

Abstract
CLC-2 is a hyperpolarization-activated, inwardly rectifying chloride channel. Although the properties of the CLC-2 channel have been well characterized, its function in vivo is not well understood. We have found that channels encoded by the Caenorhabditis elegans CLC-2 homolog clh-3 regulate the activity of the spontaneously active hermaphrodite-specific neurons (HSNs), which control the egg-laying behavior. We identified a gain-of-function mutation in clh-3 that increases channel activity. This mutation inhibits egg laying and inhibits HSN activity by decreasing its excitability. Conversely, loss-of-function mutations in clh-3 lead to misregulated egg laying and an increase in HSN excitability, indicating that these channels modulate egg laying by limiting HSN excitability. clh-3-encoded channels are not required for GABAA-receptor-mediated inhibition of the HSN. However, they require low intracellular chloride for HSN inhibition, indicating that they inhibit excitability directly by mediating chloride influx. This mechanism of CLH-3-dependent modulation may be conserved in other neurons in which the driving force favors chloride influx.
Introduction
Members of the CLC superfamily of anion transport proteins have been identified in all phyla. Two subfamilies of CLC proteins (defined by the vertebrate CLC-3/CLC-4/CLC-5 and CLC-6/CLC-7 genes) are composed of Cl−/H+ exchangers involved in acidification and chloride homeostasis in intracellular organelles. Members of the third subfamily (CLC-1/CLC-2/CLC-Ka/Kb) are voltage-gated chloride channels localized primarily at the plasma membrane (Zifarelli and Pusch, 2007; Jentsch, 2008; Duran et al., 2010).
CLC-2 is a broadly expressed, hyperpolarization-activated, inwardly rectifying chloride channel. Although its physiological role is not well understood, CLC-2 is highly expressed in the nervous system and has been suggested to regulate neuronal excitability. Some studies have suggested that CLC-2 provides an efflux pathway for chloride that is necessary to maintain synaptic inhibition mediated by the GABAA receptor (GABAAR; Madison et al., 1986; Staley, 1994; Smith et al., 1995; Rinke et al., 2010). CLC-2 channels have also been proposed to control excitability directly by providing a background conductance that mediates chloride influx and modifies excitability by affecting the input resistance of the neuron (Rinke et al., 2010; Ratté and Prescott, 2011). Understanding the precise function of CLC-2 could have important implications for understanding how neuronal excitability is controlled.
The Caenorhabditis elegans homologs of CLC-2 are encoded by clh-3 (Schriever et al., 1999; Nehrke et al., 2000; Denton et al., 2004). Like CLC-2, the clh-3-encoded channel variants CLH-3a and CLH-3b are swelling- and hyperpolarization-activated inwardly rectifying chloride channels. CLH-3b is expressed in C. elegans oocytes, where it is activated during meiotic maturation and plays a role in regulating ovulation (Rutledge et al., 2001). The in vivo role of the CLH-3a variant is not known. clh-3-encoded channels are also expressed in a single pair of neurons, the hermaphrodite-specific neurons (HSNs; Schriever et al., 1999; Nehrke et al., 2000), but their function there has not been characterized.
The HSNs play a central role in the egg-laying behavior of C. elegans and as such provide a useful system to investigate the function of clh-3-encoded channels in the nervous system in vivo. The HSNs excite directly both the vulval muscles and the VC motor neurons, which also regulate egg laying (Schafer, 2005; Zhang et al., 2008), by releasing acetylcholine, serotonin and multiple neuropeptides (Desai and Horvitz, 1989; Lickteig et al., 2001; Nathoo et al., 2001; Kim and Li, 2004). It is likely that the activity of the egg-laying circuit is initiated by the HSNs, because they exhibit spontaneous, rhythmic calcium oscillations that persist even when all synaptic inputs to the HSN are severed (Zhang et al., 2008). The molecular basis for this spontaneous activity and its modulation by external cues is not understood.
Here we elucidate the role of clh-3-encoded channels in the HSNs. We identify a gain-of-function mutation in clh-3 that activates the channels by altering their voltage dependence and inhibits egg laying by suppressing HSN activity. Conversely, loss of clh-3 leads to hyperactive egg laying, indicating that clh-3-encoded channels modulate the activity of the HSNs. Optogenetic and genetic epistasis experiments suggest that clh-3-encoded channels reduce HSN excitability directly by mediating chloride influx.
Materials and Methods
C. elegans strains.
Worm strains were maintained between 20 and 22°C on nematode growth medium (NGM) plates seeded with the OP50 strain of Escherichia coli. The N2 Bristol strain of C. elegans was used as the wild-type reference. The following mutations were used: abts-1(ok1566) I, egl-42(n995), egl-42(n996) II, clh-3(ok763) II, clh-3(ok768) II, kcc-2(vs132) IV, and egl-1(n986 and n487) V. The integrated arrays ljIs19 [pcat-1::YC2.1] X and wzIs6 [pegl-6::ChR2::YFP] X were crossed into the mutants for calcium imaging and channel rhodopsin experiments, respectively. Hermaphrodites were used for all experiments.
Plasmids and transgenic strains.
For overexpression of wild-type clh-3, the clh-3 gene including the 727 bp region upstream of the start codon of clh-3a was amplified from N2 genomic DNA in two overlapping fragments. The two fragments were injected into N2 (each at 15 ng/μl), along with the coinjection marker pmyo-2::gfp (at 5 ng/μl) and 1 kb ladder (at 125 ng/μl) to create the array ljEx479. For overexpression of mutant clh-3, the same procedure was used except that clh-3 gene was amplified from genomic DNA isolated from egl-42(n995) mutants to create the array ljEx480. As a control, only the coinjection marker pmyo-2::gfp (at 5 ng/μl) and 1 kb ladder (at 125 ng/μl) were injected to create the array ljEx476.
For expression of wild-type and mutant clh-3a and clh-3b cDNA isoforms, constructs were created using the Multisite Gateway Three Fragment Vector Construction Kit (Invitrogen). cDNAs were cloned into pDONR221 and recombined with P4-P1R pENTRY clones containing either the 3.3 kb clh-3 promoter region (described in Nehrke et al., 2000) or the 3.5 kb egl-6 promoter region (described in Emtage et al., 2012), both of which are strongly expressed in the HSN, and a P2R-P3 pENTRY clone containing the unc-54 3′-UTR. The clh-3b(stop n995) constructs are missing the g at position 98 of the cDNA, which introduces a stop codon and shifts the reading frame. These constructs were generated using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) and confirmed by sequencing. All cDNA constructs were injected into N2 (at 15 ng/μl), along with the coinjection marker pmyo-2::gfp (at 5 ng/μl) and 1 kb ladder (at 130 ng/μl) to generate arrays ljEx620, ljEx621, ljEx622, ljEx623 and ljEx624 for pclh-3::clh-3a, pclh-3::clh-3a(n995), pclh-3::clh-3b, pclh-3::clh-3b(n995), and pclh-3::clh-3b(stop n995), respectively, and ljEx625, ljEx626, ljEx627, ljEx628, and ljEx629 for pegl-6::clh-3a, pegl-6::clh-3a(n995), pegl-6::clh-3b, pegl-6::clh-3b(n995), and pegl-6::clh-3b(stop n995), respectively.
Behavioral assays.
Egg-laying assays were performed essentially as described previously (Hart, 2006). For most assays, worms were picked at the early L4 stage, left at 20°C, and assayed 39 h afterward. Egg-retention and drug assays were performed in 96-well, flat-bottom plates. For egg-retention assays, single worms were dissolved in 50 μl of a 20% NaOCl solution in a well and the number of eggs that had been retained per worm was counted. For drug assays, single worms were allowed to lay eggs for 1 h in 50 μl of M9 buffer, serotonin creatinine sulfate complex (at 7.5 mg/ml in M9 buffer), fluoxetine hydrochloride (at 0.5 mg/ml in M9), or imipramine (at 0.75 mg/ml in M9) and the number of eggs laid per worm per hour was determined. All genotypes were assayed on at least three different days. For determining egg-laying rates on and off food, worms were placed singly onto plates with or without food and left to lay eggs for an hour. The number of eggs laid per worm per hour was determined. For determining the stage of eggs laid, 30 worms were transferred to a plate and left to lay eggs for 30 min. The stages of the eggs laid were determined immediately afterward. The experiment was repeated until >100 eggs were examined per genotype. Muscimol assays were performed as described previously (Tanis et al., 2009). At least five pools of five animals were tested for each genotype on plates made with two different batches of muscimol.
Tracking assays.
Worms were picked at the L4 stage, left at 22°C, and assayed 16–18 h afterward. Tracking was performed on 35 mm, low-peptone NGM plates that had been freshly seeded with 20 μl of OP50 using Worm Tracker 2.0 (Yemini et al., 2013). The videos were captured at 5 fps and each worm was tracked for 6 consecutive hours. The frames during which egg-laying events took place were determined manually and the intervals between egg-laying events were analyzed as described previously (Waggoner et al., 1998).
Calcium imaging.
HSN imaging was performed essentially as described previously (Zhang et al., 2008) using the integrated Cameleon ljIs19 [pcat-1::YC2.1] X with a few modifications. Briefly, adult worms were immobilized with 2-octyl cyanoacrylate glue (either Nexaband S/C veterinary glue or Dermabond topical skin adhesive) on an agarose pad (2% agarose in 10 mm HEPES, pH 7.1, 13 mOsm) and covered with the same HEPES buffer. A Zeiss Axioskop 2 upright microscope equipped with a Hamamatsu Orca R2 CCD camera, a Hamamatsu W-View emission image splitter, and a Uniblitz Shutter (Vincent Associates) was used to acquire the images with Metavue 7.6 (Molecular Devices). YFP and CFP fluorescence intensities were determined using the custom software Jmalyze (Shyn et al., 2003) and calcium spikes were detected and analyzed using the peak-finding software MiniAnalysis (Synaptosoft).
Channel rhodopsin experiments.
Worms were transferred to retinal plates at the L4 stage, left at 20°C, and assayed 24 h later as adults. Retinal plates were made by seeding NGM plates with OP50 mixed with all-trans retinal. Then, 1000 μl of OP50 culture was mixed with 4 μl of a 100 mm all-trans retinal stock (in EtOH) and plates were seeded with 160 μl. Control plates were made by substituting the all-trans retinal with EtOH. To stimulate the HSNs, worms expressing wzIs6 [pegl-6::ChR2::YFP] were illuminated for 5 s with 1 mW/mm2 blue light (440–460 nm) using LEDS (Luxeon III LXHL-PR09; Lumileds) controlled by a Mindstorms LEGO Intelligent NXT Brick.
Transfection and whole-cell patch-clamp recording of HEK293 cells.
Human embryonic kidney (HEK293) cells were cultured in 35-mm-diameter tissue culture plates in Eagle's minimal essential medium (MEM; Invitrogen) containing 10% fetal bovine serum (Hyclone Laboratories), nonessential amino acids, sodium pyruvate, 50 U/ml penicillin, and 50 μg/ml streptomycin. After reaching 40–50% confluency, cells were transfected using FuGENE 6 (Roche) with 1.0 μg of GFP, 1.5 μg of CLH-3a or CLH-3b, and 1.5 μg of functional or kinase dead GCK-3 ligated into pcDNA3.1. Point mutations were generated using a QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies). All mutations were confirmed by DNA sequencing.
After transfection, cells were incubated at 37°C for 24–30 h. Approximately 2 h before patch-clamp experiments, cells were detached from growth plates by exposure to 0.25% trypsin containing 1 mm EDTA (Invitrogen) for 45 s. Detached cells were suspended in MEM, pelleted by centrifugation, resuspended in fresh MEM, and then plated onto poly-l-lysine-coated coverslips. Plated coverslips were placed in a bath chamber mounted onto the stage of an inverted microscope. Cells were visualized by fluorescence and differential interference contrast (DIC) microscopy.
Transfected HEK293 cells were identified by GFP fluorescence and patch clamped using a bath solution containing the following (in mm): 90 NMDG-Cl, 5 MgSO4, 1 CaCl2, 12 HEPES free acid (titrated to pH 7.0 with CsOH), 8 Tris, 5 glucose, 95 sucrose, and 2 glutamine (pH 7.4, 310 mOsm), and a pipette solution containing the following (in mm): 116 NMDG-Cl, 2 MgSO4 20 HEPES, 6 CsOH, 1 EGTA, 2 ATP, 0.5 GTP, and 10 sucrose (pH 7.2, 275 mOsm). Cells were swollen by exposure to a hypotonic (250 mOsm) bath solution that contained no added sucrose. Experimental protocols were performed on at least two independently transfected groups of cells. Whole-cell currents are reported relative to whole-cell capacitance to correct for differences in cell membrane area.
Patch electrodes were pulled from 1.5 mm outer diameter silanized borosilicate microhematocrit tubes; electrode resistance ranged from 4 to 8 MΩ. Currents were measured with an Axopatch 200B (Molecular Devices) patch-clamp amplifier. Electrical connections to the patch-clamp amplifier were made using Ag/AgCl wires and 3M KCl/agar bridges. Data acquisition and analysis were performed using pClamp 8 software (Molecular Devices). HEK cells were visualized continuously during patch-clamp experiments by video-enhanced DIC microscopy. Cells that exhibited spontaneous swelling or shrinkage were discarded.
Quantification of CLH-3a and CLH-3b current properties.
Because CLH-3a and CLH-3b channels are inwardly rectifying and active only at hyperpolarized voltages, we used current-to-voltage plots (Figs. 3B, 4B,C) to estimate channel activation voltage, as we have described previously (Denton et al., 2004; Falin et al., 2009). The kinetics of hyperpolarization-induced activation of CLH-3a and CLH-3b are complex and vary with experimental conditions (Denton et al., 2005; He et al., 2006). To simplify presentation and interpretation of activation kinetics, time constants are not used. Instead, the time required for the whole-cell current to reach 50% activation when the membrane voltage is stepped from 0 mV to −100 mV for 1 s is quantified. This time is defined as the 50% rise time.
Figure 3.
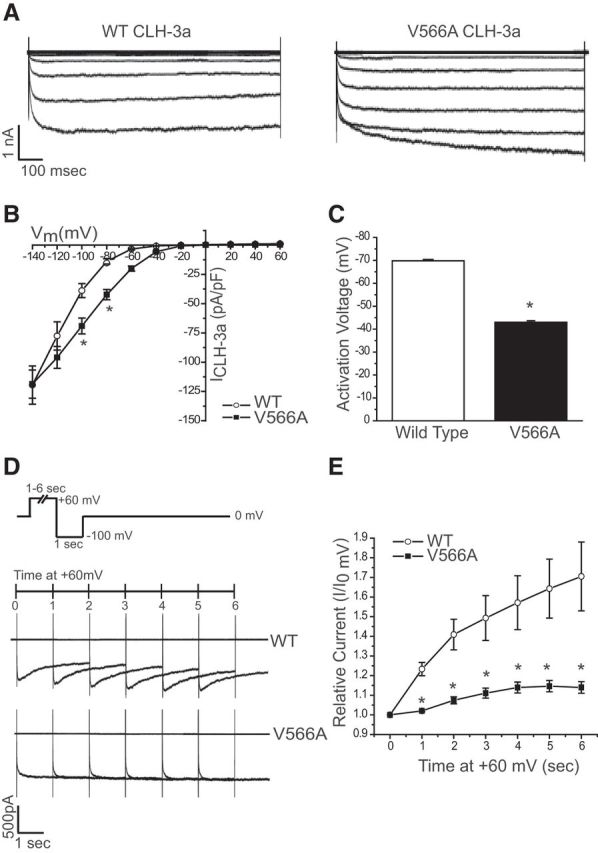
Functional properties of wild-type and V566A CLH-3a channels. A, Representative current traces. B, Current-to-voltage relationships. *p < 0.01 compared with wild-type CLH-3a. Statistical analyses were performed only at voltages more hyperpolarized than the activation voltage of wild-type channels. C, Activation voltages. *p < 0.0001 compared with wild-type CLH-3a. D, Examples of whole-cell current traces of wild-type and V566A CLH-3a channels. Membrane potential was held at 0 mV and then stepped to +60 mV for 1–6 s before currents were activated by a 1 s hyperpolarizing step in membrane voltage to −100 mV. Cells were at 0 mV for 10 s after each hyperpolarizing voltage step. E, Time dependency of pre-depolarization at +60 mV on peak wild-type and V566A current amplitude at −100 mV. Peak current amplitude was measured between 100 and 200 ms and between 850 and 950 ms after onset of the −100 mV test pulse in wild-type CLH-3a and the V566A mutant, respectively. *p < 0.01 compared with wild-type CLH-3a. Values shown in B, C, and E are means ± SEM. n = 5–8.
Figure 4.
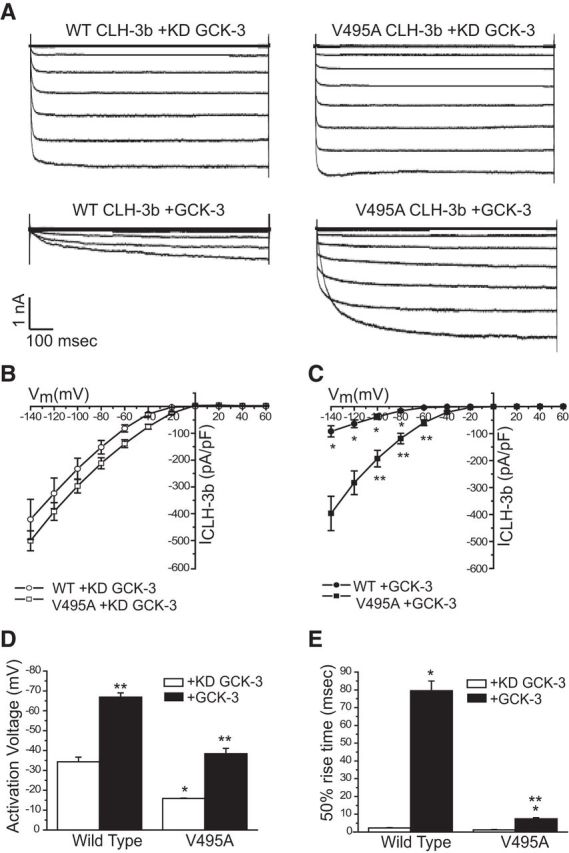
Functional properties of wild-type and V495A CLH-3b coexpressed with either functional or kinase dead (KD) GCK-3. A, Representative current traces. B, C, Current-to-voltage relationships. *p < 0.001 and **p < 0.01 compared with channels coexpressed with KD GCK-3. Statistical analyses were performed only at voltages more hyperpolarized than the activation voltage of channels coexpressed with GCK-3. D, Activation voltages. *p < 0.001 compared with wild-type CLH-3b coexpressed with KD GCK-3; **p < 0.0001 compared with channels expressed with KD GCK-3. E, The 50% rise times, defined as the time required for whole-cell current to reach 50% activation when membrane voltage is stepped from 0 mV to −100 mV for 1 s. *p < 0.001 compared with channels coexpressed with KD GCK-3; **p < 0.00001 compared with wild-type channels coexpressed with GCK-3. Values shown in B–E are means ± SE. n = 4–6.
Statistical analyses.
All statistical analyses were performed using GraphPad Prism, except for permutation tests, which were performed using MATLAB (MathWorks). ANOVA with Dunnett's multiple-comparison test was used to analyze the data presented in Figures 1, D–F, 2, A–C, and 8B. The Mann–Whitney test was used in Figure 5, B and C. The χ2 test was used in Figures 2D and 8C. The Student's t test was used in Figures 3 and 4. A one-tailed permutation test with 100,000–1,000,000 permutations was used to analyze the data presented in Figures 6 and 7A.
Figure 1.
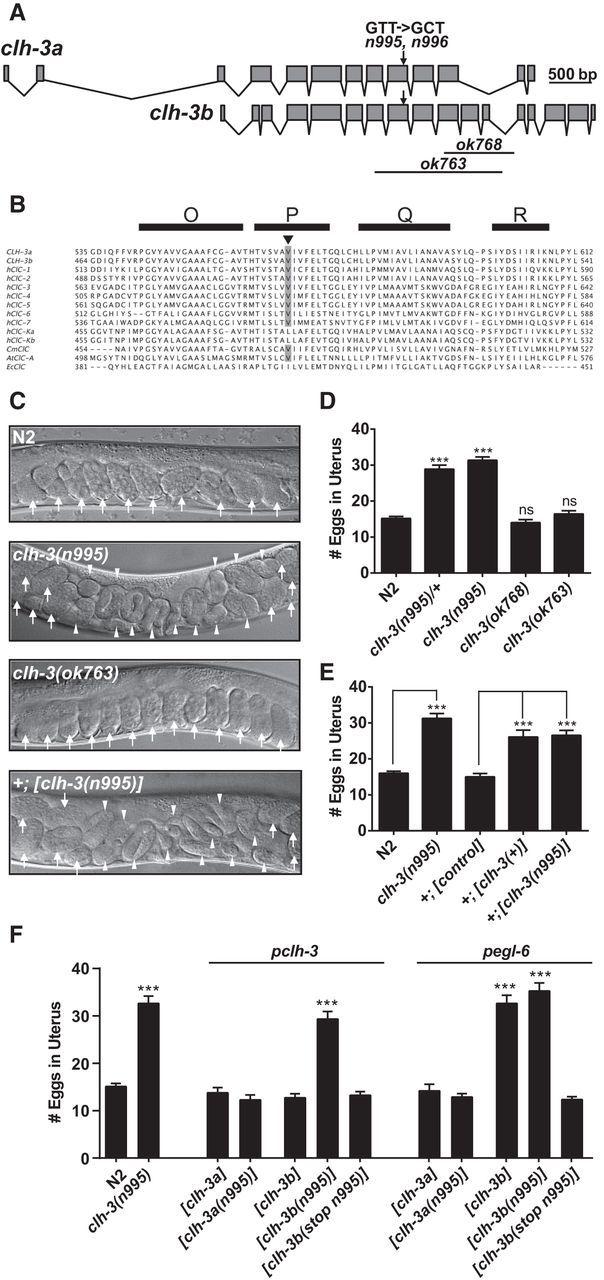
The n995 mutation is a gain-of-function mutation in clh-3. A, Gene structure of the clh-3 locus. clh-3 encodes two isoforms; the a isoform is shown on top and the b isoform on bottom. The n995/n996 mutation is a T to C substitution resulting in a V566A substitution in CLH-3a and a V495A substitution in CLH-3b. B, Alignment of membrane embedded α-helices O–R of CLH-3a and CLH-3b with human (h), Cyanidioschyzon merolae (Cm), Arabidopsis thaliana (At), and Escherichia coli (Ec) CLC proteins. The n995 mutation substitutes an alanine for a highly conserved valine residue (highlighted) in the P-helix. C, DIC images of young adult worms all at the same age. Arrows indicate embryos; arrowheads indicate late-stage embryos (>400 cells). clh-3(n995) mutants and wild-type animals overexpressing clh-3(n995) engage in egg laying less frequently than the wild-type, which leads to the accumulation of twice as many eggs as the wild-type and clh-3(ok763) mutants, many of which reach >400 cells before they are laid. D–F, Quantification of number of eggs retained in the uterus of adults. Bars represent the means ± SEM. ***p < 0.001 compared with N2 or as indicated; n>36 for each genotype or as indicated. D, clh-3(n995) is a dominant mutation that leads to the accumulation of eggs. E, Overexpression of both wild-type and mutant clh-3 leads to the accumulation of eggs. F, Expression of the clh-3b isoform, but not the clh-3a isoform, in the HSN leads to the accumulation of eggs. Introducing a stop codon early in the clh-3b coding sequence abrogates the effect, indicating that it is dependent on production of the CLH-3b protein. More than 10 independent transgenic lines were examined for each construct and one representative line is shown. n = > 24 for each transgenic line.
Figure 2.
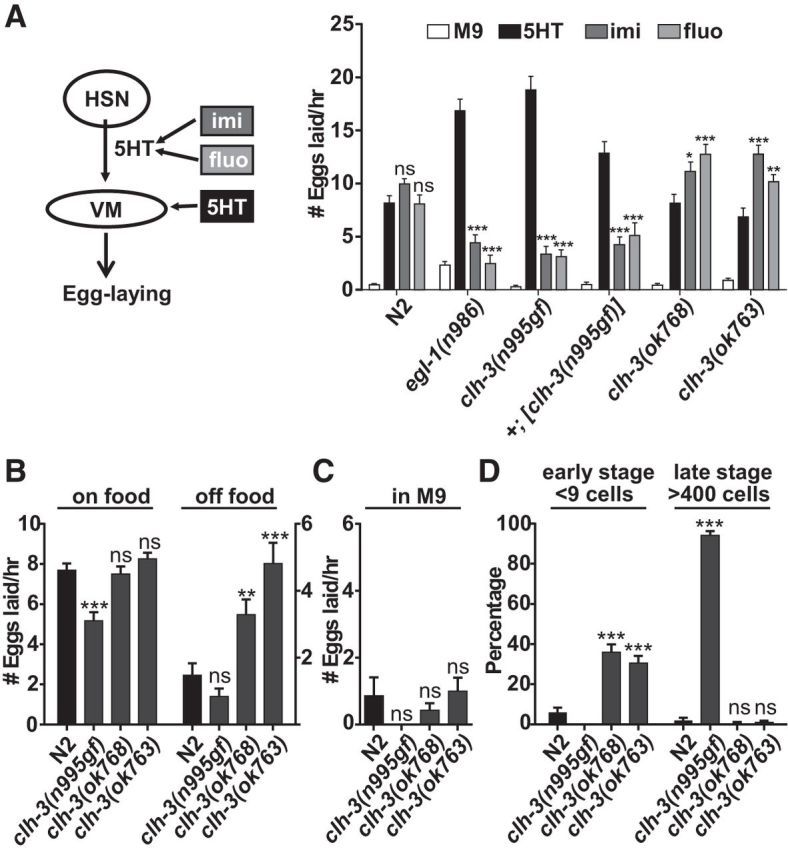
CLH-3 regulates egg laying by regulating HSN function. A, Pharmacology assay for HSN function. The HSNs release serotonin (5HT), which directly stimulates the vulval muscles (VM) and promotes egg laying. In the wild-type, egg laying can be promoted by exogenous 5HT as well as imipramine (imi) and fluoxetine (fluo), which inhibit the reuptake of 5HT. In mutants lacking the HSN or HSN function, egg laying can only be promoted by exogenous 5HT (Trent et al., 1983). In clh-3(n995gf) mutants and wild-type worms overexpressing clh-3(n995gf), egg laying can be robustly stimulated by exogenous 5HT, but significantly less so by imipramine or fluoxetine, indicating a defect in HSN function. clh-3(ok763 and ok768) mutants lay more eggs in the presence of imipramine or fluoxetine than 5HT. Bars represent the means ± SEM. n = > 36 for each genotype and treatment. For statistical analyses, the effects of fluo and imi treatment are compared with the effect of 5HT treatment on that genotype. B, Comparison of egg-laying rates in the presence and absence of food. In the wild-type, egg laying is inhibited in the absence of food. Although clh-3(n995gf) mutants lay fewer eggs than the wild-type in the presence of food, clh-3(ok763 and ok768) mutants lay more eggs than the wild-type in the absence of food. Bars represent the means ± SEM. n = > 15 for each genotype on food and n = > 36 off food. For statistical analyses, mutants are compared with the wild-type under the same condition. C, Comparison of egg laying in M9 buffer. In the wild-type, egg laying is inhibited by high osmolarity. Egg laying is similarly inhibited in clh-3 mutants. Bars represent the means ± SEM. n = > 22 for each genotype. For statistical analyses, mutants are compared with the wild-type. D, Quantification of eggs laid at early stages (at less than the nine-cell stage) and at late stages (at more than the 400-cell stage). clh-3(n995gf) mutants lay fewer eggs than the wild-type at early stages and more eggs than the wild-type at late stages; clh-3(ok763 and ok768) mutants lay more eggs than the wild-type at early stages. Bars represent the proportion ± SE of the proportion. n = > 100 eggs examined for each genotype. For all comparisons, *p < 0.05, **p < 0.001, and ***p < 0.001.
Figure 8.
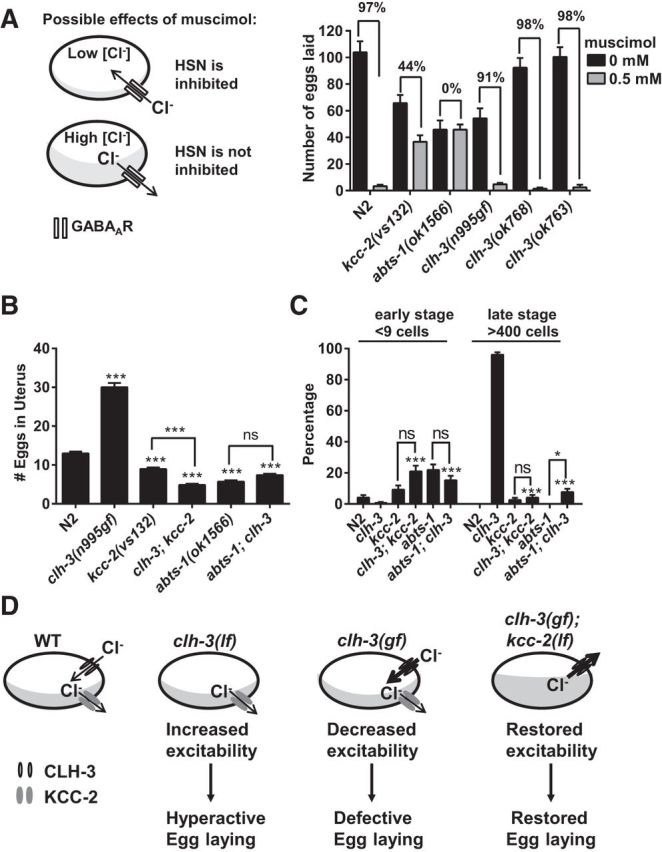
CLH-3 is not required for chloride efflux and mediates chloride influx. A, The effect of GABAAR activation can vary from inhibitory to excitatory. When intracellular levels of chloride are low, GABAAR activation leads to chloride influx, which inhibits via hyperpolarization or shunting inhibition; when intracellular levels of chloride are high, GABAAR activation leads to chloride efflux, which can become excitatory. In C. elegans, the GABAAR agonist muscimol can be used to determine if intracellular chloride levels are altered in the HSN (Tanis et al., 2009). In the wild-type, muscimol inhibits the HSN via activation of the GABAAR UNC-49. Mutants defective in chloride extrusion have elevated intracellular chloride levels and are resistant to the inhibitory effects of muscimol. In contrast to the chloride-extruding transporters kcc-2 and abts-1, clh-3 mutants are not resistant to muscimol, indicating that they are not required for chloride efflux from the HSN. Bars represent the means ± SEM of > 5 experiments per genotype. B, C, Genetic interactions between clh-3(n995) and chloride-extruding transporters kcc-2 and abts-1. B, kcc-2 and abts-1 mutants retain fewer eggs than the wild-type. These mutations are epistatic to clh-3(n995), which leads to an accumulation of more eggs than the wild-type. Bars represent means ± SEM. n = > 36 for each genotype. For statistical analyses, single mutants are compared with wild-type and the double mutants are compared with clh-3(n995) unless otherwise indicated. C, Quantification of eggs laid at early stages (at less than the nine-cell stage) and at late stages (at more than the 400-cell stage). kcc-2 and abts-1 mutations completely suppress the egg-laying phenotypes of clh-3(n995) mutants such that the double mutants are now indistinguishable from the single mutants, with the exception of abts-1; clh-3, which lays slightly more late stage eggs than abts-1. Bars represent the proportion ± SE. n = > 120 eggs examined for each genotype. For statistical analyses, the single mutants are compared with wild-type (for all comparisons, the single mutants were indistinguishable from wild-type) and the double mutants are compared with clh-3(n995) unless otherwise indicated. For all comparisons, *p < 0.05 and ***p < 0.001. D, Model for the role of CLH-3 channels. CLH-3 normally participates in a background conductance that mediates chloride influx in the HSN. In the clh-3(lf) mutants, this conductance is absent, which makes the HSNs more excitable and leads to a hyperactive egg-laying phenotype. In the clh-3(gf) mutants, the activity of the CLH-3 channel is altered in such a way that it leads to an increase in the chloride current through the channel, which inhibits the excitability of the HSN and leads to an egg-laying-defective phenotype. Mutation of the chloride extruders kcc-2 and abts-1 increases [Cl]i, reverses the driving force for chloride flow through the channel, reverses the effects of increasing the activity of the channel, and thereby restores egg laying. This indicates that the driving force normally promotes chloride influx through the CLH-3 channels.
Figure 5.
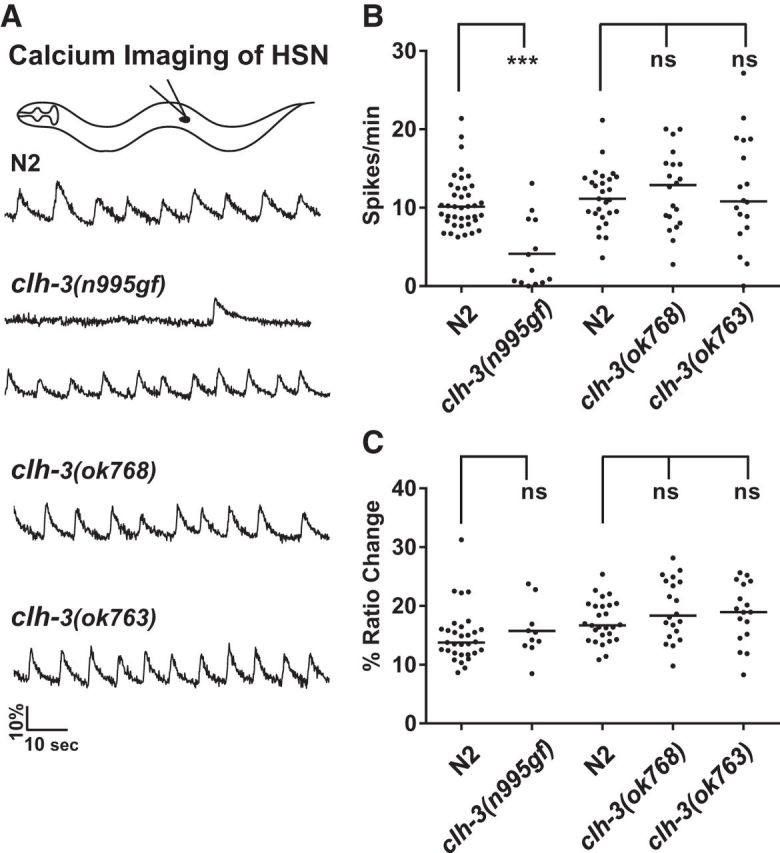
Calcium imaging reveals that CLH-3 inhibits HSN activity. A, Sample traces. The HSNs exhibit spontaneous rhythmic activity (Zhang et al., 2008). Most clh-3(n995gf) mutants have greatly reduced HSN activity, whereas some retain wild-type levels. B, C, Quantification of calcium spike parameters. The average number of spikes/minute and average percentage ratio change of each spike was determined for each recording. Each point represents the value obtained from one recording obtained from one animal. n = > 13 recordings, which corresponds to 145 spikes for clh-3(n995) and > 750 spikes for the other genotypes. Mutants were compared with the wild-type assayed at the same time. The clh-3(n995gf) mutation significantly reduces the spike frequency. None of the mutations alter spike parameters such as ratio change (as shown), area of spike, rise time, or decay time (data not shown).
Figure 6.
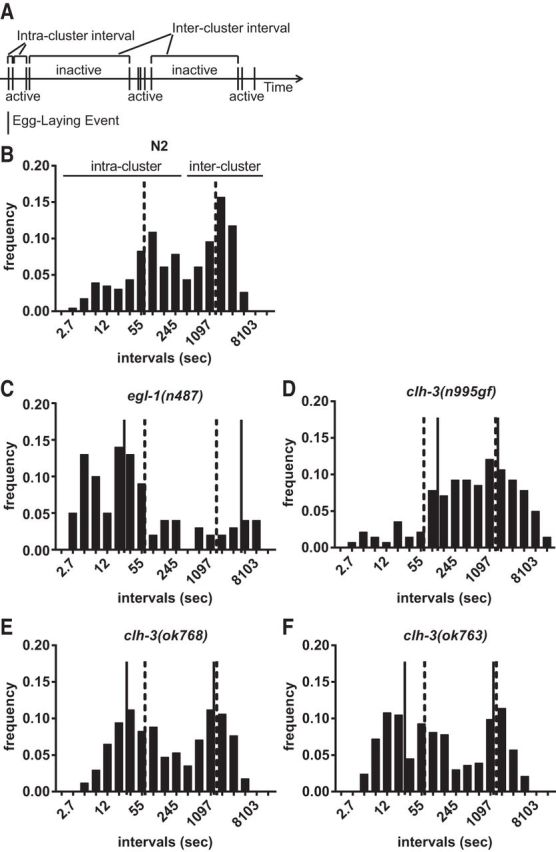
CLH-3 regulates the temporal pattern of egg laying. A, Schematic representation of the temporal pattern of egg laying. Animals alternate between active periods during which eggs are laid and inactive periods during which no eggs are laid (Waggoner et al., 1998). B–F, Histograms of intervals between egg-laying events. Intervals are plotted on a natural log scale on the x-axis and relative frequencies are plotted on the y-axis. The graphs show a bimodal distribution, with the first peak corresponding to the intervals between events in the active periods (the intracluster intervals) and the second peak corresponding to the intervals between active periods (the intercluster intervals). Intervals <400 s were considered intracluster intervals and intervals > 400 s were considered intercluster intervals. Vertical lines indicate median intervals for each peak; dashed lines indicate the median values for the wild-type; solid lines indicate the median values for the mutant in each panel. The egl-1 mutation both shortens the intracluster intervals (p = 0.00048) and lengthens the intercluster intervals (p < 0.000001). clh-3 mutations mainly affect the intracluster intervals: in clh-3(n995gf) mutants, the intervals between egg-laying events in the cluster are increased (p = 0.00142), whereas in clh-3(ok768 and ok763) mutants, the intervals are decreased (p = 0.01663 and p = 0.00062). For the long intervals, the p-values for D–F are p = 0.00093, p = 0.0702, and p = 0.29785, respectively. Sample sizes and other quantifications are shown in Table 1.
Figure 7.
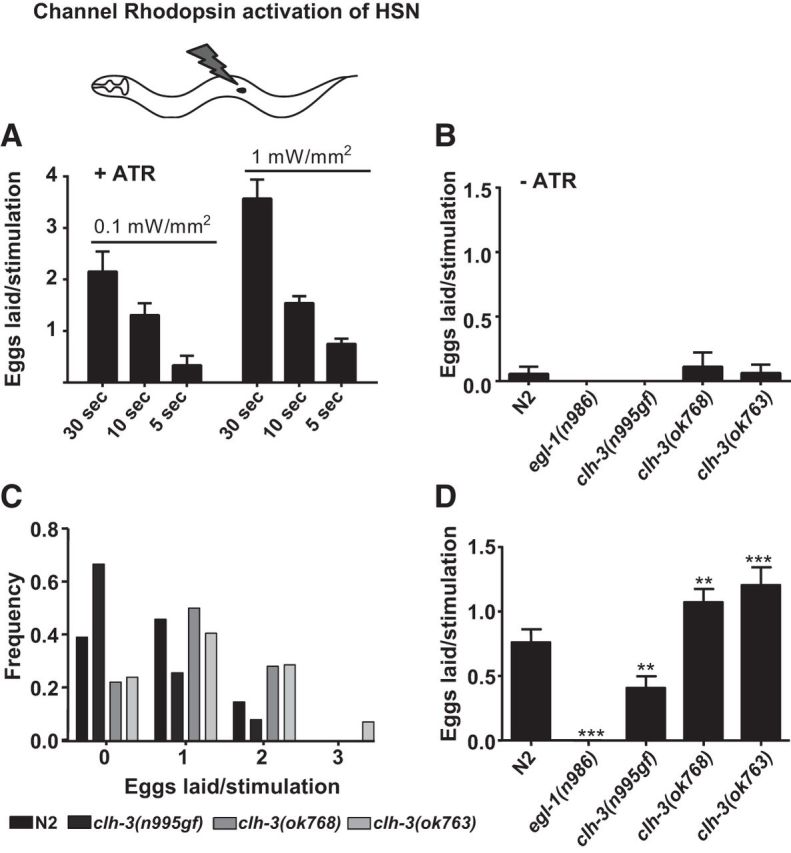
Channel rhodopsin experiments reveal that CLH-3 inhibits HSN excitability. A, The egg-laying response in the wild-type is dependent on both the intensity of the light stimulus and the duration of the light stimulus. Bars represent means ± SEM. B, The response to 5 s, 1 mW/mm2 blue light stimulation is dependent on the presence all-trans retinal, the cofactor for CHR2. Bars represent means ± SEM. More than nine stimulations were tested for each condition and genotype. C, Histogram of eggs laid per 5 s, 1 mW/mm2 blue light stimulation. Number of eggs laid per stimulation is plotted on the x-axis and the relative frequency is plotted on the y-axis. In clh-3(n995gf) mutants, stimulations more frequently do not lead to egg-laying events, whereas in clh-3(ok768 and ok763) mutants, stimulations more frequently do lead to egg-laying events, including the laying of more than one egg. n = > 42 stimulations/genotype. D, Quantification of number of eggs laid/stimulation in the presence of all-trans retinal. Bars represent means ± SEM. n = 18 stimulations for egl-1 and > 42 stimulations for the other genotypes. The response to blue light stimulation requires the HSN because egl-1 mutants do not respond. clh-3(n995gf) mutants lay fewer eggs per stimulation, whereas clh-3(ok768 and ok763) mutants lay more eggs. **p < 0.01; ***p < 0.001.
Results
egl-42(n995) mutants have a mutation in the CLC channel gene clh-3
The dominant n995 mutation was identified in a screen for mutations that disrupt the function of the HSNs (Desai and Horvitz, 1989). Because the main role of the HSNs is to promote egg laying via the release of serotonin, mutants lacking the HSNs or function of the HSNs are egg-laying defective (Egl-D). Whereas wild-type worms have a steady state of ∼14 eggs in the uterus and lay the majority eggs before they reach the 100-cell stage, HSN-defective mutants can retain two to three times as many eggs as the wild-type, some of which develop to more than the 400-cell stage before they are laid (Figs. 1, 2). In addition, HSN-defective mutants lay eggs in response to exogenous serotonin, but, unlike the wild-type, they do not lay eggs in response to drugs that prevent the reuptake of endogenously released serotonin, such as imipramine and fluoxetine (Fig. 2A).
Using a combination of three-point mapping and whole-genome sequencing, we determined that the n995 mutants carry a mutation in clh-3 (Fig. 1A), which encodes the hyperpolarization-activated, inwardly rectifying CLC-type chloride channel splice variants CLH-3a and CLH-3b. CLC channels are homodimers composed of two identical subunits, each containing 18 α-helices (designated A to R; Dutzler et al., 2002; Dutzler et al., 2003; Feng et al., 2010). Each subunit forms an independently gated pore that is opened and closed by movements of a conserved glutamate residue (Dutzler et al., 2003). A poorly understood regulatory mechanism termed “common gating” acts cooperatively on both pores and opens or closes them simultaneously, a process thought to depend on interface conformational changes (Duffield et al., 2003; Cederholm et al., 2010; Ma et al., 2011). The n995 mutation is a T to C substitution that results in a valine to alanine substitution (V566 for CLH-3a and V495 for CLH-3b) in a highly conserved residue in the P-helix (Fig. 1B), which forms part of the subunit interface. We sequenced the clh-3 gene from the other allele of egl-42, n996, and found that it contains the same T to C substitution as the n995 allele.
Because the n995 mutation is dominant (Desai and Horvitz, 1989; Fig. 1D), we could not perform rescue experiments. Instead, to confirm that this substitution causes the Egl-D phenotype, we overexpressed the mutant version of clh-3 in the wild-type. Like the n995 mutation, this leads to an egg-laying-defective phenotype (Fig. 1C,E). It also leads to an HSN-defective phenotype because egg laying in wild-type animals overexpressing mutant clh-3 is stimulated by exogenous serotonin but less robustly by fluoxetine or imipramine (Fig. 2A). Overexpressing wild-type clh-3 also leads to a similar phenotype (Fig. 1E). Together, these observations suggest that the n995 mutation is a gain-of-function (gf), hypermorphic mutation in clh-3. Consistent with the HSN-defective phenotype, clh-3 is expressed in only one neuron, the HSN (Schriever et al., 1999; Nehrke et al., 2000; and data not shown).
To determine which clh-3 isoform may be acting in the HSN, we expressed individually cDNAs encoding either the a or the b isoform of the channel. We expressed the isoforms under the clh-3 promoter, which is specific to the HSN neuron, or the egl-6 promoter, which drives strong expression in the HSN and few other neurons (Ringstad and Horvitz, 2008; Fig. 1F). We observed that only expression of the clh-3b isoform can lead to an Egl-D phenotype. Under its own promoter, expression of only the clh-3b cDNA harboring the n995 mutation confers an Egl-D phenotype, whereas under the egl-6 promoter, expression of both wild-type and mutant clh-3b confers an Egl-D phenotype. We examined >10 independent lines expressing both the wild-type and mutant versions of the clh-3a isoform under each promoter, including at higher concentrations, and none exhibited an Egl-D phenotype. These data strongly suggest that it is the CLH-3b channel that is active in the HSN.
n995gf mutation alters the properties of CLH-3a and CLH-3b and makes them hyperactive
Because the n995gf mutation appears to increase the activity of clh-3 by genetic criteria, we wanted to determine how the n995gf mutation affects the properties of the clh-3-encoded channels. To do this, we introduced the n995gf mutation into the CLH-3a and CLH-3b channel isoforms, expressed the channels in HEK293 cells, and quantified basic current properties (Figs. 3, 4). We first assessed the effect of the n995gf mutation on the CLH-3a splice variant (Fig. 3). The current-to-voltage relationships of wild-type and V566A CLH-3a were similar (Fig. 3A,B). However, the activation voltage of the V566A channel was strongly and significantly (p < 0.0001) depolarized compared with the wild-type (Fig. 3B,C). The mean 50% rise time, a measure of the rate of hyperpolarization-induced channel activation, was also significantly reduced (mean ± SE rise times for the wild-type and mutant channels were 5.8 ± 0.3 ms, n = 6, and 4.0 ± 0.2 ms, n = 8, respectively; p < 0.0002). These data indicate that the V566A mutant channels are more active than wild-type channels at less hyperpolarized membrane potentials.
Because the activity of CLH-3a channels is sensitive to pre-depolarization of the membrane, we assessed the effect of membrane depolarization on wild-type and V566A CLH-3a channels by stepping the membrane potential from 0 mV to +60 mV for 1–6 s, followed by a 1 s pulse to −100 mV to activate the channels (Fig. 3D,E). As shown previously (Denton et al., 2005), wild-type CLH-3a exhibits depolarization-induced potentiation; that is, increasing the time at which voltage is held +60 mV results in greater hyperpolarization-induced channel activation at −100 mV. In contrast, membrane depolarization has little effect on the V566A mutant. Therefore, in addition to being more active at less hyperpolarized membrane potentials, the sensitivity of V566A mutant channels to membrane depolarization is strikingly decreased compared with wild-type CLH-3a channels.
We also assessed the effect of the mutation on the CLH-3b channel splice variant (Fig. 4). CLH-3b is inactivated by the Ste20 kinase GCK-3. GCK-3 reduces basal current amplitude, hyperpolarizes channel activation voltage, and slows voltage-dependent channel activation (Denton et al., 2006; Falin et al., 2009). When the V495A mutant was coexpressed with functional GCK-3, the activation voltage was hyperpolarized ∼2-fold. However, compared with the wild-type, the effects of GCK-3 on V495A current amplitude and 50% rise time were strikingly reduced (Fig. 4). For example, GCK-3 significantly (p < 0.001) inhibited current amplitude of wild-type CLH-3b at all test voltages but had no significant (p > 0.2) effect on V495A current amplitude at −140 and −120 mV and only small effects at other test voltages (Fig. 4B,C). GCK-3 increased the 50% rise time of wild-type CLH-3b nearly 35-fold from 2.3 to 79.5 ms, whereas the 50% rise time of the V495A mutant was increased only ∼6-fold from 1.2 to 7.4 ms (Fig. 4E). These results demonstrate that V495A channels are active at more depolarized membrane potentials, activate more rapidly in response to membrane hyperpolarization, and are less sensitive to GCK-3-mediated inhibitory phosphorylation events. Together, these analyses show that the mutant channels are more active than wild-type channels by several criteria, consistent with the mutation behaving as a gain-of-function hypermorph in vivo. In addition, this analysis reveals that the conserved CLH-3a V495/CLH-3b V566 residue regulates multiple properties of the channel, including its voltage dependence.
clh-3(gf) mutations inhibit the HSN whereas clh-3(lf) mutations hyperactivate the HSN
The hypermorphic mutation and overexpression of clh-3 leads to a defect in egg laying and serotonin release from the HSN, suggesting that clh-3-encoded channels inhibit the HSN. To test this directly, we introduced an integrated transgene that expresses the calcium sensor Cameleon into the HSNs (ljIs19 [pcat-1::YC2.1]; Zhang et al., 2008) and imaged HSN activity (Fig. 5). In the wild-type, under the permissive conditions we used for calcium imaging, the HSNs exhibit spontaneous, rhythmic calcium oscillations (Zhang et al., 2008; Fig. 5A,B). We found that the clh-3(n995gf) mutants have significantly decreased HSN activity, as indicated by a decrease in the average number of calcium spikes/min. Although the majority of the clh-3(n995gf) mutants exhibit reduced activity, some of the mutants did exhibit a wild-type level of activity throughout the recording (Fig. 5A,B). These data indicate that the mutant channels inhibit the rhythmic activity of the HSN upstream of calcium entry, which is consistent with a role for clh-3-encoded channels in inhibiting the HSN.
We next characterized the loss-of-function phenotype of clh-3 by examining how ok768 and ok763, two deletion alleles of clh-3 (Fig. 1A), affect egg-laying behavior. These mutants were not significantly different from the wild-type in terms of the number of eggs they retained (Fig. 1C,D) nor in their egg-laying rate in the presence of abundant food (Fig. 2B). In HSN calcium-imaging experiments, the clh-3(ok763 and ok768) mutants also exhibited spontaneous, rhythmic calcium oscillations at a frequency and magnitude comparable to wild-type (Fig. 5). This was also the case when we imaged HSN activity under more restrictive conditions, such as in the presence of hypertonic concentrations of salt (data not shown). These results suggest that clh-3 may not play a major role in controlling HSN activity under most conditions. However, in more sensitive egg-laying assays, we did observe that the clh-3(ok768 and ok763) mutants had an egg-laying phenotype that was opposite to that of the clh-3(n995gf) mutants. For example, when we examined the developmental stage at which the eggs were laid (which indicates how long the eggs remain in the uterus before being laid and thus serves as a measure of egg-laying rate), we found that the loss-of-function mutants lay significantly more eggs at early stages than the wild-type, whereas the clh-3(n995gf) mutant lays significantly fewer early-stage eggs and significantly more late-stage eggs than the wild-type (Fig. 2D). Similarly, in the drug assays, the clh-3(ok768 and ok763) mutants lay significantly more eggs in response to serotonin reuptake inhibitors than to exogenous serotonin (Fig. 2A), whereas clh-3(n995gf) mutants lay significantly more eggs in response to serotonin than to the reuptake inhibitors. Because the wild-type is similarly stimulated by serotonin and the inhibitors, this suggests that the HSNs are more active in the clh-3(ok768 and ok763) mutants and thereby release more serotonin. Together, these data suggest that the clh-3(gf) mutations inhibit the HSN, whereas clh-3(lf) mutations lead to hyperactivity in the HSN, which is consistent with a role for clh-3-encoded channels in inhibiting the HSN.
CLH-3 is required in the HSNs for normal regulation of egg-laying behavior
The egg-laying behavior is modulated by a number of environmental conditions; in particular, hypertonic concentrations of salt or the absence of food strongly inhibit egg laying in wild-type animals. Like the wild-type, clh-3 mutants showed strong inhibition of egg laying in the hypertonic solution M9 (Fig. 2C). In contrast, the clh-3(ok768 and ok763) mutants lay eggs at high rates in the absence of food, whereas the clh-3(n995gf) mutant laid eggs at low rates even in the presence of food (Fig. 2B). This suggests that clh-3 may modulate HSN activity in response to specific environmental cues, such as the presence of food.
Because the HSNs control the temporal pattern of egg laying, we also examined how the clh-3 mutants affect the timing of the egg-laying behavior. In the presence of a thin bacterial lawn, worms alternate between an active egg-laying state, during which eggs are laid in clusters, and an inactive state, during which eggs are retained (Waggoner et al., 1998; Zhou et al., 1998; Fig. 6A). Both the onset of the active phase and egg laying within the active phase model as Poisson processes, with time constants in wild-type animals of ∼20 min and 1 min, respectively. We found that in the clh-3(lf) mutants, the intracluster time constants were significantly decreased compared with the wild-type, whereas in the clh-3(n995gf) mutant, they were significantly increased (Fig. 6B–D; Table 1). These results suggest that CLH-3 acts in the HSN to inhibit its activity mainly during the active phase of egg laying. Interestingly, the clh-3(n995gf) phenotypes contrast with mutants that completely lack HSN neurons. As described previously (Waggoner et al., 1998), egl-1 mutants have dramatically increased intercluster time constants and decreased intracluster time constants (Fig. 6B; Table 1). This suggests that the clh-3(gf) mutation alters the timing of HSN activity rather than completely inhibiting it.
Table 1.
Estimates of egg-laying parameters
| Hours obs. | Events obs. | Eggs laid/h | p | λ1 | 1/λ1 | λ2 | 1/pλ2 | |
|---|---|---|---|---|---|---|---|---|
| N2 | 54 | 239 | 7.43 | 0.419 | 0.017 | 59.17 | 0.0018 | 1325 |
| clh-3(n995gf) | 60 | 151 | 4.35 | 0.401 | 0.007 | 144.9 | 0.0014 | 1783 |
| clh-3(ok768) | 30.8 | 170 | 9.42 | 0.505 | 0.031 | 32.26 | 0.0017 | 1165 |
| clh-3(ok763) | 54 | 344 | 9.19 | 0.526 | 0.034 | 29.33 | 0.0017 | 1118 |
Analysis of the temporal pattern of egg-laying events obtained from tracking data. The number of hours of video analyzed (hours obs.), number of egg-laying events observed (events obs.), and number of total eggs laid per hour are indicated. As described in Waggoner et al. (1998), p is the probability that, after a given egg-laying event, another egg will be laid before the animal enters the inactive state. λ1 and λ2 are the rate constants for the active and inactive egg-laying states, respectively. 1/λ1 represents the intracluster time constant (in seconds) and 1/pλ2 represents the intercluster time constant (in seconds).
clh-3 regulates HSN excitability
How might wild-type clh-3 inhibit HSN activity? Because clh-3 encodes chloride channels, we reasoned that it might affect HSN activity by affecting HSN excitability. To test this, we crossed the clh-3 mutants with an integrated transgenic line that expresses Channelrhodopsin-2 (ChR2), the blue-light-activated cation channel (Nagel et al., 2005), in the HSNs (wzIs6 [pegl-6::ChR2]; Leifer et al., 2011; Emtage et al., 2012). In wild-type worms, egg laying is robustly stimulated by ChR2 activation (Leifer et al., 2011; Fig. 7). The magnitude of the response, as indicated by both the percentage of stimulations resulting in egg-laying events and the number of eggs laid per stimulation, is dependent on both the strength and duration of the light stimulus (Fig. 7A). The response is also completely dependent on the addition of all-trans retinal, the cofactor for ChR2, to the plates (Fig. 7B), as well as the presence of the HSNs (Fig. 7D). We observed that the clh-3(n995gf) mutant laid significantly fewer eggs per stimulation than the wild-type and blue light stimulation elicited an egg-laying event significantly less frequently in mutant animals than in wild-type. Conversely, the clh-3(ok768 and ok763) mutants laid significantly more eggs than the wild-type and blue light stimulation elicited egg-laying events, including the laying of multiple eggs, more frequently than for the wild-type (Fig. 7C,D). Together, these data support a role for the clh-3-encoded channels in inhibiting HSN excitability: increased channel activity inhibits HSN excitability whereas loss of the channel promotes HSN excitability.
CLH-3-encoded channels regulate HSN excitability directly via an inward chloride current
How might clh-3-encoded channels regulate HSN excitability? One possibility is that the channels affect intracellular chloride concentrations ([Cl−]i) and thereby affect excitability via the GABAAR. The effect of GABAAR activation can vary from excitatory to inhibitory depending on [Cl−]i (Farrant and Kaila, 2007); when intracellular levels of chloride are low, GABAAR activation leads to a hyperpolarizing inward chloride current that is inhibitory; when intracellular levels of chloride are high, GABAAR activation leads to chloride efflux, which can be excitatory (Fig. 8A). It has been suggested that CLC-2 channels might inhibit neuronal excitability indirectly by mediating chloride efflux necessary for GABAAR-mediated inhibition (Staley, 1994; Smith et al., 1995; Rinke et al., 2010). To determine whether the clh-3-encoded channels act in this way, we tested the mutants' response to the GABAAR agonist muscimol. In the wild-type, muscimol inhibits the HSN, indicating that GABAAR activation normally leads to chloride influx in the HSN. When pathways of chloride efflux are lost, such as in kcc-2 and abts-1 mutants, which lack the chloride-extruding K+-Cl− cotransporter (KCC2) and the Na+-driven anion exchanger (NDAE), respectively, the HSNs become resistant to the inhibitory effects of muscimol (Tanis et al., 2009; Bellemer et al., 2011). In contrast to these known Cl−-efflux-defective mutants, clh-3(ok763 and ok768) mutants are as inhibited by muscimol as the wild-type (Fig. 8A). These findings indicate that clh-3 is not required for GABAAR-mediated inhibition and do not support a requirement for these channels in reducing chloride concentrations in the HSN. Therefore, the main mechanism by which clh-3-encoded channels inhibit the HSN is unlikely to be via the GABAAR.
Another possibility is that the channels affect excitability directly by mediating chloride influx (Madison et al., 1986; Rinke et al., 2010; Ratté and Prescott, 2011). If this were the case, we reasoned that if we were to reverse the driving force on which the clh-3-mediated inhibition depends, the inhibitory effects of clh-3 on HSN activity would be suppressed. To test this, we turned again to the mutants of the chloride extruders kcc-2 and abts-1, which presumably have increased intracellular chloride levels, and crossed them with the clh-3(n995gf) mutant. We found that both of these mutations completely suppressed the egg-laying-defective phenotypes of the clh-3(n995gf) mutant (Fig. 8B,C). Whereas the clh-3(n995gf) mutants retain significantly more eggs than the wild-type, the kcc-2 and abts-1 mutants retain significantly fewer eggs. In fact, the kcc-2 and abts-1 mutations are completely epistatic to clh-3(n995gf) and the kcc-2; the clh-3 double mutant even retains significantly fewer eggs than kcc-2. Because this measure of egg laying can be dependent on brood size, which is reduced in these mutants (data not shown), we also looked at the stages of eggs laid by the mutants, a brood-size-independent measure of egg laying. As described previously, the clh-3(n995gf) mutant lays eggs at later stage than the wild-type (Fig. 2D); we found that the kcc-2 and abts-1 mutations completely suppress this phenotype (Fig. 8C). Therefore, increasing [Cl−]i appears to decrease or even reverse the driving force for chloride flow through clh-3-encoded channels, indicating that the driving force normally promotes chloride influx (Fig. 8D). Consistent with this, we also found that the clh-3(n995gf) mutants are partially resistant to low concentrations of muscimol. For example, although egg laying in the wild-type worms is already maximally (98%) inhibited by 10 μm muscimol, the clh-3(n995gf) mutants are only partially (65%) inhibited at this concentration. This supports a role for clh-3 channels in chloride influx, because muscimol resistance is brought about by increasing [Cl−]i. Together, these observations suggest that clh-3-encoded channels regulate HSN excitability directly via an outward current (chloride influx).
Discussion
Here, we have identified a role for clh-3 in the C. elegans nervous system. We have shown that the voltage-dependent chloride channels encoded by clh-3 regulate egg laying in C. elegans by controlling the excitability of the HSN neuron. Loss of the channels increases egg laying, whereas increasing CLH-3b channel activity and channel overexpression decreases HSN activity and inhibits egg laying; therefore, the normal function of the clh-3-encoded channels is to inhibit HSN function. Optogenetic experiments indicate that this occurs through an inhibition of HSN excitability.
Our results suggest a mechanism by which CLC-2/CLH-3a/b channels could affect neuronal excitability. Many previous studies have favored the possibility of an indirect role by which CLC-2 provides a chloride efflux pathway that helps maintain the chloride gradient required for GABAA-receptor-mediated inhibition (Madison et al., 1986; Staley, 1994; Smith et al., 1995; Rinke et al., 2010). However, we have shown clearly here that clh-3 is not required for GABAAR-mediated inhibition of the HSN. Although the HSN is inhibited by the GABAAR agonist muscimol, and this effect requires both the GABAA receptor and the chloride extruders KCC-2 and ABTS-1 (Bellemer et al., 2011), it does not require clh-3-encoded channels (Fig. 8A). Therefore, the CLH-3 channels are not necessary for chloride efflux from the HSN and their effect on HSN excitability is unlikely to be via a modulation of the response to GABAAR activation.
Our data are consistent with a direct role for clh-3 in inhibiting neuronal excitability via chloride influx. We observe that the clh-3(n995gf) phenotypes can be reversed by loss-of-function mutations in kcc-2 and abts-1, which increase [Cl−]i (Fig. 8), indicating that the driving force normally promotes chloride influx via clh-3 channels. This supports a recent modeling study that suggested that, despite its inward rectification properties, CLC-2 is capable of controlling neuronal excitability directly by mediating chloride influx under most physiological conditions (Ratté and Prescott, 2011). Therefore, clh-3-encoded channels could inhibit HSN activity by hyperpolarizing or maintaining the polarized membrane potential. Alternatively, these channels may control the excitability of the HSN by contributing to the background conductance and thereby affecting the membrane resistance of the neuron. Indeed, CLC-2 is active at the resting potential and therefore contributes substantially to the background conductance in hippocampal pyramidal neurons from rats (Madison et al., 1986). Moreover, a study of Clcn2−/− mice showed that CLC-2 also contributes to the background conductance in CA1 pyramidal cells from mice and that loss of this conductance leads to increased input resistance and thereby increases neuronal excitability (Rinke et al., 2010). Similarly, chloride influx mediated by clh-3-encoded channels could contribute to the background conductance of the HSN and thereby modulate its excitability. Therefore, we speculate that although CLC-2/CLH-3a/b channels can mediate chloride efflux under experimental conditions, their main role in vivo may be to limit excitability directly by mediating chloride influx.
The clh-3 mutations have particular effects on the egg-laying behavior that could suggest possible biological roles for this family of channels in the nervous system. Whereas increased channel activity and overexpression leads to strong egg-laying-defective phenotypes (Figs. 1, 2), loss of the channels leads to more specific egg-laying-constitutive phenotypes. For example, the loss-of-function mutants do not exhibit the classic egg-laying-constitutive phenotype of retaining fewer eggs than the wild-type (Fig. 1), and in contrast to egg-laying-constitutive mutants with more general neurotransmission defects (Schafer et al., 1996), egg laying is still inhibited by high salt (Fig. 2C). However, clh-3 loss-of-function mutants lay an increased number of early-stage eggs (Fig. 2D), suggesting that the HSNs are indeed hyperactive. They also exhibit abnormal timing of egg-laying events, because they have a higher frequency of egg-laying events within the active phase (Fig. 6). Although we are unable to image HSN activity under conditions in which switching between active and inactive egg-laying phases could be observed, we infer that clh-3 mutations may alter the activity of the HSN within the active state, resulting in an altered pattern of egg laying within the active phase and the laying of early-stage eggs.
clh-3 is also required for the modulation of egg laying by food. Unlike wild-type animals, which strongly reduce their rate of egg laying in the absence of food, clh-3 loss-of-function mutants lay eggs at high rates both on and off food (Fig. 2B). This suggests that these channels may be required to modulate HSN activity in specific contexts, such as in response to food. Consistent with this idea, a pathway involving the CO2-sensing BAG neurons, neuropeptides encoded by flp-17, a Go-coupled neuropeptide receptor (EGL-6), and an inwardly rectifying potassium channel (IRK-1; Ringstad and Horvitz, 2008; Emtage et al., 2012) also negatively regulates egg laying through the HSN. However, this pathway affects the latency of response to ChR2-mediated activation of the HSN (Emtage et al., 2012), whereas the clh-3-encoded channels affect the magnitude (and not the latency) of the response (Fig. 7 and data not shown). Possibly, the EGL-6/IRK-1 pathway and the clh-3 pathway are required to modulate different properties of the HSN or its activity in response to different cues.
One way the clh-3 pathway could be activated under particular conditions is by phosphorylation of the channels. In oocytes, the activity of CLH-3b is strongly inhibited by phosphorylation by the GCK-3 kinase (Denton et al., 2005) and activated by the GLC-7α/β serine/threonine phosphatases (Rutledge et al., 2002). The activity of CLC-2 is also regulated by phosphorylation (Rutledge et al., 2002) and by PKC (Madison et al., 1986). In the future, identifying the signaling pathways upstream of clh-3 in the HSN might provide a window into the contexts in which this family of channels acts to modulate neural activity.
Why are the HSNs the only neurons in C. elegans that express clh-3? The HSNs are unique among C. elegans neurons in that they exhibit spontaneous rhythmic activity that is generated cell autonomously (Zhang et al., 2008). Although CLH-3 channels are not necessary for this rhythmic activity, strong activation of the channel can suppress it (as in the n995gf mutant). It is possible that these channels contribute to the properties of the neuron that allow for spontaneous rhythmicity, for example, by modulating the membrane conductance. In the future, it would be interesting to investigate whether these channels also modulate spontaneous neural activity in other organisms.
Footnotes
This work was supported by the National Institutes of Health (Grant R01 DK51610 to K.S; Grant DA018341 to W.R.S.); the Medical Research Council (Grant MC-A022-5PB91 to W.R.S. and fellowship to R.B.); and the European Molecular Biology Organization (fellowship to R.B.). Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (Grant P40 OD010440). We thank Alexander Boyanov and Oliver Hobert for whole-genome sequencing data and analysis; Laura Grundy for help with tracking; Victoria Butler for help with assays; Andre Brown for help with statistical analyses; Niels Ringstad for providing the FQ10 egl-6::ChR2 strain; and Jizhe Hao, Marios Chatzigeorgiou, and Jerod Denton for many helpful discussions.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Bellemer A, Hirata T, Romero MF, Koelle MR. Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J. 2011;30:1852–1863. doi: 10.1038/emboj.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederholm JM, Rychkov GY, Bagley CJ, Bretag AH. Inter-subunit communication and fast gate integrity are important for common gating in hClC-1. Int J Biochem Cell Biol. 2010;42:1182–1188. doi: 10.1016/j.biocel.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Denton J, Nehrke K, Rutledge E, Morrison R, Strange K. Alternative splicing of N- and C-termini of a C. elegans ClC channel alters gating and sensitivity to external Cl- and H+ J Physiol. 2004;555:97–114. doi: 10.1113/jphysiol.2003.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton J, Nehrke K, Yin X, Morrison R, Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J Gen Physiol. 2005;125:113–125. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton J, Nehrke K, Yin X, Beld AM, Strange K. Altered gating and regulation of a carboxy-terminal ClC channel mutant expressed in the Caenorhabditis elegans oocyte. Am J Physiol Cell Physiol. 2006;290:C1109–1118. doi: 10.1152/ajpcell.00423.2005. [DOI] [PubMed] [Google Scholar]
- Desai C, Horvitz HR. Caenorhabditis elegans mutants defective in the functioning of the motor neurons responsible for egg laying. Genetics. 1989;121:703–721. doi: 10.1093/genetics/121.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield M, Rychkov G, Bretag A, Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. J Gen Physiol. 2003;121:149–161. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- Emtage L, Aziz-Zaman S, Padovan-Merhar O, Horvitz HR, Fang-Yen C, Ringstad N. IRK-1 potassium channels mediate peptidergic inhibition of Caenorhabditis elegans serotonin neurons via a G(o) signaling pathway. J Neurosci. 2012;32:16285–16295. doi: 10.1523/JNEUROSCI.2667-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falin RA, Morrison R, Ham AJ, Strange K. Identification of regulatory phosphorylation sites in a cell volume- and Ste20 kinase-dependent ClC anion channel. J Gen Physiol. 2009;133:29–42. doi: 10.1085/jgp.200810080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AC. Behavior. WormBook. 2006:1–67. (The C. elegans Research Community ed) [Google Scholar]
- He L, Denton J, Nehrke K, Strange K. Carboxy terminus splice variation alters ClC channel gating and extracellular cysteine reactivity. Biophys J. 2006;90:3570–3581. doi: 10.1529/biophysj.105.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Kim K, Li C. Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. J Comp Neurol. 2004;475:540–550. doi: 10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- Leifer AM, Fang-Yen C, Gershow M, Alkema MJ, Samuel AD. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat Methods. 2011;8:147–152. doi: 10.1038/nmeth.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickteig KM, Duerr JS, Frisby DL, Hall DH, Rand JB, Miller DM., 3rd Regulation of neurotransmitter vesicles by the homeodomain protein UNC-4 and its transcriptional corepressor UNC-37/groucho in Caenorhabditis elegans cholinergic motor neurons. J Neurosci. 2001;21:2001–2014. doi: 10.1523/JNEUROSCI.21-06-02001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Rychkov GY, Bykova EA, Zheng J, Bretag AH. Movement of hClC-1 C-termini during common gating and limits on their cytoplasmic location. Biochem J. 2011;436:415–428. doi: 10.1042/BJ20102153. [DOI] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA. Phorbol esters block a voltage-sensitive chloride current in hippocampal pyramidal cells. Nature. 1986;321:695–697. doi: 10.1038/321695a0. [DOI] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Nathoo AN, Moeller RA, Westlund BA, Hart AC. Identification of neuropeptide-like protein gene families in Caenorhabditis elegans and other species. Proc Natl Acad Sci U S A. 2001;98:14000–14005. doi: 10.1073/pnas.241231298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Begenisich T, Pilato J, Melvin JE. Into ion channel and transporter function. Caenorhabditis elegans ClC-type chloride channels: novel variants and functional expression. Am J Physiol Cell Physiol. 2000;279:C2052–2066. doi: 10.1152/ajpcell.2000.279.6.C2052. [DOI] [PubMed] [Google Scholar]
- Ratté S, Prescott SA. ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci. 2011;31:15838–15843. doi: 10.1523/JNEUROSCI.2748-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Horvitz HR. FMRFamide neuropeptides and acetylcholine synergistically inhibit egg-laying by C. elegans. Nat Neurosci. 2008;11:1168–1176. doi: 10.1038/nn.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke I, Artmann J, Stein V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci. 2010;30:4776–4786. doi: 10.1523/JNEUROSCI.6299-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge E, Bianchi L, Christensen M, Boehmer C, Morrison R, Broslat A, Beld AM, George AL, Greenstein D, Strange K. CLH-3, a ClC-2 anion channel ortholog activated during meiotic maturation in C. elegans oocytes. Curr Biol. 2001;11:161–170. doi: 10.1016/S0960-9822(01)00051-3. [DOI] [PubMed] [Google Scholar]
- Rutledge E, Denton J, Strange K. Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7alpha/beta phosphatases. J Cell Biol. 2002;158:435–444. doi: 10.1083/jcb.200204142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer WR. Egg-laying. WormBook. 2005:1–7. doi: 10.1895/wormbook.1.38.1. (The C. elegans Research Community ed) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer WR, Sanchez BM, Kenyon CJ. Genes affecting sensitivity to serotonin in Caenorhabditis elegans. Genetics. 1996;143:1219–1230. doi: 10.1093/genetics/143.3.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriever AM, Friedrich T, Pusch M, Jentsch TJ. CLC chloride channels in Caenorhabditis elegans. J Biol Chem. 1999;274:34238–34244. doi: 10.1074/jbc.274.48.34238. [DOI] [PubMed] [Google Scholar]
- Shyn SI, Kerr R, Schafer WR. Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr Biol. 2003;13:1910–1915. doi: 10.1016/j.cub.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Smith RL, Clayton GH, Wilcox CL, Escudero KW, Staley KJ. Differential expression of an inwardly rectifying chloride conductance in rat brain neurons: a potential mechanism for cell-specific modulation of postsynaptic inhibition. J Neurosci. 1995;15:4057–4067. doi: 10.1523/JNEUROSCI.15-05-04057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K. The role of an inwardly rectifying chloride conductance in postsynaptic inhibition. J Neurophysiol. 1994;72:273–284. doi: 10.1152/jn.1994.72.1.273. [DOI] [PubMed] [Google Scholar]
- Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci. 2009;29:9943–9954. doi: 10.1523/JNEUROSCI.1989-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner LE, Zhou GT, Schafer RW, Schafer WR. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 1998;21:203–214. doi: 10.1016/S0896-6273(00)80527-9. [DOI] [PubMed] [Google Scholar]
- Yemini E, Jucikas T, Grundy LJ, Brown AE, Schafer WR. A database of Caenorhabditis elegans behavioral phenotypes. Nat Methods. 2013;10:877–879. doi: 10.1038/nmeth.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chung SH, Fang-Yen C, Craig C, Kerr RA, Suzuki H, Samuel AD, Mazur E, Schafer WR. A self-regulating feed-forward circuit controlling C. elegans egg-laying behavior. Curr Biol. 2008;18:1445–1455. doi: 10.1016/j.cub.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou GT, Schafer WR, Schafer RW. A three-state biological point process model and its parameter estimation. IEEE Transactions on Signal Processing. 1998;46:2698–2707. doi: 10.1109/78.720372. [DOI] [Google Scholar]
- Zifarelli G, Pusch M. CLC chloride channels and transporters: a biophysical and physiological perspective. Rev Physiol Biochem Pharmacol. 2007;158:23–76. doi: 10.1007/112_2006_0605. [DOI] [PubMed] [Google Scholar]