

Abstract
The NMDA-type glutamate receptor (NMDAR) is essential for synaptogenesis, synaptic plasticity, and higher cognitive function. Emerging evidence indicates that NMDAR Ca2+ permeability is under the control of cAMP/protein kinase A (PKA) signaling. Whereas the functional impact of PKA on NMDAR-dependent Ca2+ signaling is well established, the molecular target remains unknown. Here we identify serine residue 1166 (Ser1166) in the carboxy-terminal tail of the NMDAR subunit GluN2B to be a direct molecular and functional target of PKA phosphorylation critical to NMDAR-dependent Ca2+ permeation and Ca2+ signaling in spines. Activation of β-adrenergic and D1/D5-dopamine receptors induces Ser1166 phosphorylation. Loss of this single phosphorylation site abolishes PKA-dependent potentiation of NMDAR Ca2+ permeation, synaptic currents, and Ca2+ rises in dendritic spines. We further show that adverse experience in the form of forced swim, but not exposure to fox urine, elicits striking phosphorylation of Ser1166 in vivo, indicating differential impact of different forms of stress. Our data identify a novel molecular and functional target of PKA essential to NMDAR-mediated Ca2+ signaling at synapses and regulated by the emotional response to stress.
Introduction
NMDARs are glutamate-gated ion channels and are enriched at excitatory synapses, where they are strategically positioned to play a crucial role in regulation of synaptic function (Cull-Candy and Leszkiewicz, 2004; Lau and Zukin, 2007; Traynelis et al., 2010). A unique feature of NMDARs is their high permeability to Ca2+. Ca2+ influx through NMDARs is essential for synaptogenesis, plasticity of neural circuitry, and higher cognitive functions, such as learning and memory. NMDARs are tetrameric assemblies of GluN1 and GluN2 subunits, which cotranslationally assemble in the endoplasmic reticulum to form functional channels. In adult hippocampus and neocortex, GluN2A and 2B are the primary GluN2 subunits expressed at synapses (Gray et al., 2011). The precise subunit composition governs the physiological and pharmacological properties of NMDARs. GluN2B-containing receptors exhibit slower decay kinetics (Yashiro and Philpot, 2008), carry more Ca2+ per unit charge (Sobczyk et al., 2005), and preferentially tether to the synaptic plasticity protein CaMKII (Barria and Malinow, 2005; Halt et al., 2012). In addition, GluN2B expression can enhance the magnitude of LTP, promote hippocampal-based learning, plasticity-associated spine growth, and dendritic patterning critical to information processing (Tang et al., 1999; von Engelhardt et al., 2008).
Synaptic NMDARs localize to the postsynaptic density (PSD) where they are structurally organized and spatially restricted in a large macromolecular signaling complex comprised of scaffolding and adaptor proteins, which physically link receptors to kinases, phosphatases, and other downstream signaling proteins (Sheng and Hoogenraad, 2007). Protein kinase A (PKA) is a major kinase known to regulate NMDAR trafficking and gating. At synaptic sites, PKA is kept in close proximity to NMDARs by association of the PKA regulatory subunit with A-kinase anchoring proteins (AKAPs), such as AKAP9 and AKAP5 (Sanderson and Dell'Acqua, 2011).
Emerging evidence reveals that PKA signaling represents a fundamental mechanism by which NMDAR-mediated Ca2+ influx is modulated in neurons. Direct activation of PKA promotes NMDAR Ca2+ permeability, Ca2+ signaling in dendritic spines, and induction of LTP at Schaffer collateral to CA1 pyramidal cell synapses (Skeberdis et al., 2006). Consistent with this, extracellular signals that modulate cAMP bidirectionally regulate Ca2+ permeation through NMDARs and Ca2+ transients in spines in a PKA-dependent manner. Whereas GABAB and dopamine D2 receptors negatively couple to the cAMP/PKA cascade and inhibit NMDAR-mediated Ca2+ signaling in spines, 2A-type adenosine receptors positively engage PKA signaling, enhance NMDAR Ca2+ permeation, and Ca2+ signaling in spines (Chalifoux and Carter, 2010; Higley and Sabatini, 2010). In addition, activation of β-adrenergic receptors enhances NMDA currents (Raman et al., 1996), and activation of D1/D5 dopamine receptors facilitates induction of LTP at CA1 synapses in a PKA-dependent manner (Otmakhova and Lisman, 1996).
Whereas the functional impact of PKA on NMDAR-dependent Ca2+ signaling is well characterized, the molecular target remains unknown. Using a combination of site-directed mutagenesis, electrophysiology, two-photon microscopy, glutamate uncaging, and Ca2+ imaging, we identify Ser1166 in the carboxy-terminal tail of GluN2B as a novel molecular target of PKA phosphorylation critical to Ca2+ signaling in dendritic spines and regulated by the emotional response to stress.
Materials and Methods
Animals.
All animal procedures were in accordance with guidelines approved by the Albert Einstein, Harvard University, and University of California Institutional Animal Care and Use Committee and federal guidelines.
Constructs.
GluN1–1a and GluN2B(WT) in pRK5 were a kind gift from M.D. Ehlers (Duke University, Durham, North Carolina). The S929/930A and S1166A GluN2B mutants were generated using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions and verified by DNA sequencing. The following primers from 5′ to 3′ were used for GluN2B(S929/930A): forward, GACTTCATCCGCCGAGAGGCAGCCGTCTACGACATCTCTG, and reverse, CAGAGATGTCGTAGACGGCTGCCTCTCGGCGGATGAAGTC. The following primers from 5′ to 3′ were used for GluN2B(S1166A): forward, GACTTCAAGCGAGATGCGGTCAGTGGAGGTG, and reverse, CACCTCCACTGACCGCATCTCGCTTGAAGTC.
In vitro phosphorylation assay.
To assess PKA-dependent phosphorylation of NMDARs, HEK293 cells were transfected with WT and mutant GluN2B with Lipofectamine 2000 (Invitrogen) as described previously (Lin et al., 2004). Forty-eight to 72 h after transfection, lysates were immunoprecipitated with an antibody to GluN2B (Millipore), and immunocomplexes were incubated with PKA-C (Sigma). Samples were run on 3–8% Tris-acetate gels (Invitrogen) for subsequent autoradiography and Western blotting (adapted from Leonard and Hell, 1997).
Whole-cell patch-clamp recording.
Primary cultures of hippocampal neurons were generated from embryonic day (E)18 Sprague Dawley rats and transfected at 5–7 days in vitro (DIV) with WT or mutant GluN2B and GFP by means of calcium phosphate as described previously (Jiang et al., 2004). To minimize NMDAR-induced toxicity, cells were maintained in the presence of the NMDAR antagonist 2R-amino-5-phosphonovaleric acid (AP5) (Tocris Bioscience) from the time of transfection until recording. Whole-cell patch-clamp recordings were obtained from hippocampal neurons at 11–15 DIV as described previously (Skeberdis et al., 2006). The external solution contained the following (in mm): 145 NaCl, 2.5 KCl, 2 CaCl2, 10 glucose, 10 HEPES buffer, pH 7.4, 0.5 μm TTX (Ascent Scientific) to block action potentials and 200 nm Zn2+ to preferentially inhibit GluN2A-containing NMDARs (Paoletti et al., 1997; Rodenas-Ruano et al., 2012). Glass electrodes (2–4 mΩ) were filled with internal solution containing the following (in mm): 140 CsMeSO3, 1 EGTA, 0.1 CaCl2, 0.5 MgCl2, 10 HEPES, pH 7.3 with CsOH, 4 Mg-ATP, 0.4 Na2-GTP, and 5 phosphocreatine. Voltage-clamp recordings were performed at a holding potential of −60 mV using an Axopatch 200A amplifier; data were filtered at 2 kHz and digitized at 10 kHz. NMDA currents were elicited by application of NMDA (100 μm + 10 μm glycine) in the presence or absence of PKA-C (Calbiochem, Millipore) (250 nm; see Fig. 1; or in Fig. 2, the PKA inhibitor H-89 (10 μm) (Sigma). Rapid agonist application was achieved by stimulating a single cell repeatedly with agonist under computer control from a multibarrel array fed by gravity, which achieved changes in solutions in <100 ms (Lin et al., 2004). Given the speed of this system, the peak as measured may represent an underestimate of the actual peak. Nevertheless, the comparison of the peak current before and after PKA application (and between WT and S1166 mutant) is likely to be valid. The height of the solution column did not change significantly over 10 applications, thereby eliminating any systematic change in the perfusion rate. Time course data represent averaged values of the peak phase of NMDA-elicited currents at each time point for 5–12 cells per condition; values for cells infused with PKA-C were normalized to the corresponding value for control cells infused with recording solution. Data analysis was performed with Clampfit software.
Figure 1.
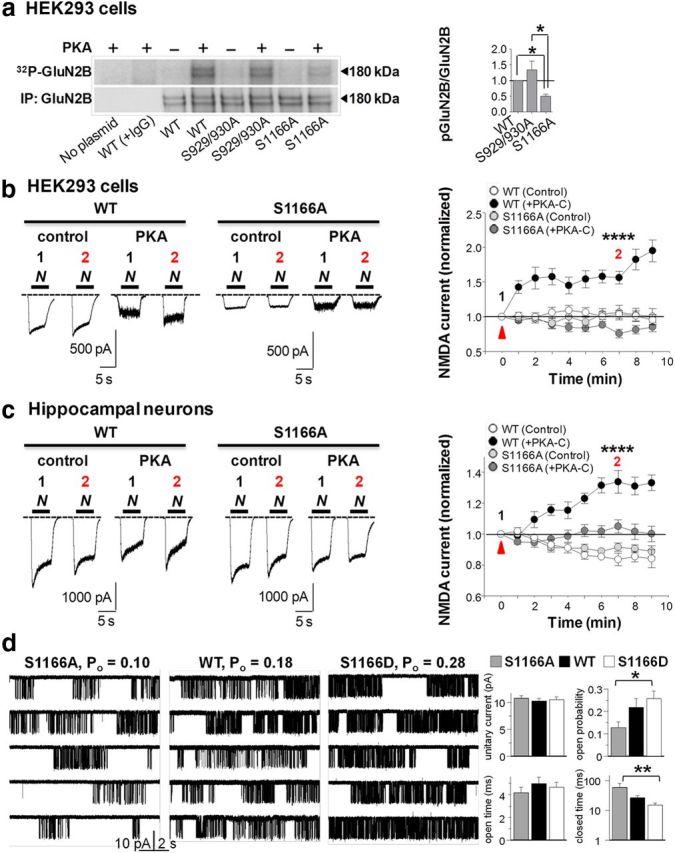
Ser1166 in GluN2B is a direct target of PKA phosphorylation critical to PKA potentiation. a, HEK293 cells expressing WT or mutant GluN2B subunits were immunoprecipitated and incubated with PKA-C in the presence of γ-32P-ATP. Top, Autoradiogram depicting incorporation of γ-32P into GluN2B. Bottom, Western blot probed with an antibody to GluN2B. Right, Summary data. Values for mutant receptors were normalized to values for WT receptors. b, PKA potentiation of NMDA-evoked currents is abolished in HEK293 cells expressing GluN1 with GluN2B(S1166A); bar above sample records represents duration of NMDA+glycine application (N). Left, Sample traces at 0 (1) and 7 (2) min. Right, Summary time course data of the peak NMDAR current, defined as the initial maximum current. Potentiation is defined as the ratio of the peak current recorded from the same cell at 7 min versus 0 min. Values for WT and mutant receptors in the presence (PKA-C) and absence (Control) of PKA-C were normalized to values for WT receptors in the absence of PKA-C (control). WT+PKA-C, n = 9 cells; S1166A+PKA-C, n = 11 cells; WT control, n = 7 cells; S1166A control, n = 6 cells from 4 to 9 independent experiments. c, PKA potentiation of NMDA-evoked currents is abolished in hippocampal neurons expressing GluN2B(S1166A). Left, Representative traces. Right, Summary time course data as in b. WT+PKA-C, n = 5 cells; S1166A+PKA-C, n = 8 cells; WT control, n = 5 cells; S1166A control, n = 5 cells from 3 or 4 independent experiments. d, Far left, Representative traces for individual GluN1/GluN2B(S1166A) (left), WT (center), and S1166D (right) channels. Low gating activity of S1166A relative to that of WT is restored with the phosphomimetic mutation, S1166D. Single-channel activity was recorded with cell-attached pipettes as inward currents through one S1166A, WT, or S1166D channel expressed in a HEK293 cell. Far right, Summary of single-channel parameters for S1166A (gray, n = 14), WT (black, n = 19), and S1166D (white, n = 16). The apparent differences in Po and closed time values for either the S1166A or S1166D mutant versus WT were not significant, consistent with variability in the basal phosphorylation state of WT GluN1/2B receptors. *p < 0.05. **p < 0.01. ****p < 0.0001. Here and in all figures, error bars indicate SEM.
Figure 2.

Ser1166 is critical to modulation of NMDAR Ca2+ permeability and NMDA-evoked Ca2+ rises by PKA. Representative traces (a) and current–voltage relationship (b) for NMDA-elicited currents recorded from HEK293 cells expressing GluN1 with WT and mutant GluN2B(S1166A) in the absence and presence of H-89. H-89 induced a negative shift in reversal potential in cells expressing WT (ΔErev = −6.79 mV), but not GluN2B(S1166A) (ΔErev = 0.46 mV). c, Summary data depicting reversal potential (Erev); WT, n = 6 cells; S1166A, n = 6 cells from 3 independent experiments. *p = 0.0207; n.s., Not significant. d–f, PKA potentiation of NMDA-evoked rises in Ca2+ is diminished in HEK293 cells expressing GluN1 with GluN2B(S1166A). d, Representative images of basal and NMDA-evoked Ca2+ rises before (control), during (H-89), and after washout of H-89 (recovery) for cells expressing GluN1 with WT or S1166A receptors. e, Time course data of cells in d. f, Left, Summary data for NMDA-evoked Ca2+ rises. Right, Summary data expressed as percentage inhibition by H-89; WT, n = 7 cells; S1166A, n = 5 cells from 5 independent experiments. *p = 0.0165. **p < 0.01. Scale bar, 30 μm.
Recording from HEK293 cells was performed at 1–2 d after transfection as described for neurons with the following exceptions: The external solution consisted of the following (in mm): 145 NaCl, 5.4 KCl, 10 HEPES, 11 glucose, and 2 CaCl2, pH 7.4. The internal solution was as follows (in mm): 145 KCl, 5.5 EGTA, 4 Mg-ATP, and 10 HEPES, pH 7.25. Currents were elicited by application of NMDA (300 μm + 10 μm glycine). PKA-C was infused to a final concentration of 100 nm. Cells were washed for 1 min to allow recovery to basal levels before the next stimulation.
Single-channel recording and analyses.
Currents were recorded and analyzed with the cell-attached patch-clamp technique as described previously (Kussius et al., 2009). The pipette solution contained the following (in mm): 150 NaCl, 2.5 KCl, 1 EDTA, and 10 HEPES and NMDAR agonists glutamate (1 mm) and glycine (0.1 mm). The pipette voltage was clamped at +100 mV (membrane potential ∼−120 mV), and inward sodium currents were recorded and amplified (Axopatch 200B), low pass filtered (10 kHz), and sampled (40 kHz, NIDAQ board) into digital files. Only records originating from one-channel patches were retained for analyses. The selected records were idealized with the SKM algorithm in QuB software to calculate average unitary current amplitude, open probability, and durations for open and closed intervals for each record. Statistical significance was evaluated by ANOVA, followed by Tukey's post hoc analysis.
Ca2+ imaging in HEK293 cells.
To image NMDA-evoked Ca2+ signals, cells were loaded with the Ca2+ indicator Fura-2 AM (Invitrogen) in external recording solution or DMEM (1 h at 37°C). NMDA-evoked Ca2+ rises were elicited by application of NMDA (100 μm + 10 μm glycine) 2 min before, during, and 10 min after application of the PKA inhibitor H-89 (10 μm). Cells were washed for 10–15 min to allow Ca2+ levels to return to baseline before each stimulation with NMDA and glycine. The ratio of the Ca2+ signal at 340 to 380 nm was calculated using MetaFluor software.
Slice culture preparation.
Organotypic hippocampal slice cultures were prepared from 6- to 7-d-old mice as described previously (Carter and Sabatini, 2004). Two days later, DNA constructs were biolistically transfected with a Helios Gene Gun. Bullets were loaded with 50 μg of WT GluN2B or GluN2B(S1166A) and tdTomato (30 μg) to assess efficiency of transfection and identify transfected cells.
Two-photon laser glutamate uncaging.
For two-photon laser uncaging experiments, synaptic Ca2+ transients were measured and uncaging-evoked EPSCs (uEPSCs) were simultaneously recorded at individual spines from tdTomato+ CA1 neurons expressing wild-type (WT) or mutant GluN2B(S1166A) before and after glutamate photolysis in organotypic hippocampal slice cultures at 10–14 d after transfection. Photolysis of MNI glutamate was performed by focal illumination with a 0.5 ms pulse of light at 720 nm at ∼0.5 μm from the target spine head. In all conditions, Mg2+-free ACSF (in mm) 127 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 25 glucose included 20 μm NBQX to block AMPAR-mediated currents, 1 μm TTX to block action potentials, and 2.5 mm MNI glutamate to elicit laser-induced glutamate release. Glass electrodes (2–3.5 mΩ) were filled with internal solution containing the following (in mm): 130 CsMeSO4, 10 HEPES, 1.8 MgCl2, 4 Na2ATP, 0.3 NaGTP, and 8 sodium creatine phosphate,10 CsCl2, 300 μm Fluo-5F, 20 μm AlexaFluor-594, pH 7.3. Voltage-clamp recordings were performed at a holding potential of −70 mV using an Axopatch 200A amplifier; data were filtered at 2 kHz and digitized at 10 kHz. Intracellular Ca2+ imaging and glutamate uncaging were performed on a custom microscope, as described previously (Carter and Sabatini, 2004). Neurons were filled via the patch electrode for 15 min before imaging.
For isoproterenol-treated cells, 10 μm isoproterenol was added 5 min after break-in. Fluo-5F (green) and AlexaFluor-594 (red) were excited using 840 nm light to monitor Ca2+ signals and spine morphology, respectively. To measure Ca2+ transients in spines, green and red fluorescence was acquired during 500 Hz line scans across a spine and a neighboring (control) spine. Reference frame scans were taken between acquisitions to correct for spatial drift. Ca2+ signals were quantified as increases in green fluorescence from baseline normalized to the average red fluorescence (ΔG/R) and represented as percentage of the G/R ratio measured in saturating Ca2+ (Gsat/R).
Imaging and physiology data were acquired using a National Instruments data acquisition boards and custom software written in MATLAB as described previously (Pologruto et al., 2003). Off-line analysis was performed using custom routines written in MATLAB and Igor Pro. For each analyzed dendritic spine, the amplitude of NMDAR-mediated uEPSCs was calculated by averaging all uEPSC traces and determining the peak in the 40 ms time window after uncaging. Only spines from cells with Rs < 25 mΩ were considered for analysis. For the same spine population, measurements of ΔG/Gsat were calculated by averaging all Ca2+ transients for each spine and subsequently determining the peak of the average trace by averaging the 20–50 ms after uncaging time window. All graphs and data are expressed as mean ± SEM, and significance was assessed using unpaired, two-tailed Student's t test.
Slice treatments.
Acute hippocampal slices were prepared as described previously (Lu et al., 2007) and treated with vehicle (H2O or EtOH), 1 μm isoproterenol, 1 μm isoproterenol + propranolol for 5 min, 10 μm SKF81297, or 10 μm SKF81297 + 5 μm SCH23390 (0.5–15 min). After treatment, slices were shock frozen and extracted with 1% deoxycholate (DOC) in buffer containing the following (in mm): 50 Tris pH 8.5, 10 EDTA, 10 EGTA, and 135 NaCl, and cleared by ultracentrifugation (Leonard and Hell, 1997). All buffers included the following protease and phosphatase inhibitors: 1.5 μm pepstatin A, 2.1 μm leupeptin, 0.3 μm aprotinin, 0.2 mm PMSF, 1 mm p-nitrophenylphosphate, 25 mm NaPPi, 25 mm NaF, and 2 μm microcystin-LR. Immunoprecipitation for GluN2B and GluA1 was as described previously (Lu et al., 2007). Samples were run on 7.5% polyacrylamide gels followed by quantitative immunoblotting with antibodies against pSer1166-GluN2B (PhosphoSolutions), GluN2B, pSer897-GluN1, GluN1, pSer845-GluA1, and GluA1 as described previously (Lu et al., 2007). Chemiluminescent immunosignals were digitized by scanning films and quantified with Adobe Photoshop.
Fox urine assay.
Postnatal day (P30) rats were placed in a cage containing a 50 ml tube with kimwipes soaked in fox urine (Wildlife Research Supplies) or 0.9% saline for 5 min, and immediately anesthetized with isoflurane and decapitated as described previously (Liu et al., 2010). Hippocampi were isolated and processed for immunoprecipitation with GluN2B and GluA1 antibodies and Western blot analysis as above.
Injections and forced swim assay.
P24-P32 rats were subjected to forced swim in a 4 L beaker chamber filled to 3 L at a temperature of 16–22°C for 5 min. The chamber was cleaned after every one or two rats. One hour prior to forced swim, rats were anesthetized with isoflurane, stereotaxically injected into the hippocampal CA1 with the PKA inhibitor Rp-8-Br-cAMPS (25 nmol, 4 μl) (Enzo Life Sciences) or with saline. The coordinates for injection into the hippocampus were −3 anterior/posterior, −2 medial/lateral, and −4 dorsal/ventral from bregma. Immediately after forced swim, rats were anesthetized with isoflurane, decapitated, and the hippocampi transferred to ice-cold PBS containing protease and phosphatase inhibitor cocktails as above. Hippocampi were microdissected around the injection site, flash-frozen in liquid N2, and stored at −80°C. For Western blot analysis, hippocampi were processed as described above.
Surface biotinylation assay.
To assess surface expression of GluN2B WT and mutant constructs, HEK293 cells were transfected with GluN1 and WT or mutant GluN2B constructs with Lipofectamine 2000 as above. Forty-eight to 72 h after transfection, total and surface extracts were isolated and processed for Western blots as described previously (Lin et al., 2004). Membranes were probed with antibodies directed to GluN1, GluN2B, and β-actin (loading control) and bands quantified as above.
Statistical analyses.
Data are represented as mean ± SEM. All statistical analyses were performed using Prism software (GraphPad). Significance was defined as p < 0.05.
Results
PKA phosphorylates GluN2B at Ser1166
To identify the molecular target of PKA relevant to regulation of NMDAR Ca2+ permeability and Ca2+ signaling in spines, we used NetPhosK and scansite.mit.edu software set at medium stringency algorithms to identify putative PKA consensus sequences in the GluN2B carboxy-tail. The GluN2B carboxy-terminal tail sequence harbors three sites that conform to a PKA consensus phosphorylation motif (K/R-K/R-X-(X)-S/T): Ser929, Ser930, and Ser1166. We used site-directed mutagenesis to mutate these residues singly or together to the nonphosphorylatable residue alanine. To examine the ability of PKA to phosphorylate GluN2B at these sites, we expressed GluN2B(WT), GluN2B(S929/930A), or GluN2B(S1166A) in HEK293 cells and immunoprecipitated cell lysates with an antibody directed to GluN2B. Immunocomplexes were incubated with the recombinant, constitutively active catalytic subunit of PKA (PKA-C) or vehicle in the presence of γ-32P-labeled ATP, and samples were subjected to autoradiography and Western blotting (Fig. 1a). In HEK293 cells, GluN2B subunits migrate as doublets, corresponding to the glycosylated and nonglycosylated forms of the receptor (Petralia et al., 1994). Background phosphorylation (defined as phosphorylation in the absence of PKA-C) of WT and mutant receptor subunits was negligible (Fig. 1a). PKA-C induced striking phosphorylation of WT and mutant GluN2B(S929/930A) receptors (Fig. 1a; Table 1). In contrast, PKA-induced phosphorylation of mutant GluN2B(S1166A) receptors was greatly diminished but not abolished (Fig. 1a; Table 1). The small residual signal may reflect additional sites of PKA phosphorylation on GluN2B, at least under the conditions of this assay. WT and mutant receptors exhibited comparable levels of total GluN2B abundance, assessed by Western blot analysis (Fig. 1a), and surface expression, assessed by surface biotinylation assays (data not illustrated). These results indicate that Ser1166 of GluN2B is a main site of PKA phosphorylation.
Table 1.
| Figure | N | Mean ± SEM | p | Statistical test |
|---|---|---|---|---|
| 1a | 4 experiments | WT: 1.000 ± 0.0 | *, 0.0218 | One-way ANOVA with Dunn's post-test |
| S929/930A: 1.306 ± 0.284 | ||||
| S1166A: 0.4616 ± 0.0802 | ||||
| 1b | WT = 9 cells | WT: 1.584 ± 0.0763 | ****, < 0.0001 | One-way ANOVA with Tukey's post-test (WT vs S1166A, WT vs WT (control), and WT vs S1166A (control)) |
| S1166A = 11 cells | S1166A: 0.9028 ± 0.0234 | |||
| WT (control) = 7 cells | WT (control): 1.025 ± 0.0126 | |||
| S1166A (control) = 6 cells | S1166A (control): 0.9935 ± 0.0152 | |||
| 1c | WT = 5 cells | WT: 1.073 ± 0.013 | ****, < 0.0001 | One-way ANOVA with Tukey's post-test (WT vs S1166A, WT vs WT (control), and WT vs S1166A (control)) |
| S1166A = 8 cells | S1166A: 0.8984 ± 0.0145 | |||
| WT (control) = 5 cells | WT (control): 0.8991 ± 0.0220 | |||
| S1166A (control) = 5 cells | S1166A (control): 0.9249 ± 0.0132 | |||
| 1d | WT = 20 cells | WT: I ,10.4 ± 0.3; Po, 0.22 ± 0.04; MOT, 5.0 ± 0.5; MCT, 27.6 ± 4.5 | Po: *, 0.0317 | One-way ANOVA with Tukey's post-test (Po, S1166A vs S1166D; MCT, S1166A vs S1166D) |
| S1166A = 14 cells | S1166A: I, 10.9 ± 0.4; Po, 0.13 ± 0.02; MOT, 4.2 ± 0.5; MCT, 61.2 ± 19.3 | MCT: **, 0.0097 | ||
| S1166D = 16 cells | S1166D: I, 10.6 ± 0.5; Po, 0.26 ± 0.03; MOT, 4.7 ± 0.4; MCT, 15.7 ± 2.1 | |||
| 2c | WT = 6 cells | WT: ΔErev = −6.79 mV | *, 0.0207 | Two-tailed paired t test for WT vs S1166A |
| S1166A = 6 cells | S1166A: ΔErev = 0.46 mV | |||
| 2f | WT = 7 cells | WT % inhibition: 59.14 ± 7.20% | *, 0.0165 | Two-tailed unpaired t test for % inhibition by H-89 of WT vs S1166A; one-way ANOVA with Dunn's post-test (WT control vs H-89) |
| S1166A = 5 cells | S1166A % inhibition: 20.20 ± 12.58% | **, 0.0030 | ||
| 3e | WT (control) = 23 cells | WT (control): 17.03 ± 1.58 | *, < 0.001 | Two-tailed unpaired t test for WT (control) vs WT (ISO) |
| WT (ISO) = 26 cells | WT (ISO): 29.34 ± 2.21 | |||
| S1166A (control) = 22 cells | S1166A (control): 18.09 ± 1.94 | |||
| S1166A (ISO) = 22 cells | S1166A (ISO): 20.25 ± 1.95 | |||
| 3f | WT (control) = 24 cells | WT (control): −9.677 ± 1.048 | *, < 0.001 | Two-tailed unpaired t test for WT (control) vs WT (ISO) |
| WT (ISO) = 27 cells | WT (ISO): −16.69 ± 1.58 | |||
| S1166A (control) = 22 cells | S1166A (control): −11.15 ± 1.05 | |||
| S1166A (ISO) = 22 cells | S1166A (ISO): −10.50 ± 1.86 |
*p < 0.05;
**p < 0.01;
****p < 0.0001.
PKA potentiation of NMDA-evoked currents is abolished in cells expressing GluN2B(S1166A)
To address whether Ser1166 is a functional target of PKA, we undertook several experimental approaches. First, we examined the impact of Ser1166 on PKA-dependent potentiation of NMDA-elicited currents (300 μm with 10 μm glycine) recorded from HEK293 cells expressing GluN1 together with WT or mutant GluN2B(S1166A) receptors and GFP to enable visualization of transfected cells (Fig. 1b). HEK293 cells are relatively small, geometrically simple, and more nearly isopotential than are neurons and enable examination of NMDAR function in a system expressing a homogeneous population of NMDARs (see Materials and Methods). In these experiments, the same cell was stimulated repeatedly with agonist (once per minute; 5 s per application) for a total of 10 applications under computer control before and after action of PKA-C. PKA-C was delivered directly into the cell via the patch pipette at the time of break-in (t = 0 min). Potentiation was defined as the ratio of peak currents recorded from the same cell at two different time points (7 vs 0 min). In cells expressing WT NMDARs, PKA-C potentiated the peak phase of NMDA-elicited currents by ∼1.5-fold (Fig. 1b; Table 1). In contrast, in cells expressing GluN1/GluN2B(S1166A) receptors, the peak to steady-state ratio of the basal current was markedly reduced, and PKA potentiation was abolished (Fig. 1b; Table 1). The peak phase of NMDA-evoked currents recorded from cells expressing WT or mutant receptors and treated with vehicle was not detectably altered within the time course of the experiment.
Next, we addressed whether Ser1166 is a molecular target of PKA relevant to potentiation of NMDA currents by PKA in primary cultures of hippocampal neurons, a more physiologically relevant system to assess NMDAR function. NMDARs and the machinery required for proper NMDAR targeting and function, such as NMDAR-interacting proteins and AKAPs, are highly expressed in neurons and enriched in dendritic spines (Lau and Zukin, 2007; Sanderson and Dell'Acqua, 2011). Toward this end, we transfected hippocampal neurons at 5–7 DIV with WT or mutant GluN2B(S1166A) together with GFP and performed recording from GFP+ neurons at 11–15 DIV. Thirty minutes before recording, we applied the adenylyl cyclase inhibitor SQ22536 (100 μm) to deplete basal levels of cAMP and reduce endogenous PKA activity. In neurons expressing WT or mutant NMDARs and treated with vehicle, NMDA-evoked responses exhibited rundown (Fig. 1c), consistent with previous findings (Skeberdis et al., 2006). Direct delivery of PKA-C via the patch pipette potentiated the peak phase of NMDA-elicited currents by ∼1.3-fold (Fig. 1c; Table 1). In neurons expressing WT GluN1 with mutant GluN2B(S1166A), PKA potentiation was essentially abolished (Fig. 1c; Table 1), indicating the ability of overexpression of mutant GluN2B to outcompete endogenous GluN2B and GluN2A for assembly with GluN1 and to dominate NMDA-elicited currents.
The results thus far indicate a role for Ser1166 in potentiation of NMDA currents by PKA, but they do not address the mechanism by which this occurs at the single-channel level. Toward this end, we recorded activity in patches containing only a single channel from HEK293 cells expressing GluN1 together with WT or mutant GluN2B receptors. The nonphosphorylatable mutant, S1166A, exhibited an open probability that was markedly reduced (Po, 0.13 ± 0.02, n = 14) relative to that of the phosphomimetic mutation S1166D (0.26 ± 0.03, n = 16, p = 0.0317; Fig. 1d; Table 1). The WT receptor exhibited an open probability (0.22 ± 0.04, n = 20) that was intermediate in value but did not differ statistically from that of S1166A or S1166D (p > 0.05, ANOVA) (Fig. 1d; Table 1), consistent with the substantial variability in GluN2B channel activity noted previously (Amico-Ruvio and Popescu, 2010; Borschel et al., 2012; Maki et al., 2012) and with the concept that WT GluN2B-containing NMDARs are a combination of both phosphorylated and nonphosphorylated receptor species. The large difference in activity between the S1166A and the S1166D mutants is largely attributable to a change in mean closed durations, as neither mean open duration nor unitary current amplitude was altered (Fig. 1d; Table 1). As with Po values, mean closed durations of WT NMDARs (28 ± 5 ms) were intermediate in value but did not differ significantly between that of S1166A (61 ± 19 ms) and S1166D (16 ± 2) mutant receptors (p > 0.05, ANOVA).
Ser1166 is critical to the PKA-dependent shift in NMDAR reversal potential
The results thus far indicate that Ser1166 of GluN2B is critical to PKA potentiation of NMDA-elicited currents, but they do not address a direct role for Ser1166 in regulation of NMDAR Ca2+ permeability by PKA. Toward this end, we undertook two experimental approaches. First, we examined current–voltage relations for HEK293 cells expressing GluN1 together with WT or mutant GluN2B(S1166A) receptors in the absence and presence of H-89. If, indeed, PKA facilitates Ca2+ permeation through NMDARs, inhibitors of PKA should cause a negative shift in the reversal potential (Erev) of the NMDAR current–voltage relationship (I/V curve) (Skeberdis et al., 2006). HEK293 cells are well suited for these experiments because of the relatively small contribution of voltage-gated conductance. NMDA-elicited currents were recorded at holding potentials ranging from −60 to 60 mV, in 20 mV steps. In cells expressing WT NMDARs, the I/V curve was slightly outwardly rectifying and the NMDA reversal potential (Erev) was ∼10 mV (Fig. 2a,b), the known reversal potential for NMDARs. The noise in the responses reflects the stochastic gating of single NMDA channels. Noise levels increased during NMDA application and were greatly reduced at potentials near the NMDA ΔErev. Moreover, the direction of the NMDA-elicited current reversed at the known reversal potential for NMDARs, consistent with an NMDAR-mediated response. Application of H-89 triggered a marked reduction in slope conductance and shifted the Erev of NMDA currents in the negative direction (ΔErev = −6.79 mV; Fig. 2a–c; Table 1), indicative of a decrease in the ratio of Ca2+ to monovalent cation permeability (PCa/Pmonovalent) in corroboration of our earlier work (Skeberdis et al., 2006). In cells expressing GluN2B(S1166A), H-89 produced a much smaller reduction in the slope conductance and did not detectably alter the reversal potential (ΔErev = 0.46 mV; Fig. 2a–c; Table 1). We obtained similar results when we fit only the points closer to the Erev at −20, 0, and 20 mV. These findings document that loss of a single residue abolishes the impact of PKA on NMDAR Ca2+ permeability.
Ser1166 of GluN2B is critical to PKA-dependent potentiation of NMDA-evoked Ca2+ rises
Second, we examined the impact of Ser1166 on PKA-dependent NMDAR-mediated Ca2+ rises. Toward this end, we imaged NMDA-evoked Ca2+ rises in HEK293 cells expressing GluN1 together with WT or mutant GluN2B(S1166A) receptors. Cells were loaded with the Ca2+ indicator Fura-2 AM; and after a brief (15–20 s) stable baseline, Ca2+ transients were elicited by a single application (60 s) of NMDA (100 μm with 10 μm glycine), and images were taken before (control), during (H-89), and after washout (recovery) of H-89. Application of NMDA and glycine elicited a rise in Ca2+ (Fig. 2d,e). In cells expressing WT NMDARs, H-89 markedly inhibited NMDA-evoked Ca2+ rises (Fig. 2d–f; Table 1). After washout of the inhibitor, NMDA-evoked Ca2+ rises exhibited substantial recovery. In cells expressing GluN2B(S1166A), inhibition by H-89 was greatly diminished (Fig. 2d–f; Table 1). Collectively, these findings strongly suggest that Ser1166 is important to regulation of NMDAR Ca2+ signaling by PKA.
Ser1166 is required for regulation of NMDAR-mediated Ca2+ transients in spines
The results thus far indicate that Ser1166 is critical to regulation of NMDA-evoked Ca2+ signaling but do not distinguish between synaptic and extrasynaptic Ca2+ signaling. Ca2+ transients in the dendrites of CA1 pyramidal neurons induced by uncaging of glutamate can be restricted to a single spine and are mainly NMDAR-mediated (Yuste et al., 1999; Bloodgood and Sabatini, 2009). Moreover, spine Ca2+ is under the control of cAMP/PKA signaling (Skeberdis et al., 2006; Chalifoux and Carter, 2010; Higley and Sabatini, 2010). Whereas the functional impact of PKA on NMDARs is well established, the molecular target remains unknown. To investigate whether Ser1166 is critical to regulation of Ca2+ rises in dendritic spines by PKA, we biolistically overexpressed WT or mutant GluN2B(S1166A) in organotypic hippocampal slices. At 10–14 d after transfection, we activated glutamatergic synapses by focal glutamate uncaging and simultaneously measured Ca2+ transients by two-photon microscopy and recorded uEPSCs at individual spines before and after application of isoproterenol, a well-known activator of β-adrenergic receptors and upstream activator of PKA (Fig. 3a,b).
Figure 3.

Ser1166 of GluN2B is essential to β-adrenergic-induced Ca2+ rises in spines. a, Representative 2PLSM image of a dendritic spine from a CA1 hippocampal neuron filled with the red fluorophore AlexaFluor-594 and the green Ca2+-sensitive indicator Fluo-5F. The yellow arrowhead indicates the location of a 500 μs pulse of 720 nm laser light used to focally uncage glutamate near the spine head. White dashed line indicates the region used for line scan analysis of fluorescence. Scale bar, 1 μm. b, Red and green fluorescence in the spine head (sp) and neighboring dendrite (dend) measured in a line scan over the region indicated by the dashed line in a. Inset scale bars, 100 ms and 5% ΔG/Gsat. Yellow arrowhead indicates time of uncaging. The increase in green fluorescence indicates increased intracellular Ca2+, and the corresponding spine Ca2+ transient (white trace) is below. c, Average trace of uncaging-evoked ΔCa2+spine produced by single uncaging stimuli in neurons expressing GluN2B(WT) (left) or GluN2B(S1166A) (right) under control conditions (black) or after isoproterenol incubation (red). Mean (solid lines) and mean ± SEM (shaded regions) are shown. d, Average trace of uEPSCs produced by single uncaging stimuli in neurons expressing GluN2B(WT) (left) or GluN2B(S1166A) (right) under control conditions (black) or after isoproterenol incubation (red). e, Population data for peak ΔCa2+spine and (f) uEPSC elicited by single uncaging stimuli in neurons expressing GluN2B(WT) or GluN2B(S1166A) under control conditions (Ctrl) or after isoproterenol incubation. Red bars represent the mean of distribution ± SEM. *p < 0.001.
In slices overexpressing WT GluN2B, application of isoproterenol (10 μm, 5 min) induced a marked increase in the pharmacologically isolated, NMDA component of uEPSC amplitude and Ca2+ transients in individual dendritic spines (Fig. 3c–f; Table 1). In contrast, in slices overexpressing GluN2B(S1166A), isoproterenol did not detectably alter either the uEPSC amplitude or Ca2+ transients (Fig. 3c–f; Table 1). Thus, the loss of Ser1166 is sufficient to abolish the impact of β-adrenergic receptor (and hence PKA) activation on NMDAR-mediated Ca2+ transients and currents in individual spines. The finding that isoproterenol increases not only NMDAR-mediated Ca2+ transients, but also NMDA uEPSCs, is consistent with our earlier observation that, in the case of GluN2B-containing NMDARs, PKA modulates not only NMDAR Ca2+ signaling but also a substantial fraction of NMDA-evoked currents (Skeberdis et al., 2006). Collectively, these data indicate that Ser1166 is critical to β-adrenergic receptor-dependent potentiation of NMDAR-mediated Ca2+ signaling and uEPSCs in individual spines. Moreover, these findings, together with our findings in Figure 1c, demonstrate that GluN2B(S1166A) serves as a dominant-negative mutant in hippocampal neurons.
Activation of β-adrenergic and D1/D5 dopamine receptors promote phosphorylation of Ser1166
Our findings thus far indicate that Ser1166 of GluN2B is a molecular and functional target of PKA, but they do not address whether physiological stimuli that engage and activate PKA signaling promote phosphorylation of Ser1166. Toward this end, we generated a phospho-specific antibody directed to Ser1166 of GluN2B. To validate the antibody, we performed several control experiments. First, we documented the ability of PKA-C to phosphorylate GluN2B at Ser1166 in GST constructs (data not illustrated), as well as immunocomplexes isolated from brain lysates (Fig. 4a; Table 2) and in whole-brain lysates (Fig. 4a). Second, we showed that preincubation with the phosphopeptide (p-peptide) blocked the phospho-Ser1166 signal, indicating specificity of the antibody (Fig. 4a). Third, we showed that omission of phosphatase inhibitors to permit endogenous phosphatase activity or addition of alkaline phosphatase abolishes the phospho-Ser1166 signal (Fig. 4a; Table 2).
Figure 4.

Physiologically relevant stimuli that activate cAMP/PKA signaling promote Ser1166 phosphorylation. a, PKA-C promotes phosphorylation of GluN2B at Ser1166 after immunoprecipitation from brain lysates in the presence or absence of phosphatase (PP) inhibitors as indicated (+/−). Top, GluN2B immunocomplexes were incubated with no enzyme (control), PKA-C, or alkaline phosphatase (AP). Membranes were probed with antibodies to pSer1166-GluN2B in the absence or presence of phosphopeptide and total GluN2B to detect IP levels. Middle, Input samples (lysates). Bottom, Summary data showing pSer1166-GluN2B normalized to total GluN2B and then to control (no PKA-C); n = 3. *p = 0.0138 for control versus PKA-C. b, c, β-Adrenergic and D1/D5 dopamine receptor agonists promote phosphorylation of Ser1166 in forebrain slices. b, Top, Slices treated with vehicle or isoproterenol in the absence or presence of the β-adrenergic antagonist propranolol. Membranes were probed with antibodies to pSer1166-GluN2B, pSer897-GluN1, and pSer845-GluA1 to assess phosphorylation or to GluN2B, GluN1, or GluA1 to determine total subunit levels. Bottom, Quantification of phosphorylation levels of Ser1166, Ser897, and Ser845 normalized to total IP levels of GluN2B, GluN1, and GluA1, respectively, and then normalized to vehicle. c, Slices were treated with vehicle, the D1/D5 dopamine receptor agonist SKF81297, or with SKF81297 in the presence of the D1/D5 dopamine receptor antagonist SCH23390. GluN2B and GluA1 were immunoprecipitated and processed for immunoblotting as above. Images are from the same blot and exposure, but non-relevant lanes were removed. Bottom, Summary data as in b. *p < 0.05. ***p < 0.001. ****p < 0.0001.
Table 2.
| Figure | N | Mean ± SEM | p | Statistical test |
|---|---|---|---|---|
| 4a | 2–5 slices | Control: 0.2732 ± 0.0585 | *, 0.0138 | One-way ANOVA with Tukey's post-test (control vs PKA) |
| PKA: 1.564 ± 0.513 | ||||
| AP: 0.02005 ± 0.02396 | ||||
| PPI: 0.06882 ± 0.06091 | ||||
| 4b | 6–10 slices | S1166: vehicle, 0.2978 ± 0.04566; ISO, 2.019 ± 0.244; ISO + PROP, 0.3674 ± 0.03934 | ****, < 0.0001 | One-way ANOVA with Tukey's post-test (vehicle vs ISO and ISO vs ISO + PROP) |
| S897: vehicle, 0.2044 ± 0.03897; ISO, 1.034 ± 0.0576; ISO + PROP, 0.5836 ± 0.04153 | ****, < 0.0001 | |||
| S845: vehicle, 0.3929 ± 0.09082; ISO, 1.190 ± 0.0627; ISO + PROP, 0.6275 ± 0.09449 | ****, < 0.0001 | |||
| ***, < 0.001 | ||||
| 4c | 2–5 slices | S1166: vehicle, 0.4482 ± 0.0.04804; SKF, 0.8647 ± 0.05879; SKF + SCH, 0.4041 ± 0.1217 | *, < 0.05 | One-way ANOVA with Tukey's post-test (vehicle vs SKF81297) |
| S897: vehicle, 0.1839 ± 0.05562; SKF, 0.6249 ± 0.2622; SKF + SCH, 0.2558 ± 0.0205 | *, < 0.05 | |||
| S845: vehicle, 0.4306 ± 0.08369; SKF, 1.063 ± 0.131; SKF + SCH, 0.4389 ± 0.1382 | ||||
| 5a | 4 animals | S1166: control, 0.7940 ± 0.1259; Fox U, 0.7689 ± 0.0638 | **, 0.0030 | Two-tailed paired t test for GluA1 S845 control vs Fox U |
| S897: control, 1.000 ± 0.117; Fox U, 1.067 ± 0.074 | ||||
| S845: control, 0.3718 ± 0.1036; Fox U: 0.6740 ± 0.1125 | ||||
| 5b | 5 animals | S1166: control, 0.1226 ± 0.0198; FS, 1.073 ± 0.175 | ***, 0.0006 | Two-tailed unpaired t test for control vs FS |
| S897: control, 0.2884 ± 0.0358; FS, 1.208 ± 0.107 | ****, < 0.0001 | |||
| S845: control, 0.7569 ± 0.0874; FS, 1.415 ± 0.052 | ***, 0.0002 | |||
| 6b,c | 5 animals | S1166: Sal control , 0.8118 ± 0.1251; Sal FS, 1.543 ± 0.238; Rp-cAMPS control , 0.4344 ± 0.1228, Rp-cAMPS FS, 0.8291 ± 0.1975 | *, < 0.05 | One-way ANOVA with Tukey's post-test (S1166 and S845 Sal control vs FS; Sal FS vs Rp-cAMPS FS) |
| S897: Sal control, 1.147 ± 0.309; Sal FS, 1.508 ± 0.366; Rp-cAMPS control, 0.3963 ± 0.1025; Rp-cAMPS FS, 0.6003 ± 0.0837 | *, < 0.05 | |||
| S845: Sal control, 0.5753 ± 0.0382; Sal FS, 0.8542 ± 0.0903; Rp-cAMPS control, 0.2804 ± 0.0389 Rp-cAMPS FS, 0.4563 ± 0.0500 | ***, < 0.001 |
*p < 0.05;
**p < 0.01;
***p < 0.001;
****p < 0.0001.
The β-adrenergic (Hall, 2004; Dai et al., 2009) and D1/D5 dopaminergic (Beaulieu and Gainetdinov, 2011) signaling cascades are well-known pathways that engage and activate cAMP/PKA signaling in neurons. Application of isoproterenol (1 μm, 5 min) to forebrain slices induced robust phosphorylation of the NMDAR subunit GluN2B at Ser1166 (Fig. 4b; Table 2), which was reversed by the β-adrenergic receptor antagonist propranolol (Fig. 4b; Table 2). Isoproterenol also promoted marked phosphorylation of the NMDAR subunit GluN1 at Ser897 and AMPA receptor (AMPAR) subunit GluA1 at Ser845, two well-established PKA targets (Roche et al., 1996; Tingley et al., 1997), in a propranolol-sensitive manner (Fig. 4b; Table 2). Similarly, application of the D1/D5 dopamine receptor agonist SKF81297 (10 μm, 15 min) induced prominent phosphorylation of GluN2B at Ser1166 (Fig. 4c; Table 2), which was reversed by the D1/D5 receptor antagonist SCH23390 (Fig. 4c; Table 2). SKF81297 also induced prominent phosphorylation of GluN1 at Ser897 and GluA1 at Ser845 (Fig. 4c; Table 2). These results demonstrate that Ser1166 on GluN2B is phosphorylated in brain slices in response to physiologically relevant stimuli that activate PKA.
Phosphorylation of Ser1166 is induced by the emotional response to stress in vivo
The results thus far indicate that phosphorylation of GluN2B at Ser1166 is critical to modulation of NMDAR function by PKA, but do not address experience-driven regulation of NMDAR phosphorylation at this site. Adverse experience in the form of stress promotes the release of norepinephrine and β-adrenergic signaling (McGaugh and Roozendaal, 2002; Padgett and Glaser, 2003). We examined whether adverse experience promotes phosphorylation of GluN2B at Ser1166 in the hippocampus. A single, brief exposure to fox urine stimulates release of norepinephrine and induces freezing behavior, indicative of fear (Hu et al., 2007; Liu et al., 2010). Exposure of animals to fox urine (5 min) increased phosphorylation of GluA1 at Ser845 in the hippocampal CA1, in corroboration of others (Hu et al., 2007) (Fig. 5a; Table 2), with little or no effect on phosphorylation of the NMDAR subunits GluN2B at Ser1166 and GluN1 at Ser897 (Fig. 5a; Table 2). These findings indicate that phosphorylation of NMDARs and AMPARs in response to emotional stress is under the control of different intracellular signaling pathways in vivo.
Figure 5.

Phosphorylation of GluN2B at Ser1166 is induced by forced swim, but not exposure to fox urine, in vivo. a, Representative Western blots (top) and summary data (bottom) for hippocampal lysates from rats exposed to fox urine for 5 min. Western blots were probed with antibodies to pSer1166-GluN2B, pSer897-GluN1, and pSer845-GluA1 and then with antibodies to total GluN2B, GluN1, and GluA1. Exposure to fox urine increased phosphorylation of GluA1 at Ser845, with little or no effect on phosphorylation levels of GluN2B at Ser1166 or GluN1 at Ser897. n = 4 animals per treatment group, **p = 0.0030. b, Representative Western blots (top) and summary data (bottom) for hippocampal lysates from rats exposed to a single episode of forced swim for 5 min. Western blots were probed with antibodies as above for exposure to fox urine. Forced swim induced striking phosphorylation of GluN2B at Ser1166, GluN1 at Ser897, and GluA1 at Ser845. Values for phosphorylated protein were normalized to the corresponding value for total protein and then to the corresponding value for control animals. n = 5 animals per treatment group. ***p = 0.0006 (S1166 control vs FS). ***p = 0.0002 (S845 control vs FS). ****p < 0.0001.
We next examined the impact of forced swim, another well-established paradigm of emotional as well as mild physical stress (Anisman et al., 2001), on NMDAR phosphorylation in the hippocampus. A single, brief exposure to forced swim stimulates release of norepinephrine (Yoshitake et al., 2004) and increases the magnitude of LTP at synapses onto dentate gyrus granule cells (Korz and Frey, 2003). Exposure of animals to forced swim (5 min) induced striking phosphorylation of GluN2B at Ser1166 and (to a lesser extent) phosphorylation of GluN1 at Ser897 (Fig. 5b; Table 2). Forced swim also induced a relatively modest phosphorylation of GluA1 at Ser845. The finding that adverse experience in the form of forced swim, but not exposure to fox urine, facilitates phosphorylation of GluN2B at Ser1166 suggests that different forms and/or intensities of stress can induce differential phosphorylation of PKA targets in a receptor- and site-specific manner.
To identify the signaling pathway that mediates stress-induced phosphorylation of GluN2B at Ser1166, we injected rats with the PKA inhibitor Rp-8-Br-cAMPS or saline, directly into the hippocampal CA1 at 1 h prior to forced swim and microdissected the small region of CA1 directly around the injection site (Fig. 6a). Forced swim induced an approximately twofold increase in phosphorylation of GluN2B at Ser1166 and a modest increase in phosphorylation of GluN1 at Ser897. Rp-8-Br-cAMPS markedly reduced phosphorylation of GluN2B at Ser1166 (and, to a lesser extent, GluN1 at Ser897) under both control and forced swim conditions (Fig. 6b; Table 2). These findings are consistent with the concept that both basal and experience-dependent phosphorylation of Ser1166, at least in part, is mediated by cAMP/PKA signaling. Rp-8-Br-cAMPS also reduced basal and forced swim-induced phosphorylation of GluA1 at Ser845 (Fig. 6c; Table 2).
Figure 6.
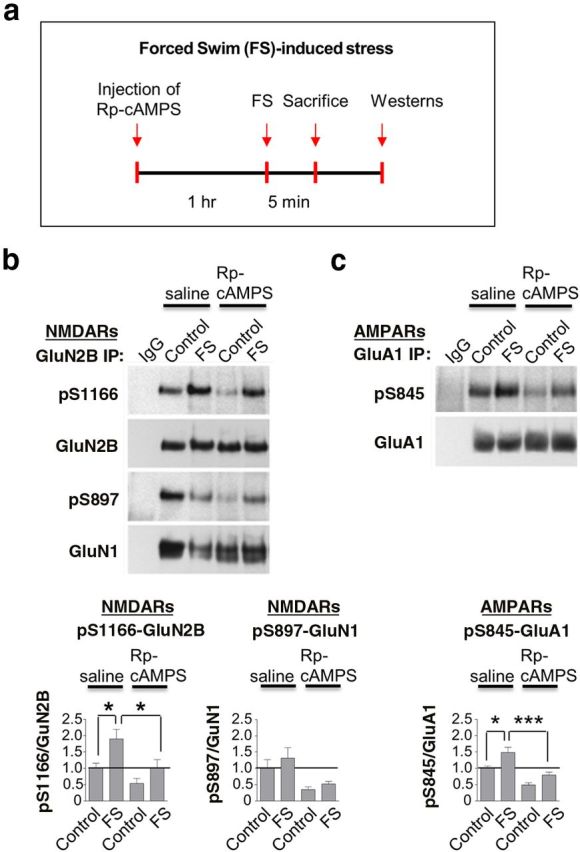
Forced swim (FS)-induced phosphorylation of Ser1166 is PKA-dependent. a, Schematic of experimental paradigm for injections and FS. b, c, Top, Representative Western blots. Bottom, Summary data for lysates of hippocampal CA1 from rats injected with Rp-8-Br-cAMPS or saline 1 h before a single episode of FS for 5 min. Western blots were probed with antibodies to pSer1166-GluN2B, pSer897-GluN1, pSer845-GluA1, and total GluN2B, GluN1, and GluA1, respectively. Rp-8-Br-cAMPS markedly reduced FS-induced phosphorylation of GluN2B at Ser1166 and GluA1 at Ser845, with little or no effect on pSer897-GluN1. Values for phosphorylated protein were normalized to the corresponding value for total protein and then to the corresponding value for control animals. n = 5 animals per treatment group. *p < 0.05. ***p < 0.001.
Discussion
The present study identifies Ser1166 in the NMDAR subunit GluN2B as a novel molecular and functional target of PKA phosphorylation critical to potentiation of NMDAR Ca2+ signaling at synapses and regulated by the emotional response to stress. We show that physiologically relevant stimuli that engage and activate cAMP/PKA signaling, and are important to the acquisition and consolidation of memory (McGaugh and Roozendaal, 2002; Hu et al., 2007), regulate phosphorylation of NMDARs and Ca2+ signaling in spines. Accordingly, activation of β-adrenergic and D1/D5 dopamine receptors stimulates phosphorylation of GluN2B at Ser1166 in brain slices. Moreover, activation of β-adrenergic receptors potentiates Ca2+ transients and uncaging-evoked synaptic currents in individual spines of neurons expressing WT, but not mutant S1166A, NMDARs. Thus, loss of this single phosphorylation site is sufficient to abolish β-adrenergic-dependent potentiation of NMDA currents and Ca2+ transients, linking phosphorylation of NMDARs to Ca2+ signaling in spines. This is significant in that central noradrenergic signaling mediates emotional arousal and the emotional response to stress (McGaugh and Roozendaal, 2002; Padgett and Glaser, 2003). We further show that adverse experience in the form of forced swim, but not exposure to fox urine, elicits striking phosphorylation of Ser1166 in a PKA-dependent manner in vivo, revealing experience-dependent phosphorylation of this site in a physiologically relevant context. These findings indicate that different stress paradigms differentially regulate NMDAR phosphorylation and, by inference, Ca2+ permeation and Ca2+ signaling.
cAMP/PKA signaling represents a pivotal mechanism by which NMDAR-mediated Ca2+ signaling is modulated in neurons under physiological and pathological conditions. PKA promotes NMDAR Ca2+ permeability, Ca2+ rises in spines, and induction of LTP at CA1 synapses (Skeberdis et al., 2006). Moreover, extracellular signals that modulate cAMP bidirectionally regulate Ca2+ permeation through NMDARs and spine Ca2+ in a PKA-dependent manner (Chalifoux and Carter, 2010; Higley and Sabatini, 2010). Although these studies establish a functional role for PKA, they do not address the molecular target. In the present study, we reveal a previously unrecognized mechanism by which activation of major signaling pathways, known to couple positively to cAMP/PKA and play a critical role in learning and memory, can alter Ca2+ signaling in activated dendritic spines. Although not addressed by the present manuscript, phosphorylation of the carboxy-terminal tails of channels is known to affect channel properties, including ion selectivity. Moreover, phosphorylation events that regulate channel activity typically occur in either the N-terminal or C-terminal domains of the channel and not in the pore itself (Levitan and Kaczmarek, 2002). By analogy, phosphorylation of Ser1166 may induce a delocalized conformational change. Alternatively, S1166 might form part of a recognition motif for an as yet unidentified protein, which in some capacity regulates the selectivity pore within the NMDA channel.
A novel finding of the present study is that emotional stress in the form of a single exposure to forced swim, but not exposure to fox urine, elicits striking phosphorylation of Ser1166 in a PKA-dependent manner in vivo, revealing experience-driven regulation of phosphorylation at this site. Notably, these effects persist as late as P32, the oldest age examined, consistent with the concept that regulation of NMDAR Ca2+ permeation and signaling in spines (and GluN2B expression) are fundamental mechanisms relevant throughout the life span of the animal. Although exposure to fox urine promotes PKA-dependent phosphorylation of GluA1 at Ser845 (Hu et al., 2007) and induces transcription of the AMPAR subunit GluA2 in cerebellar stellate cells (Liu et al., 2010), it does not alter the phosphorylation status of NMDARs. At hippocampal CA1 synapses, phosphorylation of GluA1 at Ser845 facilitates synaptic delivery of GluA1-containing AMPARs and lowers the threshold for LTP (Oh et al., 2006; Hu et al., 2007); and at cerebellar synapses, transcription of GluA2 drives a switch in AMPAR phenotype and Ca2+ signaling (Liu et al., 2010). Whereas phosphorylation of GluN2B at Ser1166 potentiates NMDAR function and Ca2+ signaling in dendritic spines, with little or no effect on NMDAR trafficking (Skeberdis et al., 2006), phosphorylation of GluN1 at Ser897 promotes forward trafficking of NMDARs from the endoplasmic reticulum to the cell surface (Scott et al., 2003). These findings reinforce the notion that different forms of stress can promote PKA-dependent phosphorylation of different glutamate receptors and/or receptor-associated proteins at different sites. Although both exposure to fox urine and a single episode of forced swim elevate noradrenergic signaling, they differ in their characteristics and severity. Whereas exposure to fox urine, an olfactory stimulus, is a psychological form of stress, forced swim stress has both psychological and physical components (Anisman et al., 2001). Forced swim also promotes surface expression of GluN2B and enhances NMDAR EPSCs at synapses of the prefrontal cortex (Yuen et al., 2009).
In conclusion, our data provide evidence for a mechanism by which physiologically relevant stimuli, important to emotional arousal and learning and memory, regulate NMDAR Ca2+ permeation and Ca2+ signaling in spines. Whereas other protein kinases that phosphorylate NMDARs, such as Src (Yu et al., 1997), Fyn (Prybylowski et al., 2005), PKC (Lu et al., 1999; Lan et al., 2001), and casein kinase 2 (Sanz-Clemente et al., 2010), regulate NMDAR trafficking and gating in response to neuronal activity, PKA is unique in that it regulates NMDAR-mediated Ca2+ permeation. We identify Ser1166 on GluN2B as a novel molecular and functional target of PKA essential to Ca2+ signaling in spines and regulated by the emotional response to stress. Importantly, different forms of adverse experience differentially regulate phosphorylation of this site. These findings add a new dimension to our understanding of how experience influences synaptic proteins and function. Given that NMDARs and PKA are expressed widely throughout the brain, phosphorylation of Ser1166 represents a potentially powerful mechanism to adjust synaptic function. NMDARs are critical to many forms of synaptic plasticity and to higher cognitive functions, such as learning and memory, and their dysregulation is implicated in neuropsychiatric disorders, such as schizophrenia, stroke, Parkinson's disease, and Huntington's disease. Thus, our findings have important ramifications for NMDARs not only under normal, but also pathological, conditions.
Footnotes
This work was supported by National Institutes of Health grants to R.S.Z., J.W.H., and B.L.S.; HHMI (B.L.S.); and the F.M. Kirby Foundation (R.S.Z). R.S.Z. is the F.M. Kirby Chair in Neural Repair and Protection. We thank Drs. Lynn Raymond, Michael V.L. Bennett, and Pablo E. Castillo and the members of the Zukin laboratory for their helpful comments on the manuscript; and Dr. Sylvia O. Suadicani for her expert guidance in calcium imaging in HEK cells.
The authors declare no competing financial interests.
References
- Amico-Ruvio SA, Popescu GK. Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys J. 2010;98:1160–1169. doi: 10.1016/j.bpj.2009.12.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisman H, Hayley S, Kelly O, Borowski T, Merali Z. Psychogenic, neurogenic, and systemic stressor effects on plasma corticosterone and behavior: mouse strain-dependent outcomes. Behav Neurosci. 2001;115:443–454. doi: 10.1037/0735-7044.115.2.443. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. NMDA receptor-mediated calcium transients in dendritic spines. Biology of the NMDA receptor. In: Van Dongen AM, editor. Frontiers in neuroscience. Boca Raton, FL: CRC; 2009. [PubMed] [Google Scholar]
- Borschel WF, Myers JM, Kasperek EM, Smith TP, Graziane NM, Nowak LM, Popescu GK. Gating reaction mechanism of neuronal NMDA receptors. J Neurophysiol. 2012;108:3105–3115. doi: 10.1152/jn.00551.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA. Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron. 2011;71:1085–1101. doi: 10.1016/j.neuron.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA. Beta-adrenergic receptors and their interacting proteins. Semin Cell Dev Biol. 2004;15:281–288. doi: 10.1016/j.semcdb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Jiang M, Deng L, Chen G. High Ca(2+)-phosphate transfection efficiency enables single neuron gene analysis. Gene Ther. 2004;11:1303–1311. doi: 10.1038/sj.gt.3302305. [DOI] [PubMed] [Google Scholar]
- Korz V, Frey JU. Stress-related modulation of hippocampal long-term potentiation in rats: involvement of adrenal steroid receptors. J Neurosci. 2003;23:7281–7287. doi: 10.1523/JNEUROSCI.23-19-07281.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius CL, Kaur N, Popescu GK. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci. 2009;29:6819–6827. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- Levitan IB, Kaczmarek LK. The neuron: cell and molecular biology. Ed 3. Oxford: Oxford UP; 2002. [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. 2004;24:10138–10148. doi: 10.1523/JNEUROSCI.3159-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabó G, Zukin RS, Liu SJ. A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat Neurosci. 2010;13:223–231. doi: 10.1038/nn.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki BA, Aman TK, Amico-Ruvio SA, Kussius CL, Popescu GK. C-terminal domains of N-methyl-d-aspartic acid receptor modulate unitary channel conductance and gating. J Biol Chem. 2012;287:36071–36080. doi: 10.1074/jbc.M112.390013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/S0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol. 2003;24:444–448. doi: 10.1016/S1471-4906(03)00173-X. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/S0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/S0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chavez AE, Cossio MJ, Castillo PE, Zukin RS. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci. 2012;15:1382–1390. doi: 10.1038/nn.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT, Roche KW. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron. 2010;67:984–996. doi: 10.1016/j.neuron.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Ehlers MD. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology. 2003;45:755–767. doi: 10.1016/S0028-3908(03)00250-8. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake T, Wang FH, Kuteeva E, Holmberg K, Yamaguchi M, Crawley JN, Steiner R, Bartfai T, Ogren SO, Hokfelt T, Kehr J. Enhanced hippocampal noradrenaline and serotonin release in galanin-overexpressing mice after repeated forced swimming test. Proc Natl Acad Sci U S A. 2004;101:354–359. doi: 10.1073/pnas.0307042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A. 2009;106:14075–14079. doi: 10.1073/pnas.0906791106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]