

Abstract
We have previously shown that the botanical drug candidate PBI-05204, a supercritical CO2 extract of Nerium oleander, provides neuroprotection in both in vitro and in vivo brain slice-based models for focal ischemia (Dunn et al., 2011). Intriguingly, plasma levels of the neurotrophin BDNF were increased in patients treated with PBI-05204 in a phase I clinical trial (Bidyasar et al., 2009). We thus tested the hypothesis that neuroprotection provided by PBI-05204 to rat brain slices damaged by oxygen-glucose deprivation (OGD) is mediated by BDNF. We found, in fact, that exogenous BDNF protein itself is sufficient to protect brain slices against OGD, whereas downstream activation of TrkB receptors for BDNF is necessary for neuroprotection provided by PBI-05204, using three independent methods. Finally, we provide evidence that oleandrin, the principal cardiac glycoside component of PBI-05204, can quantitatively account for regulation of BDNF at both the protein and transcriptional levels. Together, these findings support further investigation of cardiac glycosides in providing neuroprotection in the context of ischemic stroke.
Introduction
Stroke is the fourth-leading cause of death in the U.S., with almost 800,000 new cases occurring each year and further increases expected with the aging population (National Stroke Association, 2010). Although major advances have been made in reducing stroke risk, treatments are still lacking that provide direct neuroprotection and/or neuroresuscitation, despite >50 drug candidates having been tested to date in late-stage clinical trials (Suwanwela and Koroshetz, 2007; Zaleska et al., 2009). Thus, the only FDA-approved drug for stroke remains the intravenous “clot buster” recombinant tissue plasminogen activator (Albers et al., 2011).
We have previously reported that the botanical drug candidate PBI-05204, a defined supercritical CO2 extract of Nerium oleander, provides significant neuroprotection to neural tissues damaged by oxygen-glucose deprivation (OGD), as occurs in ischemic stroke (Dunn et al., 2011). Critically, we demonstrated that PBI-05204 can still provide significant neuroprotection even when administrated with a 4–6 h delay following OGD, and that the neuroprotective activity of PBI-05204 is blood–brain barrier (BBB)-penetrant. Finally, we showed that this neuroprotection is mediated, at least in major part, by the cardiac glycoside molecule oleandrin. In fact, this principal bioactive and BBB-penetrant constituent of PBI-05204 was the original impetus for the study, as oleandrin is closely related in structure to neriifolin, which we had previously identified as the strongest hit from a high-throughput and hypothesis-neutral chemical biology screen for neuroprotective agents using a brain slice-based model of focal ischemia (Wang et al., 2006). However, the mechanism(s) by which cardiac glycosides provide neuroprotection has remained unknown.
We report here that the neuroprotection provided by PBI-05204 appears to be mediated through augmentation of endogenous levels of the neurotrophin brain-derived neurotrophic factor (BDNF). We show that inhibition of BDNF signaling via its cognate receptor, TrkB, blocks the neuroprotection provided by PBI-05204, and that in turn recombinant BDNF itself is able to provide significant neuroprotection to OGD-treated brain slices. Finally, we provide evidence that augmentation of BDNF levels by PBI-05204 can be qualitatively and quantitatively accounted for by its principal cardiac glycoside component, oleandrin, at both the protein and gene transcriptional levels. Together with positive safety data already established in a phase I clinical trial (Bidyasar et al., 2009), these results support continued investigation of PBI-05204 as a drug lead candidate providing neuroprotection in stroke and potentially other traumatic and/or neurodegenerative conditions of the CNS.
Materials and Methods
Compounds.
PBI-05204 was provided by Phoenix Biotechnology and is an ethanol-modified supercritical CO2 extract of Nerium oleander (Newman et al., 2008). Pure oleandrin was purchased from ChromaDex. Stock solutions were made in DMSO and diluted into brain slice culture medium to a final DMSO concentration of 0.1% for all conditions. The BDNF-neutralizing monoclonal antibody mAb#9 was provided by the Developmental Studies Hybridoma Bank at the University of Iowa; a matching preimmune IgG2 isotype control was purchased from BioLegend.
Brain slice preparation and transfection.
Coronal brain slices (250 μm thick) from postnatal day 10 Sprague-Dawley rat pups of either sex (Charles River) were prepared and placed in acute organotypic culture as previously described (Braithwaite et al., 2010; Dunn et al., 2011). Animals were killed in accordance with NIH guidelines and under Duke IACUC approval and oversight. Brain tissues were sliced in ice-cold artificial CSF (ACSF), and subjected to OGD by suspension in glucose-free, N2-bubbled ACSF containing low O2 (<0.5%) for 5.5 min, after which control and OGD-treated brain slices were plated in interface configuration on top of culture medium (Neurobasal A medium supplemented with 15% heat-inactivated horse serum, 10 mm KCl, 10 mm HEPES, 100 U/ml penicillin/streptomycin, 1 mm sodium pyruvate, and 1 mm l-glutamine) set in 0.5% reagent-grade agarose. One hour after cutting, brain slices were biolistically transfected with DNAs encoding yellow fluorescent protein (YFP) and/or TrkB.T1 receptor as previously described (Yacoubian and Lo, 2000). Brain slice explants were incubated for 24 h under 5% CO2 at 37°C. PBI-05204 or oleandrin was added to culture medium at the time of brain slice explantation.
Assessment of neuroprotection.
Twenty-four hours after their preparation, brain slice explants were assessed for neuroprotection by scoring numbers of healthy pyramidal neurons in the cortical regions of each brain slice as previously described (Wang et al., 2006; Dunn et al., 2011). Briefly, pyramidal neurons were identified by their positions and orientations within the cortex and were scored as healthy if exhibiting: (1) a clear and continuously labeled cell body with no obvious signs of shrinkage or collapse, (2) a fully and continuously labeled apical dendrite of normal length, and (3) >2 clear basal dendrites >2 cell body diameters long. All experimental runs were done using single litters of animals (males and females), with brain slices from the littermates pooled and N = 12–18 brain slices randomly assigned to each experimental condition. Data from at least three independent experimental runs are presented as mean numbers of healthy cortical neurons per brain slice, or as a percentage increase over those surviving in the OGD condition ± SD.
Statistical analysis.
For all neuroprotection assays, statistical significance was tested using ANOVA followed by Dunnett's post hoc comparison test at the 0.01 confidence level.
Western blotting.
Lysates from brain slice explants were prepared as previously described (Dunn et al., 2011). Total TrkB or EGF receptor protein levels were assessed by immunoblotting whole brain slice lysates using a pan-Trk antibody (C-14, 1:3000; Santa Cruz Biotechnology) or EGF receptor antibody (C74B9, 1:1000; Cell Signaling Technology), respectively. Phospho-TrkB or phospho-EGF receptor levels were quantified by immunoprecipitation against phosphotyrosine (4G10, 3.5 μg/ml; Millipore) with protein-G agarose, followed by immunoblotting with C-14 or C74B9, respectively. Equivalent total protein amounts were loaded in each lane as determined by BCA protein assay (Pierce).
ELISA.
Brain slices were washed three times with PBS, flash frozen in liquid N2 and stored at −80°C until use. Samples were thawed on ice and homogenized in cold lysis buffer containing 100 mm Tris/HCl pH 7, 2% BSA, 1 m NaCl, 4 mm EDTA, 2% Triton X-100, and 0.1% sodium azide supplemented with Complete Mini protease inhibitor cocktail tablets (Roche) and 17 μg/ml PMSF. Following centrifugation at 14,000 × g for 30 min., supernatants were collected and assayed by BDNF ELISA as per manufacturer's protocol (ChemiKine BDNF sandwich ELISA kit; EMD Millipore).
RNA harvesting and real-time PCR.
RNA was harvested from frozen brain slices (see ELISA, above) using Absolutely RNA miniprep kits (Agilent Technologies/Stratagene). Aliquots (500 ng) of RNA were used for reverse transcription with oligo dT primers and Superscript II (Invitrogen). Resulting cDNA samples were used for quantitative PCR of gene transcripts using Bullseye EvaGreen-low ROX (MidSci) on a ViiA 7 real-time PCR instrument (Applied Biosystems) and intron-spanning Bdnf primers (Aid et al., 2007; McDowell et al., 2010). Each sample was measured in triplicate, with GAPDH levels used to normalize for RNA recovery and sample processing.
Results
PBI-05204 increases BDNF levels and activates TrkB receptors
The impetus for this study was the intriguing observation that plasma levels of BDNF were increased in patients treated with PBI-05204 in a phase I clinical trial (R. Newman, personal communication; Bidyasar et al., 2009). We thus asked whether PBI-05204 could also increase BDNF in brain slice explants as a potential mechanism for the neuroprotection it provides against OGD that we had previously reported (Dunn et al., 2011). As can be seen in Figure 1A, treatment of brain slice explants with PBI-05204 for 24 h did in fact significantly increase BDNF protein levels in a concentration-dependent manner. The effective concentration range for PBI-05204 corresponded well to that we previously established for neuroprotection (Dunn et al., 2011). As a control, we treated brain slices with the pan-caspase inhibitor Boc-D-FMK to show that elevations in BDNF levels were not simply a secondary consequence of general neuroprotection (see Fig. 2G). Figure 1B shows that treatment with the cardiac glycoside oleandrin, the principal known bioactive component of PBI-05204 (Newman et al., 2008), also increased BDNF expression in brain slices in a parallel concentration range given its ∼3% relative abundance in PBI-05204 (Dunn et al., 2011).
Figure 1.

PBI-05204 treatment increases BDNF protein levels and activation of TrkB receptors. A, Brain slices were treated with varying concentrations of PBI-05204 for 2 d, after which tissue lysates were analyzed by ELISA for total BDNF protein content. The pan-caspase inhibitor Boc-D-FMK (“Boc”) also provides neuroprotection (Fig. 2G) but had no effect on BDNF levels. B, The cardiac glycoside oleandrin, the principal bioactive component of PBI-05204, also increased BDNF expression in brain slices in a concentration-dependent manner. C, Representative immunoblot showing PBI-05204 induction of TrkB autophosphorylation in the same concentration range as for BDNF induction in A. In the second lane, 200 ng/ml BDNF is the positive control. D, Densitometric analysis of relative levels of TrkB phosphorylation (“Phospho/total TrkB”) induced by PBI-05204 averaged over six independent runs. *p < 0.05 by Student's t test.
Figure 2.
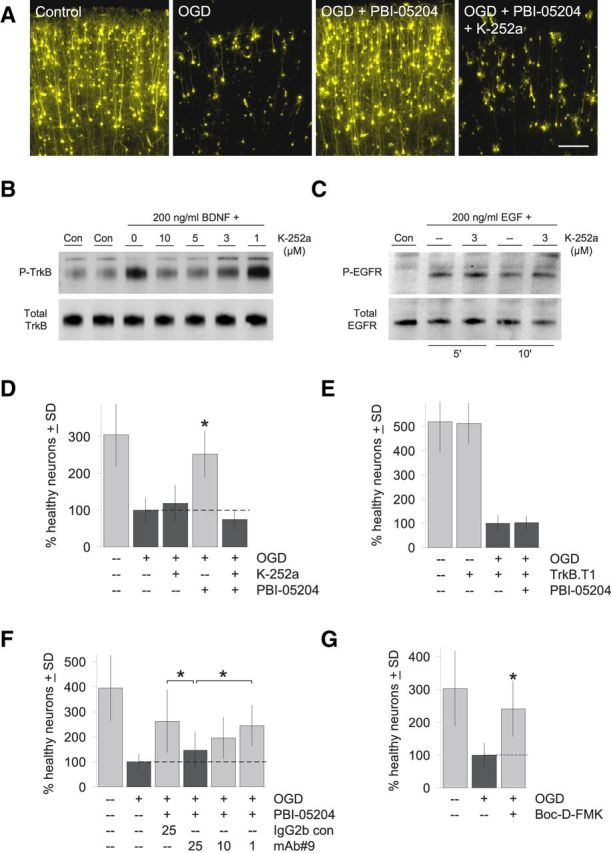
TrkB receptor activation is required for the neuroprotective effects of PBI-05204. A, Representative photomicrographs showing that neurodegeneration induced by transient OGD can be quantitatively rescued by 23 μg/ml PBI-05204 (compare first three panels). Neuroprotection by PBI-05204 is completely blocked by a minimal concentration of K-252a (3 μm) that inhibits the majority of TrkB autophosphorylation (“P-TrkB”) induced by 1 h of treatment with 200 ng/ml BDNF as shown in the representative immunoblot in B. Scale bar, 200 μm. C, In contrast, 3 μm K-252a had no effect on the activation and autophosphorylation of EGFR by 5 or 10 min. of treatment with 200 ng/ml EGF under the identical experimental conditions. D, Quantification of brain slice assays showing that 3 μm K-252a completely blocks the neuroprotection provided by 23 μg/ml PBI-05204 (compare fourth and fifth bars) but has no significant effect on general neuronal health even in OGD-treated brain slices (compare second and third bars). y-Axis shows the percentage increase in numbers of healthy cortical neurons per brain slice relative to the OGD condition. E, Cotransfection with dominant-negative, truncated TrkB.T1 also blocks neuroprotection by 23 μg/ml PBI-05204. Although TrkB.T1 has no effect on control brain slices (light bars), it completely blocks the neuroprotective effects of PBI-05204 (dark bars). F, Finally, the BDNF-neutralizing antibody mAb#9 (Kolbeck et al., 1999) was used as a third independent method to show that disrupting BDNF-TrkB signaling inhibits the neuroprotective activity of 23 μg/ml PBI-05204. mAb#9 was applied to brain slice explants at the concentrations shown (in μg/ml). Significant blockade of the neuroprotective effects of PBI-05204 compared with the preimmune IgG2 isotype control was observed in the concentration range previously reported for mAb#9 (Deogracias et al., 2012). G, Neuronal degeneration induced by transient OGD was rescuable by the pan-caspase inhibitor Boc-D-FMK (100 μm). D–G, Averages of three independent runs are shown; *p < 0.01 by ANOVA followed by Dunnett's post hoc comparison test.
We further showed that increases in BDNF expression induced by PBI-05204 resulted in increased activation of endogenous TrkB receptors as monitored by autophosphorylation (Ip et al., 1993; Fig. 1C). Apparent increases in TrkB phosphorylation were relatively modest, even for the positive control treated for 1 h with 200 ng/ml recombinant BDNF, due to high levels of basal TrkB autophosphorylation in brain slice explants reflecting active ambient BDNF signaling as we have previously shown (McAllister et al., 1997). Nevertheless, significantly increased levels of TrkB phosphorylation could be induced by PBI-05204 treatment in the same concentration range as was observed in Figure 1A for increasing BDNF levels (Fig. 1D).
TrkB receptor activation is required for neuroprotection by PBI-05204
That PBI-05204 treatment increases BDNF protein levels in brain slices as well as downstream activation of their cognate TrkB receptors led to the hypothesis that neuroprotection provided by PBI-05204 to brain slices damaged by OGD is mediated by BDNF. To test this hypothesis, we asked whether downstream activation of TrkB receptors by BDNF is required for neuroprotection by PBI-05204. First, we took a pharmacological approach using an inhibitor of high-affinity neurotrophin Trk receptors, the kinase inhibitor K-252a (Berg et al., 1992; Tapley et al., 1992). Figure 2B shows that 3 μm is a minimal concentration of K-252a that inhibits the majority of Trk receptor activation by BDNF (20 min pretreatment with K-252a at the indicated concentrations followed by 1 h of incubation with 200 ng/ml BDNF as in Fig. 1C; note that concentrations of pharmacological agents generally have to be increased in brain slice culture media to promote tissue penetration compared with conventional dissociated cultures). K-252a at 3 μm under identical treatment conditions did not affect signaling through a parallel receptor tyrosine kinase pathway (EGF receptor after 5 or 10 min of treatment with EGF reflecting the faster kinetics of this pathway; Fig. 2C), suggesting that the selectivity of K-252a for TrkB receptors is retained at this concentration. We found that this concentration of K-252a completely inhibited the neuroprotective effects of PBI-05204 in OGD-treated brain slices (Fig. 2A,D), but had no deleterious effects on either control brain slices transfected with YFP but not subjected to OGD (100 ± 18% vs 86 ± 22% healthy neurons for control vs K-252a-treated; mean ± SD, difference not significant) or those that had been subjected to OGD (Fig. 2D, compare second and third bars).
Next, we used a second method to confirm that TrkB activation is necessary for the neuroprotection provided by PBI-05204. In these experiments, we cotransfected along with YFP a naturally occurring truncated isoform of TrkB, termed TrkB.T1, that is lacking the kinase domain and thereby acts as a dominant negative when recruited to dimerize with full-length TrkB receptors by BDNF ligand (Biffo et al., 1995; Eide et al., 1996). Taking advantage of the near 100% cotransfection linkage rate of biolistics (Braithwaite et al., 2010), we had previously shown that transfection of TrkB.T1 into brain slice explants profoundly alters neuronal responsiveness to BDNF (Yacoubian and Lo, 2000). Here, we show in Figure 2E that transfection of TrkB.T1 completely inhibited the neuroprotective effects of PBI-05204 in OGD-treated brain slices, confirming the results described above using a pharmacological Trk receptor antagonist.
Finally, we used a third independent method to show that BDNF-TrkB signaling is required for the neuroprotective activity of PBI-05204, namely, a highly selective function-blocking antibody against BDNF itself, mAb#9 (Kolbeck et al., 1999; Deogracias et al., 2012). As can be seen in Figure 2F, mAb#9 inhibited the ability of PBI-05204 to protect against OGD treatment in a concentration-dependent manner, whereas a preimmune IgG2 isotype control had no significant effect.
These experiments demonstrated with three independent methods that BDNF-TrkB signaling is necessary for the neuroprotective actions of PBI-05204 in OGD-treated brain slices.
BDNF is sufficient to rescue brain slices from OGD
Conversely, we showed that exogenous BDNF alone is sufficient to rescue brain slices subjected to OGD. Treatment with 200 ng/ml of recombinant BDNF, a concentration previously shown to retain Trk receptor selectivity and induce TrkB-specific effects in brain slice explants (Ip et al., 1993; McAllister et al., 1997; Yacoubian and Lo, 2000), resulted in significant rescue of neurons that otherwise would have degenerated over this period (Fig. 3A). Protection against such OGD treatment can also be provided by cotransfection with the anti-apoptotic gene Bcl-xL (Wang et al., 2006) or by application of the pan-caspase inhibitor Boc-D-FMK (Fig. 2G), suggesting that a significant portion of the neuronal cell death protected against by BDNF and PBI-05204 in OGD-treated brain slices likely involves apoptotic mechanisms.
Figure 3.

A, Exogenous BDNF is sufficient for neuroprotection against OGD. Addition of 200 ng/ml BDNF provides direct neuroprotection to OGD-treated brain slices. y-Axis shows the percentage increase in numbers of healthy cortical neurons per brain slice relative to the OGD condition. Average of three independent runs; *p < 0.01 by ANOVA followed by Dunnett's post hoc comparison test. B–D, PBI-05204 and oleandrin regulate BDNF at the gene transcriptional level. B, Time course of induction of Bdnf gene transcription by 23 μg/ml PBI-05204 as quantified by qPCR analysis of total Bdnf mRNA levels. ΔΔCt, threshold cycle. C, Use of qPCR with specific primers to distinguish among eight alternative promoters for Bdnf shows that transcription is most strongly induced by PBI-05204 at promoters I and IV, with little or no induction at promoters II, III, and V–VIII. Last pair of bars shows that increases in promoters I and IV can quantitatively account for total Bdnf mRNA induction as measured by a primer set that recognizes all Bdnf transcripts. Measurements taken 9 h after treatment with 23 μg/ml PBI-05204. D, Three micrometers of oleandrin induces Bdnf transcription at promoters I and IV to a similar extent as 23 μg/ml PBI-05204 (compare striped vs dark bars, respectively). In contrast, PBI-05204 chromatographically depleted of all cardiac glycosides (“CG-depleted”) had no effect on Bdnf gene transcription. Measurements taken 9 h after compound treatments.
PBI-05204 and oleandrin increase Bdnf transcription through selective promoters
Given that the Bdnf gene is known to be highly regulatable by extracellular signals (Lyons and West, 2011), we asked whether PBI-05204-induced increases in BDNF protein levels are driven at the gene transcriptional level. In fact, as shown in Figure 3B, PBI-05204 treatment led to a rapid increase in the transcriptional rate of the Bdnf gene as assessed by quantitative PCR (qPCR), reaching a steady-state of some 4–5 times over control levels by 3–9 h after treatment, and maintaining this level through 9–18 h.
The transcription of the Bdnf gene can be driven by at least eight distinct promoter/enhancer sequences that, through alternative splicing, all drive a common ninth exon encoding the entire BDNF protein sequence (Liu et al., 2006; Aid et al., 2007; Pruunsild et al., 2007; Lyons and West, 2011). qPCR analysis of these alternative transcripts indicated that promoters I and IV are the most highly induced by PBI-05204 treatment and could quantitatively account for the total increase in Bdnf transcription (Fig. 3C). PBI-05204 increased transcription at promoter VI only minimally, whereas promoters II, III, V, VII, and VIII all showed low basal levels of expression with little or no induction by PBI-05204.
Finally, we asked whether the action of oleandrin could account for the regulation of Bdnf by PBI-05204 at the transcriptional level. As can be seen in Figure 3D, treatment with an equivalent concentration of oleandrin corresponding to its ∼3% relative abundance in PBI-05204 drove increases in Bdnf gene expression to equivalent degrees through promoters I and IV compared with the full PBI-05204 extract. In contrast, PBI-05204 depleted of cardiac glycoside content had no effect on Bdnf transcription, at promoters I and IV or overall.
Discussion
We have demonstrated that augmentation of endogenous BDNF is both necessary and sufficient for the neuroprotective action of PBI-05204 in a brain slice model for focal ischemia, and provided evidence that oleandrin, the principal cardiac glycoside component of PBI-05204, can quantitatively account for regulation of BDNF at both the protein and transcriptional levels. The strongest regulation of Bdnf by PBI-05204 and oleandrin was via promoter IV, and to a lesser extent promoter I, strongly suggesting convergence with known pathways that regulate Bdnf transcription downstream of neuronal activity (Lyons and West, 2011). Regulation of promoter IV has been shown to be Ca2+-dependent and mediated via the transcription factors CaRF, USF1/2, and CREB; and MeCP2 involved in DNA methylation (for review, see Lyons and West, 2011).
This transcriptional profile for Bdnf regulation could plausibly be triggered by the primary target of cardiac glycosides, Na+, K+-ATPase. In heart, the inhibition of the Na+, K+-ATPase by digitalis compounds leads to secondary inhibition of the Na+/Ca2+ exchanger via erosion of the transmembrane Na+ gradient and consequent increases in intracellular Ca2+ levels (Hardman et al., 1996). In fact, we have previously reported that that OGD treatment of brain slices results in selective upregulation of the α3-subunit of the Na+, K+-ATPase, to which oleandrin exhibits preferential binding (Yang et al., 2009; Dunn et al., 2011).
However, the contribution of other potential neuroprotective mechanisms initiated through the Na+, K+-ATPase cannot be excluded, including maintaining intracellular Ca2+ “set point” implicated in neuronal survival (Lee et al., 1999, 2000); and energy conservation through ATP-sparing given that 40–70% of brain energy metabolism is allocated to powering the Na+, K+-ATPase (Hochachka, 1986; Clausen et al., 1991; Friberg and Wieloch, 2002). Finally, there is accumulating evidence that the Na+, K+-ATPase functions within a stable signalosome, and mediates signal transduction through multiple pathways including EGFR and downstream MAPK pathways (for review, see Xie and Cai, 2003; Prassas and Diamandis, 2008).
Although the neuroprotective properties of oleandrin and PBI-05204 may seem surprising, digitalis compounds such as digoxin have been in continuous clinical usage since the 18th century (Withering, 1785). We first identified the neuroprotective potential of such cardiac glycoside compounds in a large-scale, hypothesis-neutral chemical biology screen in a brain slice OGD assay (Wang et al., 2006). The strongest hit from this screen was the cardiac glycoside neriifolin which we subsequently showed could also provide benefit in two whole-animal models for stroke, including the adult middle cerebral occlusion model (Wang et al., 2006). However, penetration of the FDA-approved cardiac glycoside, digoxin, into CNS is limited by the efflux transporter P-glycoprotein expressed in the BBB (Beringer and Slaughter, 2005), and BBB penetration of neriifolin is also modest (Zhao et al., 1986). In contrast, oleandrin is readily BBB-penetrant in rodent models, as we have previously reported (Dunn et al., 2011).
Thus, our findings here that the neuroprotective actions of PBI-05204 in brain slice assays is mediated at least in part by BDNF further supports potential therapeutic applications of cardiac glycosides in ischemic stroke. BDNF is a principal neurotrophic factor during brain development as well as in the adult, and has been associated with neuroprotective benefit in a broad range of CNS traumatic and neurodegenerative disorders, including stroke (Zaleska et al., 2009; Nagahara and Tuszynski, 2011; Hartmann et al., 2012). In fact, endogenous BDNF has been implicated in neuronal survival following ischemia (Larsson et al., 1999; Ke et al., 2011), and treatment with exogenous BDNF postischemia has been shown to be neuroprotective and contribute to recovery and neural regeneration (Schäbitz et al., 2007; for review, see Zaleska et al., 2009; Rothman and Mattson, 2013).
However, the BDNF protein itself has proven problematic as a clinical agent, due to short serum half-life, poor bioavailability, minimal BBB-penetrability, and limited diffusion within CNS tissues (Thoenen and Sendtner, 2002; Longo and Massa, 2013; Lu et al., 2013). There has thus been strong interest in developing small molecule mimetics of BDNF or drug therapies that increase endogenous expression of BDNF (for review, see Obianyo and Ye, 2013; Rothman and Mattson, 2013). In contrast, cardiac glycoside drugs have been in effective clinical usage for decades (Hardman et al., 1996) and we have previously shown that oleandrin displays excellent brain penetration (Dunn et al., 2011). In addition, it is notable that PBI-05204 was well tolerated in a phase I clinical trial for a non-CNS indication (Bidyasar et al., 2009).
Interestingly, the recently FDA-approved drug fingolimod for multiple sclerosis has been shown to regulate BDNF production (Deogracias et al., 2012). In fact, fingolimod has been shown also to be effective in a range of preclinical stroke models (Campos et al., 2013) and may transpire to exert its neuroprotective effects at least in part through a BDNF mechanism. Given the wide range of neurological and neuropsychiatric disorders in which BDNF has been implicated (Longo and Massa, 2013; Lu et al., 2013), small molecule compounds such as fingolimod and oleandrin/PBI-05204 reported here may merit further investigation in ischemic stroke as well as other disorders of the CNS.
Footnotes
This work was supported in part by NIH Grant NS074379 and by funding provided by Phoenix Biotechnology.
Conflict of Interest: P. Yang and R. Newman are paid consultants of Phoenix Biotechnology.
References
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, Hurn P, Liebeskind DS, Nogueira RG, Saver JL. Stroke treatment academic industry roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–2650. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- Berg MM, Sternberg DW, Parada LF, Chao MV. K-252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- Beringer PM, Slaughter RL. Transporters and their impact on drug disposition. Ann Pharmacother. 2005;39:1097–1108. doi: 10.1345/aph.1E614. [DOI] [PubMed] [Google Scholar]
- Bidyasar S, Kurzrock R, Falchook GS, Naing A, Wheler JJ, Durand J, Yang P, Johansen MJ, Newman RA, Khan R, Hong D. A first-in-human phase I trial of PBI-05204 (oleandrin), an inhibitor of Akt, FGF-2, NF-kB, and p70S6K in advanced solid tumor patients. J Clin Oncol. 2009;27:3537. [Google Scholar]
- Biffo S, Offenhäuser N, Carter BD, Barde YA. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development. 1995;121:2461–2470. doi: 10.1242/dev.121.8.2461. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Schmid RS, He DN, Sung ML, Cho S, Resnick L, Monaghan MM, Hirst WD, Essrich C, Reinhart PH, Lo DC. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer's disease. Neurobiol Dis. 2010;39:311–317. doi: 10.1016/j.nbd.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F, Qin T, Castillo J, Seo JH, Arai K, Lo EH, Waeber C. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke. 2013;44:505–511. doi: 10.1161/STROKEAHA.112.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T, Van Hardeveld C, Everts ME. Significance of cation transport in control of energy metabolism and thermogenesis. Physiol Rev. 1991;71:733–774. doi: 10.1152/physrev.1991.71.3.733. [DOI] [PubMed] [Google Scholar]
- Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE, Barde YA. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn DE, He DN, Yang P, Johansen M, Newman RA, Lo DC. In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J Neurochem. 2011;119:805–814. doi: 10.1111/j.1471-4159.2011.07439.x. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/S0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Hardman JG, Limbird LE, Milinoff PB, Ruddon RW, Gilman AG. Goodman and Gilman's: the pharmacological basis of therapeutics. New York: McGraw-Hill; 1996. [Google Scholar]
- Hartmann D, Drummond J, Handberg E, Ewell S, Pozzo-Miller L. Multiple approaches to investigate the transport and activity-dependent release of BDNF and their application in neurogenetic disorders. Neural Plast. 2012;2012:203734. doi: 10.1155/2012/203734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW. Defense strategies against hypoxia and hypothermia. Science. 1986;231:234–241. doi: 10.1126/science.2417316. [DOI] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-C. [DOI] [PubMed] [Google Scholar]
- Ke Z, Yip SP, Li L, Zheng XX, Tong KY. The effects of voluntary, involuntary, and forced exercises on brain-derived neurotrophic factor and motor function recovery: a rat brain ischemia model. PLoS ONE. 2011;6:e16643. doi: 10.1371/journal.pone.0016643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Larsson E, Nanobashvili A, Kokaia Z, Lindvall O. Evidence for neuroprotective effects of endogenous brain-derived neurotrophic factor after global forebrain ischemia in rats. J Cereb Blood Flow Metab. 1999;19:1220–1228. doi: 10.1097/00004647-199911000-00006. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Longo FM, Massa SM. Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov. 2013;12:507–525. doi: 10.1038/nrd4024. [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Lyons MR, West AE. Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol. 2011;94:259–295. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/S0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McDowell KA, Hutchinson AN, Wong-Goodrich SJ, Presby MM, Su D, Rodriguiz RM, Law KC, Williams CL, Wetsel WC, West AE. Reduced cortical BDNF expression and aberrant memory in Carf knock-out mice. J Neurosci. 2010;30:7453–7465. doi: 10.1523/JNEUROSCI.3997-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- National Stroke Association. Stroke 101. 2010. Available at http://www.stroke.org/site/DocServer/STROKE101_ 2010.pdf?docID=4541.
- Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- Obianyo O, Ye K. Novel small molecule activators of the Trk family of receptor tyrosine kinases. Biochim Biophys Acta. 2013;1834:2213–2218. doi: 10.1016/j.bbapap.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov. 2008;7:926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007;90:397–406. doi: 10.1016/j.ygeno.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Mattson MP. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience. 2013;239:228–240. doi: 10.1016/j.neuroscience.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Steigleder T, Cooper-Kuhn CM, Schwab S, Sommer C, Schneider A, Kuhn HG. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. 2007;38:2165–2172. doi: 10.1161/STROKEAHA.106.477331. [DOI] [PubMed] [Google Scholar]
- Suwanwela N, Koroshetz WJ. Acute ischemic stroke: overview of recent therapeutic developments. Annu Rev Med. 2007;58:89–106. doi: 10.1146/annurev.med.58.070605.115306. [DOI] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci. 2002;5:1046–1050. doi: 10.1038/nn938. [DOI] [PubMed] [Google Scholar]
- Wang JK, Portbury S, Thomas MB, Barney S, Ricca DJ, Morris DL, Warner DS, Lo DC. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc Natl Acad Sci U S A. 2006;103:10461–10466. doi: 10.1073/pnas.0600930103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withering W. An Account of the floxglove and some of its medical uses with practical remarks on dropsy and other diseases. London: J and J Robinson; 1785. [Google Scholar]
- Xie Z, Cai T. Na+-K+-ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003;3:157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci. 2000;3:342–349. doi: 10.1038/73911. [DOI] [PubMed] [Google Scholar]
- Yang P, Menter DG, Cartwright C, Chan D, Dixon S, Suraokar M, Mendoza G, Llansa N, Newman RA. Oleandrin-mediated inhibition of human tumor cell proliferation: importance of Na, K-ATPase alpha subunits as drug targets. Mol Cancer Ther. 2009;8:2319–2328. doi: 10.1158/1535-7163.MCT-08-1085. [DOI] [PubMed] [Google Scholar]
- Zaleska MM, Mercado ML, Chavez J, Feuerstein GZ, Pangalos MN, Wood A. The development of stroke therapeutics: promising mechanisms and translational challenges. Neuropharmacology. 2009;56:329–341. doi: 10.1016/j.neuropharm.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Zhao KC, Zhu XY, Yi MG, Liu ZM, Song ZY. Studies on the pharmacokinetics of 3H-neriifolin in rats. Yao Xue Xue Bao. 1986;21:572–579. [PubMed] [Google Scholar]