to analyze the regrowth of single axons in vivo following laser axotomy in C. elegans offers great advantages in dissecting conserved regulators of axon regeneration. Recent studies in C. elegans have identified multiple intrinsic influences on axon regrowth capacity, including the DLK-1 MAPK cascade (Hammarlund et al., 2009; Yan et al., 2009), microtubule dynamics (Chen et al., 2011; Ghosh-Roy et al., 2012), and Notch signaling (El Bejjani and Hammarlund, 2012). Here we show that the ribosomal S6 kinase RSKS-1 acts as a cell-autonomous inhibitor of PLM axon regeneration.
S6 kinases were originally defined by their ability to phosphorylate ribosomal subunit protein S6 (Jenö et al., 1988). However, this role accounts for only some of their physiological functions (Pende et al., 2004; Meyuhas, 2008), and other S6K substrates affect a range of processes, including protein production, lipid synthesis, and synaptic plasticity (Magnuson et al., 2012). The function of S6K in axon regeneration has not previously been tested directly. We find that C. elegans RSKS-1/S6K inhibits axon regeneration cell autonomously, and that this requires its kinase activity. Axon regeneration can be broadly divided into two major phases of growth cone initiation and axon extension; RSKS-1 appears to inhibit both of these stages. Although the loss of RSKS-1 function did not bypass the requirement for the DLK-1 MAPK pathway in PLM regrowth, rsks-1(0) mutants significantly suppressed dlk-1(0) in ALM. These observations suggest DLK-1 and RSKS-1 do not act in a single linear pathway. Instead, DLK-1 activity is the main determinant of growth cone reformation; RSKS-1 may inhibit regrowth partly by modulating the DLK-1 pathway itself and via additional growth regulators such as AMPK.
We find that the axon regeneration inhibitory function of rsks-1 depends on AMPK. AMPK is a conserved sensor of intracellular energy levels; upon activation in stress conditions it triggers ATP production to mediate adaptive changes (Mihaylova and Shaw, 2011). In both C. elegans and mice, AMPK activity is increased in rsks-1(0) and S6K1−/− mutants (Aguilar et al., 2007; Selman et al., 2009). Our results suggest that AMPK is a physiologically relevant output for RSKS-1 in axonal regrowth. We speculate that the S6K/AMPK pathway regulates metabolic processes that are limiting in axon regeneration. Our finding that the AMPK-activating drug phenformin can improve axon regeneration in an AAK-2-dependent manner suggests potential therapeutic interventions for the enhancement of axon regrowth by selective manipulation of S6 kinase and AMPK signaling.
Footnotes
The authors declare no competing financial interests.
Some mutations used in this work were generated by the C. elegans Gene Knockout Consortium and were obtained from the Caenorhabditis Genetics Center, which was supported by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (Grant P40 OD010440). This work was supported by NIH Grant R01 NS057317 to A.D.C. and Y.J.; Z.W. is a research associate and Y.J. is an Investigator of the Howard Hughes Medical Institute. We thank Shohei Mitani, Malene Hansen, Simon Tuck, and Andy Dillin for strains. We also thank Malene Hansen, Michael Petraschek, and members of our laboratories for discussions.
References
- Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudière E, Foretz M, Viollet B, Diolez P, Bastin J, Benit P, Rustin P, Carling D, Sandri M, Ventura-Clapier R, Pende M. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 2007;5:476–487. doi: 10.1016/j.cmet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, Lee JK, Goldberg JL, Lemmon VP, Bixby JL. Kruppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A. 2012;109:7517–7522. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chisholm AD. Axon regeneration mechanisms: insights from C. elegans. Trends Cell Biol. 2011;21:577–584. doi: 10.1016/j.tcb.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O'Rourke S, Bowerman B, Wu Z, Jin Y, Chisholm AD. Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron. 2011;71:1043–1057. doi: 10.1016/j.neuron.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci. 2010;30:9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bejjani R, Hammarlund M. Notch signaling inhibits axon regeneration. Neuron. 2012;73:268–278. doi: 10.1016/j.neuron.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell. 2012;23:716–728. doi: 10.1016/j.devcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- Jenö P, Ballou LM, Novak-Hofer I, Thomas G. Identification and characterization of a mitogen-activated S6 kinase. Proc Natl Acad Sci U S A. 1988;85:406–410. doi: 10.1073/pnas.85.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tedeschi A, Park KK, He Z. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 2011;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- Lu P, Wang Y, Graham L, McHale K, Gao M, Wu D, Brock J, Blesch A, Rosenzweig ES, Havton LA, Zheng B, Conner JM, Marsala M, Tuszynski MH. Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell. 2012;150:1264–1273. doi: 10.1016/j.cell.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ, Dillin A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyuhas O. Physiological roles of ribosomal protein S6: one of its kind. Int Rev Cell Mol Biol. 2008;268:1–37. doi: 10.1016/S1937-6448(08)00801-0. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS One. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, Xue J, Qiao Y, Weisberg S, Roodhouse K, Sun L, Whitesides GM, Samuel A, Driscoll M. The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS Biol. 2012;10:e1001331. doi: 10.1371/journal.pbio.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997;410:78–82. doi: 10.1016/S0014-5793(97)00323-2. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaffer KL, Updike DL, Mango SE. The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol. 2008;18:1355–1364. doi: 10.1016/j.cub.2008.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Manteuffel SR, Dennis PB, Pullen N, Gingras AC, Sonenberg N, Thomas G. The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol Cell Biol. 1997;17:5426–5436. doi: 10.1128/mcb.17.9.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;18:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jin Y. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron. 2012;76:534–548. doi: 10.1016/j.neuron.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]