

Abstract
Gangliosides are widely expressed sialylated glycosphingolipids with multifunctional properties in different cell types and organs. In the nervous system, they are highly enriched in both glial and neuronal membranes. Mice lacking complex gangliosides attributable to targeted ablation of the B4galnt1 gene that encodes β-1,4-N-acetylegalactosaminyltransferase 1 (GalNAc–transferase; GalNAcT−/−) develop normally before exhibiting an age-dependent neurodegenerative phenotype characterized by marked behavioral abnormalities, central and peripheral axonal degeneration, reduced myelin volume, and loss of axo-glial junction integrity. The cell biological substrates underlying this neurodegeneration and the relative contribution of either glial or neuronal gangliosides to the process are unknown. To address this, we generated neuron-specific and glial-specific GalNAcT rescue mice crossed on the global GalNAcT−/− background [GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial)] and analyzed their behavioral, morphological, and electrophysiological phenotype. Complex gangliosides, as assessed by thin-layer chromatography, mass spectrometry, GalNAcT enzyme activity, and anti-ganglioside antibody (AgAb) immunohistology, were restored in both neuronal and glial GalNAcT rescue mice. Behaviorally, GalNAcT−/−-Tg(neuronal) retained a normal “wild-type” (WT) phenotype throughout life, whereas GalNAcT−/−-Tg(glial) resembled GalNAcT−/− mice, exhibiting progressive tremor, weakness, and ataxia with aging. Quantitative electron microscopy demonstrated that GalNAcT−/− and GalNAcT−/−-Tg(glial) nerves had significantly increased rates of axon degeneration and reduced myelin volume, whereas GalNAcT−/−-Tg(neuronal) and WT appeared normal. The increased invasion of the paranode with juxtaparanodal Kv1.1, characteristically seen in GalNAcT−/− and attributed to a breakdown of the axo-glial junction, was normalized in GalNAcT−/−-Tg(neuronal) but remained present in GalNAcT−/−-Tg(glial) mice. These results indicate that neuronal rather than glial gangliosides are critical to the age-related maintenance of nervous system integrity.
Keywords: ganglioside, glycosyltransferase, neurodegeneration, transgenic
Introduction
Gangliosides are sialylated glycosphingolipids widely expressed in vertebrate plasma membranes and intracellular compartments (Ledeen and Wu, 2011; Yu et al., 2011; Sandhoff and Harzer, 2013). They are particularly enriched in nervous system gray and white matter, in which they have roles in development and maintenance, cell–cell recognition, signal transduction, growth, and motility. They are key components of lipid raft membrane domains (Sonnino et al., 2007; Ohmi et al., 2012) and function as receptors for microbial pathogens and toxins (Fewou et al., 2013; Ravindran et al., 2013) and neuropathy-associated auto-antibodies (Willison and Yuki, 2002; Kusunoki et al., 2008).
Gangliosides are synthesized by stepwise addition of monosaccharides to ceramide by glycosyltransferases (Hakomori, 1981; Yu et al., 2004; Fig. 1B). The predominant gangliosides found in neural tissue are the complex gangliosides GM1, GD1a, GD1b, and GT1b, likely because of high activity of their synthesizing enzyme, β-1,4-N-acetylegalactosaminyltransferase 1 (GalNAc–transferase; GalNAcT, encoded by the B4galnt1 gene) in the nervous system (Dicesare and Dain, 1971). Transgenic mice with disruptions in specific glycosyltransferase genes, and thus deficient in downstream ganglioside biosynthesis, have been critical tools in advancing understanding of their roles. Mice generated with a disruption in the B4galnt1 gene are deficient in GalNAcT but are viable and appear grossly normal, indicating that complex ganglioside expression is not necessary for normal development (Takamiya et al., 1996) but later develop an age-dependent neurodegenerative phenotype characterized by weakness, ataxia, nerve degeneration and demyelination, and loss of nodal axo-glial junction adhesion and integrity (Sheikh et al., 1999; Chiavegatto et al., 2000; Susuki et al., 2007). Overexpression of the precursor simple gangliosides GM3, GD3, and 9-O-Ac(etyl)-GD3 in GalNAcT−/− mice may play a compensatory developmental role that limits the severity of the phenotype (Ngamukote et al., 2007; Furukawa et al., 2008).
Figure 1.

Expression of GalNAcT and complex gangliosides in rescue mice. A, Constructs were generated to drive GalNAcT expression in the neurons and glia of GalNAcT−/− mice by promoting the GalNAcT gene under the control of the hNFL and mPLP promoters, respectively. B, Ganglioside biosynthetic pathway. The GalNAcT enzyme is necessary for generation of complex gangliosides (surrounded by the green box). C, TLC of extracts from brains of WT, GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and GalNAcT−/−-Tg(glial) mice, sprayed with resorcinol to label gangliosides. Standards for the complex gangliosides GM1, GD1a, GD1b, and GT1b are labeled in the right lane. WT extracts contain all complex gangliosides, which are absent from GalNAcT−/− extract and restored in GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) mice. GalNAcT−/− extracts are enriched with simple gangliosides. GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) mice extracts are also enriched with simple gangliosides and additionally express the complex gangliosides but at lower levels than WT. D, Chromatograms of extracted major gangliosides in mouse brains of all genotypes confirm TLC findings. All major gangliosides can be putatively detected. The complex gangliosides GT1b, GD1b, GD1a, and GM1 are the most abundant gangliosides found in WT extracts. These are also present in rescue mice at lower levels. GD3, 9-O-Ac(etyl)-GD3, and GM3 are highly abundant in GalNAcT−/− extracts and remain at high levels in both rescue mice.
Mice lacking b-series gangliosides (GD3s−/−) are grossly normal throughout life but repair peripheral nerve poorly (Kawai et al., 2001; Okada et al., 2002). Mice whose ganglioside repertoire is restricted to GM3 (GalNAcT−/− × GD3s−/− double knock-out) develop lethal audiogenic seizures (Kawai et al., 2001), age-dependent progressive motor and cognitive deficits (Tajima et al., 2009), and sensory loss (Inoue et al., 2002). Complete ganglioside ablation is not embryonic lethal; however, from 2 weeks of age, mice undergo progressive and severe neurodegeneration resulting in death at ∼2 months (Yamashita et al., 2005). Together, these mouse data are suggestive of a more fundamental necessity for a-series gangliosides in age-related nervous system maintenance, although this is difficult to conclusively prove because a mouse with selective deficiency of a-series gangliosides has not been generated. Humans with inherited ganglioside deficiency also develop complex neurodevelopment and degenerative syndromes (Simpson et al., 2004; Boukhris et al., 2013).
Although these studies indicate that bodywide expression of simple gangliosides are sufficient to promote viability and complex gangliosides are required for nervous system maintenance, stability, and repair, it is unknown whether neuronal or glial ganglioside deficiency has the greater impact on the age-related phenotype and maintenance of the axon, myelin, and axo-glial junction. To assess the relative significance and necessity of complex ganglioside expression in neuronal and myelin-forming cells, we developed GalNAcT−/− rescue mice that selectively express gangliosides either neuronally [GalNAcT driven by the neurofilament-light (NFL) promoter; GalNAcT−/−-Tg(neuronal)] or in myelin [GalNAcT driven by the proteolipid protein (PLP) promoter; GalNAcT−/−-Tg(glial)] and analyzed them for behavioral and structural abnormalities.
Materials and Methods
Generation of transgenic mice
The generation of GalNAcT−/− mice lacking complex gangliosides has been described previously (Takamiya et al., 1996) and backcrossed seven generations on a C57BL/6 background. Transgenic mice expressing the full-length cDNA encoding GalNAcT under the control of the NFL or PLP promoter were generated by pronuclear injection to produce neuronal and glial expression, respectively (Fig. 1A). The activity of the NFL and PLP promoters are classically restricted to mature neurons and myelinating glia (oligodendrocytes and Schwann cells), respectively. GalNAcT cDNA was cloned into the pGCHNF-L vector (provided by J.-P. Julien, Laval University, Quebec, Quebec, Canada) for generating NFL–GalNAcT transgenic mice. Similarly, GalNAcT cDNA was cloned into PLP–SV40 (provided by Wendy Macklin, University of Colorado, Boulder, CO) for generating PLP–GalNAcT transgenic mice. Transgenic lines and germ-line transmitters were identified by PCR and backcrossed seven generations on a C57BL/6 background. NFL–GalNAcT and PLP–GalNAcT were then interbred with GalNAcT−/− mice to create GalNacT−/−-Tg(neuronal) and GalNacT−/−-Tg(glial) mice, respectively, that were used in all analyses. Evidence for GalNAcT enzyme activity restoration and complex gangliosides synthesis in neural tissues were confirmed by glycosyltransferase activity assays as described previously (Ruan and Lloyd, 1992; Ruan et al., 1995). Mice of either sex were killed by CO2 inhalation, and all experiments complied with United Kingdom Home Office guidelines.
Antibodies and reagents
Anti-ganglioside antibodies (AgAb) were generated as described previously (Bowes et al., 2002; Boffey et al., 2005); herein we used the monoclonal antibody MOG16 that reacts with complex gangliosides GD1b and GT1b, the latter being prominently restored in GalNacT−/−-Tg(neuronal) and GalNacT−/−-Tg(glial) mice (Fig. 1C,D). Immunostaining reagents were sourced as follows: α-bungarotoxin (BTx; Invitrogen); rabbit anti-Caspr (diluted 1:1000; gift from Elior Peles, Rehovot, Israel); rabbit anti-Kv1.1 (diluted 1:200; Alomone Labs); mouse anti-Caspr (diluted 1:500; NeuroMab); rabbit anti-Nav1.6 (diluted 1:100; Sigma); rabbit anti-neurofilament (diluted 1:750; Affinity Bioreagents); isotype-specific (IgG1, IgG2a, IgG2b, IgG3) Alexa Fluor 488- and 555-conjugated anti-mouse IgG antibodies; and Alexa Fluor 488- and 555-conjugated anti-rabbit secondary antibodies. Ringer's solution (in mm: 116 NaCl, 4.5 KCl, 1 MgCl2, 2 CaCl2, 1 NaH2PO4, 23 NaHCO3, and 11 glucose, pH 7.4) was pregassed with 95% O2/5% CO2.
Lipid analysis
Extraction and TLC.
Gangliosides were extracted from brains removed and snap frozen from WT, GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and GalNAcT−/−-Tg(glial) (2–4 months old) mice according to Williams and McCluer (1980). Briefly, lipids were extracted from homogenized tissue in 1:1 CHCl3/MeOH. Gangliosides were phase partitioned into the upper phase according to Folch et al. (1951) and then desalted using SepPak C18 cartridges. TLC was performed on high-performance TLC silica plates (Merck) in the solvent system 50:42:11 CHCl3/MeOH/0.2% CaCl2. Bands were detected with resorcinol.
Liquid chromatography tandem mass spectrometry analysis of lipids.
Extracts were separated on an Ultimate 3000 HPLC system running a 50–100% isopropanol/10 mm ammonium formate gradient at 250 μl/min. WT, GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and GalNAcT−/−-Tg(glial) rescue extracts were diluted 1:100 in methanol, and 10 μl was injected onto an Acclaim C30 2.1 × 150 mm 5 μm reverse-phase column (Thermo Fisher Scientific). Data were acquired on an LTQ Orbitrap Elite equipped with a heated electrospray ionization source at 80°C in negative ion mode. Full scans were taken from mass-to-charge ratio 500–2000 at 60,000 resolution. Collision-induced dissociation fragmentation was performed on the top five peaks in each full scan. Commercial extract and blanks were run at the beginning and end of the batch.
Data analyses.
Extracted ion chromatograms were produced for ganglioside elution by applying mass filters for the d18:1/18:0 species of the [M-H]-ions of GM1, GM2, GM3 and [M-2H]2-ions of GD1, GD3, GT1, GQ1, and 9-O-Ac(etyl)-GD3 to each dataset for WT, GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and GalNAcT−/−-Tg(glial) extracts in Xcalibur qual browser. Normalized intensities were taken for each ganglioside from extracted ion chromatograms of the d18:1/18:0 and d18:1/20:0 species. Retention times were used to verify consistency between samples. Ganglioside identifications were confirmed and isomers distinguished using preestablished fragmentation patterns.
Behavioral assessment
Rotarod.
Mice [WT, n = 27; GalNAcT−/−, n = 23; GalNAcT−/−-Tg(neuronal), n = 44; GalNAcT−/−-Tg(glial), n = 20] were placed on the beam in separate lanes of the rotarod apparatus, and the speed was fixed at 15 rpm. Mice were trained three times a day for 3 consecutive days to allow familiarization with the equipment. Latency to fall from the rod was recorded for up to 100 s each trial for three trials per day on 3 consecutive days. The nine results were then pooled for each mouse.
Hindlimb grip strength.
A rectangular metal bar was attached to a digital force gauge (Chatillon DFIS; AMETEK) and set to record the maximum tensile force generation (in Newtons). Each mouse was suspended by the tail, and tensile force generated by the forelimb grip on the longest aspect of the metal bar was measured. Total tensile force generated by both forelimbs and hindlimbs was then measured. The mouse was then gently pulled back until its grip on the bar was released. Each mouse was measured three times a day for 3 d, and the mean force generated by the hindlimb was calculated by subtracting force generated by forelimb from total force (generated by forelimb and hindlimb).
Grid walking.
This test assessed accurate paw placement and paw slips during recorded 10 min free-walking sessions on a wire grid. A paw slip consisted of a paw completely missing a rung resulting in the limb falling through the grid or an initially successful paw placement that subsequently slipped through the grid attributable to limb weakness on weight bearing. Each hindlimb forward motion counted as one step, and 500 steps were counted for each mouse.
Ultrastructural analyses
Tissue for electron microscopy was dissected from mice transcardially perfused with 5% glutaraldehyde/4% paraformaldehyde mixture. The second segment of the cervical spinal cord, the optic nerve (OpN), and sciatic nerve (SN) were processed for resin embedding (Griffiths et al., 1981). Sections were cut for both light and electron microscopy. Tissue from 2- and 12-month-old mice (n = 4 per genotype, n = 3 WT) were studied for ultrastructural analysis. Electron micrographs from transverse sections of the OpN and the ventral columns of the spinal cord at 6700× and 2700× magnification, respectively, were taken on a Jeol CX-100 Electron microscope. Images from 1 μm semithin sections stained with methylene blue/azurII were captured of the SN at 100× magnification. For quantification, a minimum of 10 electron micrographs or six digital light microscopic images per animal were taken of randomly selected fields. All measurements were made on scanned or digitally captured images using NIH ImageJ software.
Axon morphometry and quantification of axonal changes.
All axons within or touching the top and left borders of an area of interest (AOI) were counted. The axon density, number of degenerating axons, or those containing accumulations of organelles within the AOI was counted.
The volume occupied by compact myelin or axoplasm was quantified by a point counting method using an appropriately sized grid (Williams, 1977) and expressed as the ratio of the number of intercepts coinciding with the structure to the total number of intercepts.
Immunohistochemistry, imaging, and analysis
Ganglioside localization.
Triangularis sterni (TS) muscles (n = 3 per genotype, 6 months) were maintained in Ringer's solution and incubated in 100 μg/ml AgAb for 30 min at 4°C with 2 μg/ml FITC-conjugated BTx to label the neuromuscular junctions (NMJs). Spinal cord (n = 3 per genotype, 6 months) were snap frozen on removal, transversely cryosectioned at 10 μm onto 3-aminopropyltriethoxysilane (APES)-coated glass slides, and then incubated with 20 μg/ml AgAb and anti-neurofilament antibody to identify axons (1:750) in PBS for 2 h at 4°C. All preparations were washed in Ringer's solution or PBS before 20 min fixation in 4% paraformaldehyde at room temperature, followed by 10 min washes with PBS, 0.1 m glycine, and PBS. Tissues were incubated with appropriate isotype-specific fluorescently labeled secondary antibodies at 2 μg/ml for 1 h at room temperature (cord) or 5 μg/ml with 1% normal goat serum overnight at 4°C (TS), washed with PBS, and finally coverslipped, or mounted and coverslipped in the case of TS. Images were captured on a Zeiss Axio Imager Z1 with ApoTome attachment.
Nodal integrity assessment.
Fixed SN and OpN (n = 3 per genotype, 6 months) were teased into individual fibers or sectioned at 10 μm, respectively, on to APES-coated glass slides. Nerves were incubated with blocking solution (3% normal goat serum plus 0.5% Triton X-100 in PBS) for 30 min at 4°C before incubation overnight in the same solution plus primary antibodies (rabbit anti-Kv1.1 and mouse anti-Caspr). Nerves were washed in PBS and incubated for 3 h at room temperature with appropriate combination of fluorescently labeled secondary antibodies each at 2 μg/ml. Several features of nodal architecture were examined (and quantified in SN), including juxaparanodal (JPN) Kv1.1 channel immunostaining invasion into paranodes, the distance between Kv1.1-positive domains, paranodal (PN) Caspr protrusions and the length of Caspr, and Nav1.6 staining. Nodes of Ranvier (NoR) were identified in phase contrast and by Kv1.1 channel staining. At least 20 nodes per mouse were imaged and assessed using NIH ImageJ software.
Extracellular SN recordings
Nerves were mounted in a Perspex recording block across three chambers and sealed in with vacuum grease. SNs were maintained in Ringer's solution, and recordings were performed at room temperature. To measure conduction velocity (CV) and rate of rise, nerves were stimulated at 1 Hz and supramaximal voltage (Grass S88 stimulator; Grass Instruments) for 30 min. Signals were amplified (CED1902; Cambridge Electronic Design), digitized (NIDAQ-MX analog-to-digital converter; National Instruments), and analyzed using WinWCP version 4.5.2. Subsequently, paired-pulse recordings were performed on the same nerve to assess refractoriness; intervals between stimuli ranged from 3 to 30 ms. At termination, 5 μm tetrodotoxin was applied or nerve crush was performed to confirm that the recorded waveform originated from the opening of sodium channels. A minimum of 200 control waveforms were used to calculate CV and rate of rise. To determine refractoriness, the percentage amplitude of the second waveform compared with the first was calculated for every interstimulus interval and plotted. Four- to 6-month-old mice were used [n = 3 for WT; n = 4 for GalNAcT−/−-Tg(neuronal); n = 5 for GalNAcT−/− and GalNAcT−/−-Tg(glial)]; both nerves were used for recordings per mouse. A one-way ANOVA was used to compare CV and rate of rise among genotypes and two-way ANOVA to compare paired-pulse amplitudes with increasing interstimulus interval.
Statistics
Statistical analyses were performed using GraphPad Prism 6 (GraphPad Software), and statistical significance was set at p < 0.05. Data in figures are presented as mean ± SEM. Behavioral data among genotypes was compared by two-way ANOVA, followed by Tukey's post hoc tests for multiple comparisons; one-way ANOVA was applied to morphological studies and electrophysiological recordings, followed by Tukey's post hoc analysis.
Results
GalNAcT enzyme activity and complex ganglioside expression is restored in neural tissue of GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) mice
Four neuronal and four glial transgenic lines were backcrossed onto GalNAcT−/− and screened for GalNAcT enzyme activity in whole-brain homogenate and for complex ganglioside expression in neural tissue by immunohistology. One line of each was then selected for additional study based on the highest enzyme activity. In the two selected lines, whole-brain GalNAcT enzyme activity (compared with WT, 100%) was present at 5.3% in GalNAcT−/−-Tg(neuronal) and 11.7% in GalNAcT−/−-Tg(glial) and undetectable in whole-brain homogenate from GalNAcT−/− mice.
To characterize complex ganglioside profiles, ganglioside fractions were extracted from brains for resorcinol staining of TLC and accurate molecular mass identification using liquid chromatography mass spectrometry (Fig. 1C,D). TLC of brain extracts confirmed the presence of complex gangliosides in expected profiles (Fig. 1C). WT brain contains a full complement of simple and complex gangliosides, with greater expression of the complex gangliosides GD1a, GT1b, GM1, and GD1b, as also indicated by liquid chromatography mass spectrometry (Fig. 1D). In GalNAcT−/− brain, complex gangliosides are absent; instead, GM3, GD3, and 9-O-Ac(etyl)-GD3 that are upstream of GalNAcT in the biosynthetic pathway are highly enriched, as reported previously (Takamiya et al., 1996; Furukawa et al., 2008). Enrichment of these simple gangliosides is maintained in GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) brain; additionally, the complex gangliosides GD1a, GT1b, GM1, and GD1b are also present at approximately equal levels in each strain and at lower levels than in WT brain. Based on the normalized intensities from the extracted ion chromatogram of the D18:1/18:0 species for each complex ganglioside, restoration levels are 25–40% of WT whole-brain values. The lower levels of complex gangliosides seen in both GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) shown by TLC and enzyme activity assays compared with WT may be in part attributable to a dilution effect, because analyses were conducted on whole brain. The enrichment of simple gangliosides in the rescue strains is likely attributable to restricted cell specificity of the transgenic GalNAcT enzyme, with consequent buildup in the un-rescued tissue compared with the global expression of GalNAcT in WT brain.
Immunostaining of PNS and CNS tissue was performed using AgAbs to delineate cell specificity of the transgenically reinstated gangliosides (Fig. 2). Monoclonal antibodies raised against complex gangliosides were applied to spinal cord sections and ex vivo whole-mount TS nerve–muscle preparations to demonstrate appropriate expression in CNS and PNS among genotypes and compare fluorescence levels as an indication of expression levels in the target membranes. Despite lower levels of whole-brain ganglioside as determined by TLC above, AgAb immunostaining at similar levels was evident in the distal axon of the NMJ and also on spinal cord ventral column fibers in WT and GalNAcT−/−-Tg(neuronal) mice (Fig. 2). Complex ganglioside expression was absent on axons of GalNAcT−/− mice as expected. In GalNAcT−/−-Tg(glial) mice, pronounced immunostaining was observed at sites corresponding to perisynaptic Schwann cells overlying the nerve terminal (in which the PLP promoter is active; Michalski et al., 2011) and surrounding neurofilament immunoreactivity in ventral spinal cord. It is not possible to establish whether the latter immunostaining is present on the axonal membrane or the adaxonal myelin membrane at this level of resolution, because the two membranes are very closely apposed. With some monoclonal antibodies, we observed perisynaptic Schwann cell labeling at the NMJ of GalNAcT−/−-Tg(neuronal) mice, consistent with activity of the NFL promoter in non-myelinating Schwann cells (Haynes et al., 1999; data not shown). However, primary cultures of Schwann cells and oligodendrocytes from GalNAcT−/−-Tg(neuronal) mice were negative when immunostained with AgAbs to complex gangliosides compared with positive WT glial cultures (data not shown).
Figure 2.

Restoration of complex ganglioside expression in PNS and CNS tissue identified by anti-ganglioside mAb immunostaining. Complex ganglioside at the NMJ (PNS) and in the ventral spinal cord (CNS) are labeled by mouse monoclonal AgAb, followed by detection with a fluorescently labeled anti-mouse IgG antibody (green in merged images). In nerve–muscle preparations, the NMJs are identified by fluorescently conjugated BTx, which binds the AChR on the postsynaptic membrane. The axons of ventral columns in spinal cord sections are identified by anti-neurofilament antibody (red). Complex gangliosides are present on nerve fiber axolemma in WT and GalNAcT−/−-Tg(neuronal) mice in both preparations and absent in the GalNAcT−/− mice. Perisynaptic Schwann cells at the motor nerve terminal of the GalNAcT−/−-Tg(glial) mice are positive for AgAb, and AgAb immunoreactivity also surrounds the neurofilament marker in the ventral spinal cord. Scale bars, 10 μm.
Neuronal but not glial expression of GalNAcT attenuates the behavioral deficits seen in GalNAcT−/− mice
Because GalNAcT−/− mice exhibit age-dependent locomotor deficits, we performed behavioral tests to assess the impact of neuronal and glial complex ganglioside rescue on motor coordination, strength, gait, and balance. Mice of all genotypes maintained a normal lifespan; however, GalNAcT−/− and GalNAcT−/−-Tg(glial) mice developed a severe locomotor deficit with age, manifested most obviously by hindpaw clasping and impaired extension (Fig. 3A) and a markedly ataxic gait with pronounced tremor. Interestingly, old (>18 months) GalNAcT−/−-Tg(glial) mice exhibited excessive facial scratching with excoriation and hair loss, not seen in other genotypes, including GalNAcT−/− (data not shown). GalNAcT−/−-Tg(neuronal) mice appeared normal throughout life, indistinguishable from WT.
Figure 3.

Neuronal but not glial expression of complex gangliosides in GalNAcT−/− mice rescues behavioral deficiencies. A, Hindpaw extension on lifting by the tail occurs in WT mice; this is replicated in GalNAcT−/−-Tg(neuronal) mice, but abnormal clasping of the hindpaws occurs in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice. Grip strength (B), latency to fall from the rotarod (C), and number of foot slips (D) are comparable among genotypes at 2 months. There is significant deterioration with age in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice (2-way ANOVA, p < 0.001) compared with WT and GalNAcT−/−-Tg(neuronal). *** indicates significance compared with WT; ### signifies significance compared with neuronal rescue. *p < 0.05; *** and ###, p < 0.001.
On quantitative testing, hindlimb grip strength is similar in the four genotypes at 2–3 months and then significantly and progressively deteriorates (two-way ANOVA, p < 0.001; Fig. 3B) in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice from 6 months of age, as revealed by post hoc tests. GalNAcT−/− and GalNAcT−/−-Tg(glial) mice also exhibit equally poor motor performance and coordination that progresses with age as determined by a reduced latency to fall from the rotarod (two-way ANOVA, p < 0.001; Fig. 3C) and an increase in the number of foot falls on grid walking (two-way ANOVA, p < 0.001; Fig. 3D). There is no significant difference in task performance between WT GalNAcT−/−-Tg(neuronal) mice.
Morphological appearances as a function of complex ganglioside expression in neurons and glia
Ultrastructural abnormalities in GalNAcT−/− mice principally include an age-dependent increase in degenerate axon number, abnormal myelination (CNS dysmyelination and PNS demyelination), loss of innermost PN transverse bands, and axolemmal protrusions at the NoR (Sheikh et al., 1999; Sugiura et al., 2005; Susuki et al., 2007). Therefore, we assessed these parameters in groups of normal, GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and GalNAcT−/−-Tg(glial) mice at young (2 months) and aged (12 months) time points to determine whether neuronal or glial ganglioside expression was critical to the development of these pathological abnormalities and that correlated with behavioral performance. None of the parameters measured were significantly different among the four genotypes at 2 months (data not shown); however, by 12 months, significant pathological deviations in genotypes were seen (Fig. 4), confirming the age-dependent nature of neurodegeneration and corresponding to the timeframe of the development of behavioral deficiencies.
Figure 4.
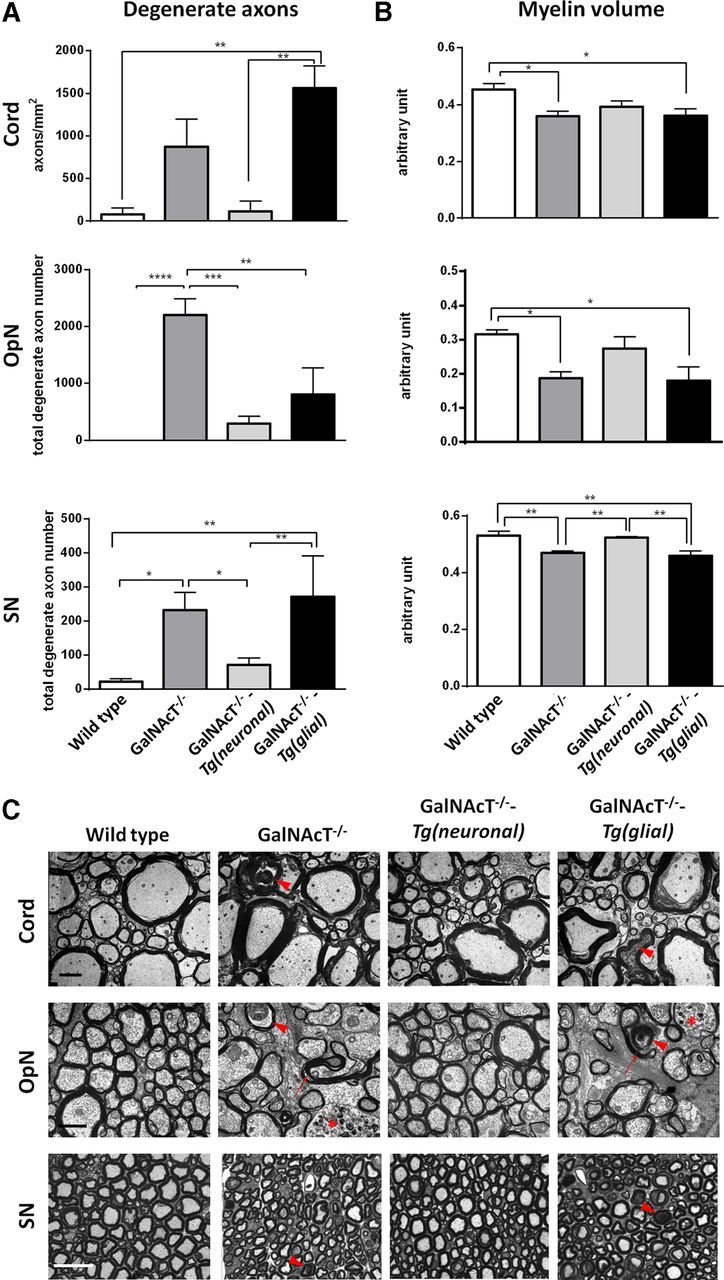
Prevention of morphological abnormalities in neuronal but not glial rescue mice. A, Degenerate axon density or number is greater in 12-month-old GalNAcT−/− mice and reaches significance in OpN and SN compared with age-matched WT mice. Neuronal expression of complex gangliosides significantly attenuates this degeneration, whereas degenerate axon density and number in glial rescue mice remains significantly greater in spinal cord and SN, respectively. B, Myelin volume is significantly reduced in all tissues studied for GalNAcT−/− and GalNAcT−/−-Tg(glial) mice compared with WT and also compared with GalNAcT−/−-Tg(neuronal) in SN. C, Representative EM and light microscopic images from transverse sections of spinal cord, OpN, and SN for all genotypes show normalization of axon and myelin in neuronal rescue mice and increased degenerate axons (indicated by red arrowheads), myelin thinning, and poorer ultrastructure in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice. Organelle-filled axons and redundant myelin occurred frequently in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice OpN and are indicated by red asterisks and arrows, respectively. One-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: Cord, 2 μm; OpN, 1 μm; SN, 25 μm.
At 12 months of age, the density (cord) or number (OpN and SN) of degenerate axons is significantly greater in both GalNAcT−/− and GalNAcT−/−-Tg(glial) SN and in OpN for GalNAcT−/− and ventral spinal cord for GalNAcT−/−-Tg(glial) compared with WT and GalNAcT−/−-Tg(neuronal) mice (one-way ANOVA, p < 0.05; Fig. 4A). In contrast GalNAcT−/−-Tg(neuronal) mice have no significant axon degeneration in any of the three sites assessed compared with WT. Degenerate axons are indicated by red arrowheads in representative images in Figure 4C. Average myelin volume varies significantly among genotypes (one-way ANOVA, p < 0.05) and also varies between neural tissues (Fig. 4B). GalNAcT−/− and GalNAcT−/−-Tg(glial) myelin volume is significantly reduced compared with WT mice in all three sites. GalNAcT−/−-Tg(neuronal) myelin volumes does not significantly differ from WT in any site. Together, these findings indicate that GalNAcT−/−-Tg(glial) mouse morphology (degenerate axon number and myelin volume) is not significantly different from GalNAcT−/−, whereas neuronal expression of gangliosides in GalNAcT−/−-Tg(neuronal) is sufficient to attenuate age-related axonal degeneration and myelin volume changes.
In qualitative observations, abnormal organelle-filled axons and redundant myelin (Fig. 4C, indicated by red asterisks and arrows, respectively) were frequently observed in OpN axons from GalNAcT−/− and GalNAcT−/−-Tg(glial) mice. Whereas myelin volume measurements were quantitatively normal in GalNAcT−/−-Tg(neuronal) mice, the myelin appeared looser than in WT, suggesting that subtle defects in compaction were not ameliorated in either this or the GalNAcT−/−-Tg(glial) mice.
Restoration of nodal architecture by neuronal expression of complex gangliosides
The aberrant distribution of nodal ion channels (Kv1.1 and Nav1.6) described previously in GalNAcT−/− mice (Susuki et al., 2007) indicates a role for complex gangliosides in the maintenance of nodal domains. In both SN (PNS) and OpN (CNS), rectification of nodal ion channel localization to discrete domains was observed after neuronal, but not glial, expression of complex gangliosides (Fig. 5). In the SN of GalNAcT−/− and GalNAcT−/−-Tg(glial) mice, immunohistology reveals significant invasion of the PN with JPN potassium channels (Kv1.1) compared with WT mice (Fig. 5A,B, indicated by orange arrows; one-way ANOVA, p < 0.05). In GalNAcT−/−-Tg(neuronal) mice, Kv1.1 localization is restored to normal. A consequence of the Kv1.1 PN invasion in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice is a reduction in the distance between JPN Kv1.1-positive domains compared with WT, whereas Caspr staining lengths remain similar (Fig. 5A–C; one-way ANOVA, p < 0.001), thereby effectively indicating an overlap in Kv1.1 and Caspr staining at the PN/JPN border. In contrast, in GalNAcT−/−-Tg(neuronal) mice, the inter-Kv1.1 distance and Caspr staining domains are significantly longer than in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice (Figure 5A–C; one-way ANOVA, p < 0.01). The number of PN protrusions filled with Caspr immunoreactivity were significantly greater in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice compared with both WT and GalNAcT−/−-Tg(neuronal) mice (Fig. 5A–C; one-way ANOVA, p < 0.05, indicated by white arrows). These protrusions do not contain neurofilament immunoreactivity, suggesting that they are attributable to discrete local disturbances in the membrane rather than alterations in neurofilament ultrastructure (data not shown). The lateral extent of Nav1.6 immunostaining increased with loss of complex gangliosides compared with WT and remained increased in both GalNAcT−/−-Tg(glial) and GalNAcT−/−-Tg(neuronal) mice (Fig. 5B,C). These results suggest a role for complementary complex ganglioside expression in closely apposed membranes and the reliance on these lipid interactions for the fine-tuning of domain organization.
Figure 5.

Restoration of normal nodal architecture by expression of complex gangliosides in neurons of GalNAcT−/− mice. A, Representative illustrative images per genotype of Caspr (green) and Kv1.1 (magenta) immunoreactivity at SN and OpN NoR in 6-month-old mice. In SN Kv1.1, invasion into the PN is indicated by orange arrows (SN and OpN) and Caspr protrusions (SN only) by white arrows. B, Invasion of the PN (identified by Caspr) with JPN marker Kv1.1 staining significantly increased in GalNAcT−/− and GalNAcT−/−-Tg(glial) mice compared with WT and GalNAcT−/−-Tg(neuronal) mice SN. Consequently, the distance between Kv1.1-positive domains significantly decreased for both genotypes. This distance was lengthened in GalNAcT−/−-Tg(neuronal) mice compared with WT. The number of PN Caspr staining protrusions significantly increased for GalNAcT−/− and GalNAcT−/−-Tg(glial) SN compared with WT and GalNAcT−/−-Tg(neuronal) levels, which were comparable. Compared with WT nerve, Caspr staining was significantly lengthened in GalNAcT−/−-Tg(neuronal) nerve. Conversely, Caspr staining length was significantly shorter in GalNAcT−/− and GalNAcT−/−-Tg(glial) SN compared with GalNAcT−/−-Tg(neuronal). The length of Nav1.6 immunostaining significantly increased in GalNAcT−/− and both rescue mice compared with WT. C, To scale, schematic representing the length of staining in each domain per genotype. One-way ANOVA, p < 0.05. * signifies significance compared with WT; # signifies significance compared with GalNAcT−/−-Tg(neuronal). * and #, p < 0.05; ** and ##, p < 0.01; *** and ###, p < 0.001. Scale bar, 10 μm.
Ultrastructural examination of longitudinal sections through WT SN NoR demonstrates individual PN loops aligning with the axon, connected by transverse bands (Fig. 5D). In all three complex ganglioside-deficient genotypes, the PN loops appear disorganized and aberrantly stacked, particularly at the nodal/PN region in GalNAcT−/−-Tg(glial) and GalNAcT−/−-Tg(neuronal) mice. These focal areas of disorganization may correspond to the sites of Caspr-filled protrusions observed by immunostaining. Whereas transverse bands between axons and PN loops are absent at the PN/JPN border of GalNAcT−/− mice, we observed that they were present here both in GalNAcT−/−-Tg(glial) and GalNAcT−/−-Tg(neuronal) mice (Fig. 6, enlargements shown in insets). The neurofilament appears normal in all genotypes in this region.
Figure 6.

Transverse bands are maintained by expression of complex gangliosides in neurons and glia of GalNAcT−/− mice. Representative longitudinal SN EM images demonstrate normal NoR ultrastructural features in WT mice, including ordered arrangement of PN loops and appropriate formation of transverse bands between the axon and glial membranes (visible in enlarged inset). Transverse bands are often absent in GalNAcT−/− mice but can be identified in both GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) mice. In GalNAcT−/− and both rescue mice, PN loops aberrantly stack and do not all make appropriate contact with the axolemma. Scale bars, 0.5 μm.
Electrophysiological examination of SN
In view of the ion channel and nodal architecture abnormalities, degenerate axons, and myelin volume loss present in ganglioside null and rescue mice, we performed ex vivo extracellular recordings on SN from the different genotypes. Subtle reductions in CV were seen in all ganglioside-deficient mice compared with WT, but this did not reach significance (Fig. 7A). There is a significant difference in rate of rise of the compound nerve action potential in all genotypes compared with WT mice (Fig. 7B,C; one-way ANOVA, p < 0.01). Paired pulse recordings show that refractoriness is not altered in GalNAcT−/−, GalNAcT−/−-Tg(glial), or GalNAcT−/−-Tg(neuronal) mice compared with WT (Fig. 7D). These results suggest that the behavioral phenotype is not accounted for by SN conduction slowing because recovery of the behavioral phenotype in neuronal rescue mice is not mirrored by an improvement in conduction. It is likely that changes to the refractory period were not detected because of very subtle invasion of the paranodes by Kv1.1 channels.
Figure 7.

Peripheral nerve functional recovery in rescue mice. Analysis of extracellular recordings from 4- to 6-month-old GalNAcT−/−, GalNAcT−/−-Tg(neuronal), and Tg(glial) SN show a reduction in CV that does not reach significance (A). B, Rate of rise of the compound nerve action potential is significantly reduced compared with WT for all other genotypes (one-way ANOVA, p < 0.05). C, Representative trace for each genotype from which analysis was performed. D, The amplitude of the second pulse per interstimulus interval in paired pulse recordings were plotted per genotype; there were no significant changes to refractory period (two-way ANOVA, p > 0.05). ** and *** signifies significance compared with WT. **p < 0.01, ***p < 0.001. Individual mice data are plotted to demonstrate the variance in the recordings among animals.
Discussion
Many neurological abnormalities associated with global complex ganglioside deficiency achieved through targeted gene knock-out have been described previously (Takamiya et al., 1996; Sheikh et al., 1999; Chiavegatto et al., 2000; Sugiura et al., 2005; Susuki et al., 2007). However, the precise cellular contribution to these phenotypes has never been investigated. Herein, through selective reintroduction of glycosyltransferase activity in a site-specific manner, we find that neuronal, and not glial, rescue of complex gangliosides is both necessary and sufficient to prevent the age-dependent neurodegenerative phenotype seen in global GalNAcT−/− deficiency states. These findings clearly demonstrate the importance of neuronally expressed GalNAcT in maintaining nervous system integrity throughout the lifespan.
Certain technical caveats may affect the interpretation of these results. The targeting strategy we adopted used NFL and PLP promoters to drive GalNAcT expression in a cell-restricted manner to neurons and myelin-forming glia, respectively. Although both vectors have been used widely to achieve this, it is recognized that, at different stages of mouse development and in different neuronal and glial subtypes, these promoter activities may be more promiscuously active than generally stated (Haynes et al., 1999; Michalski et al., 2011). For example, transient expression of neurofilament mRNA has been described in non-myelinating Schwann cells (Fabrizi et al., 1997; Sotelo-Silveira et al., 2000), thereby providing one explanation for the presence of complex ganglioside immunoreactive perisynaptic Schwann cells we observed at the GalNAcT−/−-Tg(neuronal) mouse NMJ. However, the absence of complex gangliosides in cultured Schwann cells and oligodendrocytes provides overall confidence in the validity of using the NFL promoter for this model. Furthermore, the activity of the natural GalNAcT promoter and of the enzyme itself, which is highly regulated in developmental and spatial patterns, is not recapitulated in these transgenic mice, especially when considering that the regional patterns of brain ganglioside composition will change continuously throughout life (Segler-Stahl et al., 1983; Ikarashi et al., 2011). It is also known that gangliosides can transfer between membranes by shedding and uptake (Olshefski and Ladisch, 1996; Lauc and Heffer-Lauc, 2006). Indeed, substantial levels of different gangliosides circulate in the plasma and may be derived from dietary intake or acquired transplacentally from heterozygous dams used in our breeding programs (McJarrow et al., 2009; Mitchell et al., 2012). Therefore, it is possible that glial membranes might acquire complex gangliosides from neuronal membranes in GalNAcT−/−-Tg(neuronal) mice and vice versa, regardless of their primary site of biosynthesis.
Notwithstanding these caveats, our analyses demonstrate that site-specific reintroduction of complex gangliosides has been achieved. GalNAcT−/− mice have increased expression of the simple gangliosides (Takamiya et al., 1996; Furukawa et al., 2008). Both GalNAcT−/−-Tg(neuronal) and GalNAcT−/−-Tg(glial) retained high levels of these simple gangliosides, although we do not know whether these were present in GalNAcT-deficient cells or were still accumulated in GalNAcT-replete cells containing rate-limiting levels of enzyme activity. Although it has been proposed previously that neurodegeneration in GalNAcT−/− mice might in part be attributable to a toxic gain of function, our data showing retention of very high levels of these simple gangliosides would argue that deficiency of complex gangliosides appear to be the major factor.
Complex gangliosides are present in both axons and glia (Ogawa-Goto et al., 1992; Svennerholm, 1994; Ogawa-Goto and Abe, 1998), with greater enrichment of GM1 and GD1a in axons (Ogawa-Goto and Abe, 1998) and immunostaining (Gong et al., 2002). Our immunostaining for complex gangliosides was most apparent on the axons rather than myelin in WT and GalNAcT−/−-Tg(neuronal) mice and was absent in both compartments in GalNAcT−/− mice. GalNAcT−/−-Tg(glial) perisynaptic Schwann cells at motor nerve terminals were labeled by AgAbs, and ventral column fibers were also immunopositive. If indeed complex gangliosides are more prevalent in the axonal compartment in WT mice, this could explain why a rescue of neuronal complex gangliosides restores the GalNAcT−/− phenotype to near normality, whereas recovery of gangliosides in the glial compartment has no effect.
Behavioral features associated with motor coordination and balance that are impaired in GalNAcT−/− mice (Chiavegatto et al., 2000) are normalized in GalNAcT−/−-Tg(neuronal) mice. Additionally, the characteristic tremor and ataxia observed in the GalNAcT-deficient mice that has been likened to parkinsonism (Wu et al., 2011) is also completely attenuated in GalNAcT−/−-Tg(neuronal) mice. The precise source(s) of the gross behavioral abnormalities in GalNAcT−/− mice has never been established because of the remarkably preserved gross brain architecture (Takamiya et al., 1996) combined with the multitude of cell biological functions modulated by gangliosides (Wu et al., 2005; Ohmi et al., 2009). A prominent suggestion is that complex ganglioside deficiency leads to impairment of axon–myelin stability and consequent axonal degeneration (Sugiura et al., 2005; Schnaar, 2010). The myelin structural abnormalities seen in GalNAcT−/− mice and their reversal in at least some sites in GalNAcT−/−-Tg(neuronal) mice point toward an impairment in myelination and axo-glial junction formation as at least partially responsible for the behavioral phenotype rather than being directly attributable to the rather low frequency of degenerate axons. Electrophysiologically, we only examined peripheral myelinated axons; although confirming peripheral CV slowing in GalNAcT−/− mice (Susuki et al., 2007), this was not recovered by neuronal or glial complex ganglioside rescue, indicating that peripheral nerve CV defects cannot be responsible for the clinical phenotype, although the situation may differ in the CNS, which is the more likely source of the phenotype.
Sodium channel dispersion and myelin sheath disruption lead to conduction slowing (Gutiérrez et al., 1995; Ichimura et al., 2005). The necessary spatial separation of sodium and potassium channels at the NoR is provided by PN loop contact with the axolemma and axo-glial transverse band formation (Rasband et al., 2003; Rosenbluth et al., 2003). Our immunohistological data demonstrate lengthened Nav1.6 clusters in the PNS for all ganglioside-deficient genotypes compared with WT, most apparent in GalNAcT−/−-Tg(glial) mice. This correlates with our CV data. Additionally, PN loops only form orderly attachments on the axon in WT mice and instead have stacked PN loops in all mutants, primarily seen at the nodal border. This suggests that the abnormal Nav1.6 distribution may be attributable to abnormal PN loop formation. Lengthening of sodium channel domains reported here and previously in GalNAcT−/− mice (Susuki et al., 2007) and prominently in GalNAcT−/−-Tg(glial) mice indicates a breakdown in the nodal/PN border that may be influenced by complex ganglioside interactions with ligands in both the glial and axonal membranes. Indeed, a breakdown in the axo-glial junction and subsequent lengthening of sodium channel domains and PN invasion by Kv channels is also seen in the sulfatide-deficient mouse (Ishibashi et al., 2002). Unexpectedly, despite the dogma that potassium channels mislocalize to the paranodes attributable to loss of transverse bands, here Kv1.1 invasion occurred in GalNAcT−/−-Tg(glial) mice although we observed intact transverse bands ultrastructurally, albeit in qualitative rather than quantitative observations. It is possible that the physical barrier provided by transverse bands is only one component required for Kv1.1 localization, additionally requiring specialized lipid raft associated anchoring domains that involve gangliosides (Gu and Gu, 2011). Normal refractory periods were recorded in all mice, suggesting that the Kv1.1 channel mislocalization in ganglioside deficiency states is insufficient to directly result in major functional disruption.
Our principal finding that function is restored by neuronal rather than glial GalNAcT expression indicates the requirement for an axonally expressed ligand in fine tuning the interacting molecules at axo-glial junctions that are required for maintaining myelin, axonal, and nodal integrity. The complex gangliosides GD1a and GT1b have been described as axonal ligands for myelin-associated glycoprotein (MAG) localized on the innermost wrap of the myelin sheath (Collins et al., 1997). The age-related degenerative phenotype, functional, and morphological deficits displayed by GalNAcT−/− mice parallel those found in MAG null mice and mice created from interbreeding the two genotypes (Pan et al., 2005). Restoration of MAG–ganglioside interaction in GalNAcT−/−-Tg(neuronal) mice might thus account for at least some components of the preserved phenotype.
Other abundant gangliosides, GM1 and GD1b, are likely to also have MAG-independent neuroprotective roles. Complex gangliosides, particularly GM1, are an integral member of membrane-specific microdomains, lipid rafts (Simons and Ikonen, 1997), that subserve many signaling and trafficking functions that could be disrupted by ganglioside loss, such as complement regulator dysfunction (Ohmi et al., 2009). Lipid rafts are also involved in the recruitment and stabilization of the glial adhesion molecule neurofascin 155 at the NoR (Schafer et al., 2004). Indeed, GalNAcT−/− mice have mislocalized neurofascin at the NoR (Susuki et al., 2007), and mice lacking neurofascin fail to develop normal NoR (Sherman et al., 2005), demonstrating the importance of such an interaction.
The global loss of complex gangliosides results in an obvious behavioral phenotype whose specific origin is unknown but likely attributable to multiple impairments. Here we show that the wide-ranging degenerative features and consequent behavioral phenotype can be overcome by the expression of complex gangliosides limited to neuronal membranes. Strikingly, glial expression of complex gangliosides does not have the same effect. This demonstrates the necessity for site-specific expression of complex gangliosides in maintenance of the nervous system and opens up experimental avenues for more precisely defining the role of complex gangliosides in nervous system maintenance. The discovery of human disorders of ganglioside biosynthesis (Simpson et al., 2004; Boukhris et al., 2013) and autoimmune disorders in which gangliosides are targets (Willison and Plomp, 2008) adds important clinical dimensions to this area.
Footnotes
R.M., D.Y., J.A.B., and H.J.W. are supported The Wellcome Trust Grant 092805. M.E.C. holds a Medical Research Council PhD studentship. G.R.M. holds a Guillain-Barré Syndrome Support Group UK studentship. J.C. holds a Biotechnology and Biological Sciences Research Council Doctoral Training Award studentship, and associated work was performed at the Glasgow Polyomics Facility. P.J.B. is supported by The Wellcome Trust. Ke.F., Ko.F., and Y.O. are supported by Grant-in-Aid for Scientific Research on Innovative Areas 23110008 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Boffey J, Odaka M, Nicoll D, Wagner ER, Townson K, Bowes T, Conner J, Furukawa K, Willison HJ. Characterisation of the immunoglobulin variable region gene usage encoding the murine anti-ganglioside antibody repertoire. J Neuroimmunol. 2005;165:92–103. doi: 10.1016/j.jneuroim.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Boukhris A, Schule R, Loureiro JL, Lourenço CM, Mundwiller E, Gonzalez MA, Charles P, Gauthier J, Rekik I, Acosta Lebrigio RF, Gaussen M, Speziani F, Ferbert A, Feki I, Caballero-Oteyza A, Dionne-Laporte A, Amri M, Noreau A, Forlani S, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93:118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes T, Wagner ER, Boffey J, Nicholl D, Cochrane L, Benboubetra M, Conner J, Furukawa K, Furukawa K, Willison HJ. Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in Campylobacter jejuni strains associated with Guillain-Barre syndrome. Infect Immun. 2002;70:5008–5018. doi: 10.1128/IAI.70.9.5008-5018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavegatto S, Sun J, Nelson RJ, Schnaar RL. A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp Neurol. 2000;166:227–234. doi: 10.1006/exnr.2000.7504. [DOI] [PubMed] [Google Scholar]
- Collins BE, Yang LJ, Mukhopadhyay G, Filbin MT, Kiso M, Hasegawa A, Schnaar RL. Sialic acid specificity of myelin-associated glycoprotein binding. J Biol Chem. 1997;272:1248–1255. doi: 10.1074/jbc.272.2.1248. [DOI] [PubMed] [Google Scholar]
- Dicesare JL, Dain JA. The enzymic synthesis of ganglioside. IV. UDP-N-acetylgalactosamine: (N-acetylneuraminyl)-galactosylglucosyl ceramide N-acetylgalactosaminyltransferase in rat brain. Biochim Biophys Acta. 1971;231:385–393. doi: 10.1016/0005-2760(71)90151-2. [DOI] [PubMed] [Google Scholar]
- Fabrizi C, Kelly BM, Gillespie CS, Schlaepfer WW, Scherer SS, Brophy PJ. Transient expression of the neurofilament proteins NF-L and NF-M by Schwann cells is regulated by axonal contact. J Neurosci Res. 1997;50:291–299. doi: 10.1002/(SICI)1097-4547(19971015)50:2<291::AID-JNR17>3.0.CO%3B2-B. [DOI] [PubMed] [Google Scholar]
- Fewou SN, Plomp JJ, Willison HJ. The pre-synaptic motor nerve terminal as a site for antibody-mediated neurotoxicity in autoimmune neuropathies and synaptopathies. J Anat. 2013 doi: 10.1111/joa.12088. doi: 10.1111/joa.12088. Advance online publication. Retrieved November 27, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Arsove S, Meath JA. Isolation of brain strandin, a new type of large molecule tissue component. J Biol Chem. 1951;191:819–831. [PubMed] [Google Scholar]
- Furukawa K, Aixinjueluo W, Kasama T, Ohkawa Y, Yoshihara M, Ohmi Y, Tajima O, Suzumura A, Kittaka D, Furukawa K. Disruption of GM2/GD2 synthase gene resulted in overt expression of 9-O-acetyl GD3 irrespective of Tis21. J Neurochem. 2008;105:1057–1066. doi: 10.1111/j.1471-4159.2008.05232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA. Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain. 2002;125:2491–2506. doi: 10.1093/brain/awf258. [DOI] [PubMed] [Google Scholar]
- Griffiths IR, Duncan ID, McCulloch M. Shaking pups: a disorder of central myelination in the spaniel dog. II. Ultrastructural observations on the white matter of the cervical spinal cord. J Neurocytol. 1981;10:847–858. doi: 10.1007/BF01262657. [DOI] [PubMed] [Google Scholar]
- Gu C, Gu Y. Clustering and activity tuning of Kv1 channels in myelinated hippocampal axons. J Biol Chem. 2011;286:25835–25847. doi: 10.1074/jbc.M111.219113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R, Boison D, Heinemann U, Stoffel W. Decompaction of CNS myelin leads to a reduction of the conduction velocity of action potentials in optic nerve. Neurosci Lett. 1995;195:93–96. doi: 10.1016/0304-3940(94)11789-L. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Glycosphingolipids in cellular interaction, differentiation, and oncogenesis. Annu Rev Biochem. 1981;50:733–764. doi: 10.1146/annurev.bi.50.070181.003505. [DOI] [PubMed] [Google Scholar]
- Haynes LW, Schmitz S, Clegg JC, Fooks AR. Expression of neurofilament L-promoter green-fluorescent protein constructs in immortalized Schwann cell-neuron coculture. Neurosci Lett. 1999;271:155–158. doi: 10.1016/S0304-3940(99)00510-8. [DOI] [PubMed] [Google Scholar]
- Ichimura H, Shiga T, Abe I, Hara Y, Terui N, Tsujino A, Ochiai N. Distribution of sodium channels during nerve elongation in rat peripheral nerve. J Orthop Sci. 2005;10:214–220. doi: 10.1007/s00776-004-0870-8. [DOI] [PubMed] [Google Scholar]
- Ikarashi K, Fujiwara H, Yamazaki Y, Goto J, Kaneko K, Kato H, Fujii S, Sasaki H, Fukumoto S, Furukawa K, Waki H, Furukawa K. Impaired hippocampal long-term potentiation and failure of learning in beta1,4-N-acetylgalactosaminyltransferase gene transgenic mice. Glycobiology. 2011;21:1373–1381. doi: 10.1093/glycob/cwr090. [DOI] [PubMed] [Google Scholar]
- Inoue M, Fujii Y, Furukawa K, Okada M, Okumura K, Hayakawa T, Furukawa K, Sugiura Y. Refractory skin injury in complex knock-out mice expressing only the GM3 ganglioside. J Biol Chem. 2002;277:29881–29888. doi: 10.1074/jbc.M201631200. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, Baba H. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci. 2002;22:6507–6514. doi: 10.1523/JNEUROSCI.22-15-06507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- Kusunoki S, Kaida K, Ueda M. Antibodies against gangliosides and ganglioside complexes in Guillain-Barre syndrome: new aspects of research. Biochim Biophys Acta. 2008;1780:441–444. doi: 10.1016/j.bbagen.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Lauc G, Heffer-Lauc M. Shedding and uptake of gangliosides and glycosylphosphatidylinositol-anchored proteins. Biochim Biophys Acta. 2006;1760:584–602. doi: 10.1016/j.bbagen.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Ledeen R, Wu G. New findings on nuclear gangliosides: overview on metabolism and function. J Neurochem. 2011;116:714–720. doi: 10.1111/j.1471-4159.2010.07115.x. [DOI] [PubMed] [Google Scholar]
- McJarrow P, Schnell N, Jumpsen J, Clandinin T. Influence of dietary gangliosides on neonatal brain development. Nutr Rev. 2009;67:451–463. doi: 10.1111/j.1753-4887.2009.00211.x. [DOI] [PubMed] [Google Scholar]
- Michalski JP, Anderson C, Beauvais A, De Repentigny Y, Kothary R. The proteolipid protein promoter drives expression outside of the oligodendrocyte lineage during embryonic and early postnatal development. PLoS One. 2011;6:e19772. doi: 10.1371/journal.pone.0019772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MD, Henare K, Balakrishnan B, Lowe E, Fong BY, McJarrow P. Transfer of gangliosides across the human placenta. Placenta. 2012;33:312–316. doi: 10.1016/j.placenta.2011.12.018. [DOI] [PubMed] [Google Scholar]
- Ngamukote S, Yanagisawa M, Ariga T, Ando S, Yu RK. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem. 2007;103:2327–2341. doi: 10.1111/j.1471-4159.2007.04910.x. [DOI] [PubMed] [Google Scholar]
- Ogawa-Goto K, Abe T. Gangliosides and glycosphingolipids of peripheral nervous system myelins—a minireview. Neurochem Res. 1998;23:305–310. doi: 10.1023/A:1022497114813. [DOI] [PubMed] [Google Scholar]
- Ogawa-Goto K, Funamoto N, Ohta Y, Abe T, Nagashima K. Myelin gangliosides of human peripheral nervous system: an enrichment of GM1 in the motor nerve myelin isolated from cauda equina. J Neurochem. 1992;59:1844–1849. doi: 10.1111/j.1471-4159.1992.tb11018.x. [DOI] [PubMed] [Google Scholar]
- Ohmi Y, Tajima O, Ohkawa Y, Mori A, Sugiura Y, Furukawa K, Furukawa K. Gangliosides play pivotal roles in the regulation of complement systems and in the maintenance of integrity in nerve tissues. Proc Natl Acad Sci U S A. 2009;106:22405–22410. doi: 10.1073/pnas.0912336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmi Y, Ohkawa Y, Yamauchi Y, Tajima O, Furukawa K, Furukawa K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem Res. 2012;37:1185–1191. doi: 10.1007/s11064-012-0764-7. [DOI] [PubMed] [Google Scholar]
- Okada M, Itoh M, Haraguchi M, Okajima T, Inoue M, Oishi H, Matsuda Y, Iwamoto T, Kawano T, Fukumoto S, Miyazaki H, Furukawa K, Aizawa S, Furukawa K. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem. 2002;277:1633–1636. doi: 10.1074/jbc.C100395200. [DOI] [PubMed] [Google Scholar]
- Olshefski R, Ladisch S. Intercellular transfer of shed tumor cell gangliosides. FEBS Lett. 1996;386:11–14. doi: 10.1016/0014-5793(96)00392-4. [DOI] [PubMed] [Google Scholar]
- Pan B, Fromholt SE, Hess EJ, Crawford TO, Griffin JW, Sheikh KA, Schnaar RL. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Exp Neurol. 2005;195:208–217. doi: 10.1016/j.expneurol.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Taylor CM, Bansal R. Paranodal transverse bands are required for maintenance but not initiation of Nav1.6 sodium channel clustering in CNS optic nerve axons. Glia. 2003;44:173–182. doi: 10.1002/glia.10284. [DOI] [PubMed] [Google Scholar]
- Ravindran MS, Tanner LB, Wenk MR. Sialic acid linkage in glycosphingolipids is a molecular correlate for trafficking and delivery of extracellular cargo. Traffic. 2013;14:1182–1191. doi: 10.1111/tra.12100. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Dupree JL, Popko B. Nodal sodium channel domain integrity depends on the conformation of the paranodal junction, not on the presence of transverse bands. Glia. 2003;41:318–325. doi: 10.1002/glia.10179. [DOI] [PubMed] [Google Scholar]
- Ruan S, Lloyd KO. Glycosylation pathways in the biosynthesis of gangliosides in melanoma and neuroblastoma cells: relative glycosyltransferase levels determine ganglioside patterns. Cancer Res. 1992;52:5725–5731. [PubMed] [Google Scholar]
- Ruan S, Raj BK, Furukawa K, Lloyd KO. Analysis of melanoma cells stably transfected with beta 1,4GalNAc transferase (GM2/GD2 synthase) cDNA: relative glycosyltransferase levels play a dominant role in determining ganglioside expression. Arch Biochem Biophys. 1995;323:11–18. doi: 10.1006/abbi.1995.0003. [DOI] [PubMed] [Google Scholar]
- Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci. 2013;33:10195–10208. doi: 10.1523/JNEUROSCI.0822-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Bansal R, Hedstrom KL, Pfeiffer SE, Rasband MN. Does paranode formation and maintenance require partitioning of neurofascin 155 into lipid rafts? J Neurosci. 2004;24:3176–3185. doi: 10.1523/JNEUROSCI.5427-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010;584:1741–1747. doi: 10.1016/j.febslet.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler-Stahl K, Webster JC, Brunngraber EG. Changes in the concentration and composition of human brain gangliosides with aging. Gerontology. 1983;29:161–168. doi: 10.1159/000213109. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, Wang H, Wiznitzer M, Gurtz K, Verganelaki A, Pryde A, Patton MA, Dwek RA, Butters TD, Platt FM, Crosby AH. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology. 2007;17:1R–13R. doi: 10.1093/glycob/cwl052. [Erratum (2007) 17: 1030] [DOI] [PubMed] [Google Scholar]
- Sotelo-Silveira JR, Calliari A, Kun A, Benech JC, Sanguinetti C, Chalar C, Sotelo JR. Neurofilament mRNAs are present and translated in the normal and severed sciatic nerve. J Neurosci Res. 2000;62:65–74. doi: 10.1002/1097-4547(20001001)62:1<65::AID-JNR7>3.0.CO%3B2-Z. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Furukawa K, Tajima O, Mii S, Honda T, Furukawa K. Sensory nerve-dominant nerve degeneration and remodeling in the mutant mice lacking complex gangliosides. Neuroscience. 2005;135:1167–1178. doi: 10.1016/j.neuroscience.2005.07.035. [DOI] [PubMed] [Google Scholar]
- Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, Furukawa K, Furukawa K, Rasband MN, Yuki N. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia. 2007;55:746–757. doi: 10.1002/glia.20503. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. Designation and schematic structure of gangliosides and allied glycosphingolipids. Prog Brain Res. 1994;101:XI–XIV. doi: 10.1016/S0079-6123(08)61935-4. [DOI] [PubMed] [Google Scholar]
- Tajima O, Egashira N, Ohmi Y, Fukue Y, Mishima K, Iwasaki K, Fujiwara M, Inokuchi J, Sugiura Y, Furukawa K, Furukawa K. Reduced motor and sensory functions and emotional response in GM3-only mice: emergence from early stage of life and exacerbation with aging. Behav Brain Res. 2009;198:74–82. doi: 10.1016/j.bbr.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, Sakae M, Kishikawa M, Shiku H, Furukawa K, Aizawa S. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci U S A. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA. Stereological techniques. Practical methods in electron microscopy. In: Glauert AM, editor. Quantitative methods in biology. Amsterdam: North Holland; 1977. pp. 5–84. [Google Scholar]
- Williams MA, McCluer RH. The use of Sep-Pak C18 cartridges during the isolation of gangliosides. J Neurochem. 1980;35:266–269. doi: 10.1111/j.1471-4159.1980.tb12515.x. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Plomp JJ. Anti-ganglioside antibodies and the presynaptic motor nerve terminal. Ann N Y Acad Sci. 2008;1132:114–123. doi: 10.1196/annals.1405.010. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Wang J, Wang Y, Xie X, Meyenhofer MF, Ledeen RW. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci. 2005;25:11014–11022. doi: 10.1523/JNEUROSCI.3635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Kulkarni N, Amin R, Ledeen RW. Mice lacking major brain gangliosides develop parkinsonism. Neurochem Res. 2011;36:1706–1714. doi: 10.1007/s11064-011-0437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Wu YP, Sandhoff R, Werth N, Mizukami H, Ellis JM, Dupree JL, Geyer R, Sandhoff K, Proia RL. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci U S A. 2005;102:2725–2730. doi: 10.1073/pnas.0407785102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J Lipid Res. 2004;45:783–793. doi: 10.1194/jlr.R300020-JLR200. [DOI] [PubMed] [Google Scholar]
- Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides—an overview. J Oleo Sci. 2011;60:537–544. doi: 10.5650/jos.60.537. [DOI] [PMC free article] [PubMed] [Google Scholar]