

Abstract
Background and the purpose of the study
A common approach in cancer chemotherapy is development of drugs that interrupt the mitosis phase of cell division. Dimethylenastron is a known kinesin inhibitor. In this study, six novel dimethylenastron analogues (4a-f), in which 3-hydroxyphenyl substituent has been replaced with substituted benzylimidazolyl, were synthesized through Biginelli reaction.
Methods
Six novel Biginelli compounds (4a-f) were synthesized through one step Biginelli reaction of imidazole aldehydes (3a-c), dimedone and urea or thioura. In vitro cytotoxicities of prepared compounds were investigated using MTT assay. Furthermore the ELIPA kit was implemented to study inhibitory effects of synthesized compounds on ATPase activity of kinesin by measuring of organic phosphate.
Results
Our results indicated that analogue 4c is the most toxic and analogues 4f, 4b and dimethylenasteron were less cytotoxic in compare with other analogues. On the other hand, analogue 4a, 4b, 4c and 4e showed stronger Kinesin inhibition as compared with analogue 4f and dimethylenasteron. None of synthesized compounds were as potent kinesin inhibitor as Taxol. Docking analysis revealed that hydrogen bond formation and hydrophobic interactions were the key factors affecting inhibitory effects of these compounds.
Conclusion
Newly synthesized compounds were found to have moderate to good cytotoxicity against HeLa cancer cell. Our results may be helpful in further design of dihydropyrimidine as potential anticancer agents.
Keywords: Biginelli reaction, Dihydropyrimidine, Mitotic kinesin Eg5, Dimethylenastron
Background
A common approach in cancer chemotherapy is development of drugs that interrupt the mitosis phase of cell division. The mitotic spindle is an important target in cancer chemotherapy [1]. Compounds that perturb the spindle assembly checkpoint by interfering microtubule polymerization or depolymerization, arrest the cell cycle in mitosis due to prevention of the microtubule dynamics.
Nowadays, Taxol, the undisputed star, which inhibit the depolymerisation of microtubules to disassemble the mitotic spindle during cell division, is frequently used in cancer chemotherapy [2].
For the first time in 1999, Mayer et al., [3] identified a novel cell-permeable small molecule, named monastrol. Unlike taxol, monastrol as an antimitotic agent has not exhibited neuronal cytotoxity.
Monastrol induces a mono-astral conformation of microtubules by inhibiting the mitotic kinesin Eg5 [4,5].
Exploration of monastrol, commenced a new stage in Biginelli 3,4-dihydropyrimidine-2(1H)-one chemistry. Although many researches have been devoted to reveal the anti-mitotic mechanism of monastrol in the cell cycle [6-8], there are few examples concerning the anticancer activity [9-11]. Leizerman et al.[12] described the antiproliferative effect of monastrol on AGS and HT-29 cell lines as compared with taxol. Since the antimitotic activity of monastrol is not very high, structural variants could be verified to have better activity. Russowsky et al.[13], investigated the differential antiproliferative activity of monastrol, oxo-monastrol and oxygenated analogs on seven human cancer cell lines. In another study, more potent analogs of monastrol such as dimethylenastron [14] and quinazoline-2(1H)-thione [15] (Figure 1) were provided by skeleton modifications of monastrol in the parent ring by annelation across the C-5–C-6 bond. Notable work has also been dedicated to delineate the structure-activity relationship in the monastrol derivatives [16]. In this work six novel compounds (4a-f) were synthesized through Biginelli reaction in which hydroxyphenyl at C-4 position in dimethylenastron has been replaced with substituted benzylimidazolyl (Figure 2).
Figure 1.
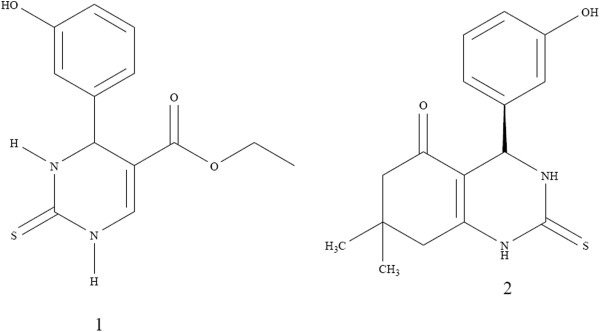
Monastrol (1) and dimethylenastron (2) structures.
Figure 2.
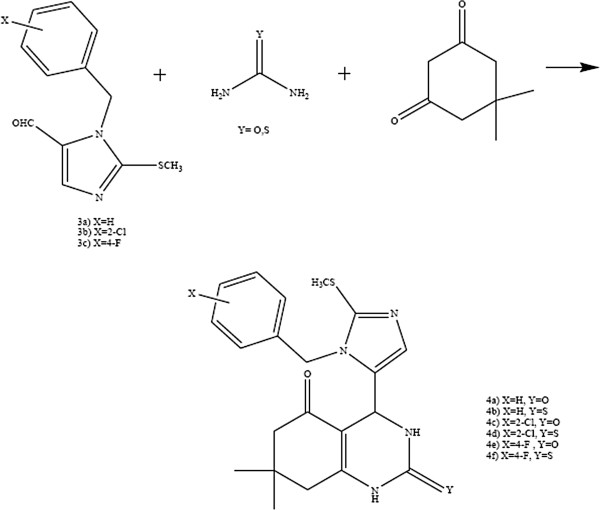
Synthesis of 4-imidazolyl tetrahydroquinazolines (4a-f) under Biginelli condition.
Methods
Chemistry
Melting points were determined using an Electrothermal Capillary apparatus and are uncorrected. 1H-NMR spectra were recorded using Bruker AC-80 NMR spectrometer. The chemical shift values are on δ scale and the coupling constant values (J) are in ppm relative to tetramethylsilane as internal standard. Errors of elemental analyses were within ±0.4% of theoretical values.
The desired compounds were synthesized by the reactions outlined in Figure 2. Imidazole aldehydes [3a-c] was synthesized as described previously [17].
General procedure for synthesis of 4a-f
The suspension of 3a-c (2 mmoles), dimedone (2 mmoles) and urea or thiourea (2.4 mmoles) in TMSCl (0.25 ml), DMF (0.8 ml) and acetonitrile (1.6 ml) was stirred for 4 h. The solid was separated by centrifugation and washed with distilled water followed by methanol. The residue was completely dried to give compound 4.
4-[1-benzyl-2-(methylthio)-1H-imidazol-5-yl)- 3,4,7,8- tetrahydro- 7,7 dimethyl- quinazoline-2,5-(1H,6H)-diones (4a)
This compound was obtained in 44% yield; mp 169°C; 1H-NMR (DMSO-d6): 9.5(s, 1H, NH), 8 (s, 1H, NH), 7.82–6.94 (m, 6H, arom, H-imidazole), 6.21–5.44 (3H, CH2N, C-H quinazoline), 2.91 -2.52 (m, 7H, SCH3, 6,8,CH2 quinazoline), 1.111-0.93 (m, 6H,CH3 quinazoline). Anal. Calcd for C21H24N4O2S: C, 63.61; H, 6.10; N, 14.13. Found: C, 63.48; H, 6.07; N, 14.07.
4-[1-benzyl-2-(methylthio)-1H-imidazol-5-yl)-1,2,3,4,7,8- hexahydro- 7,7 dimethyl-2-thio oxoquinazoline-5-(6H)-one (4b)
This compound was obtained in 36% yield; mp 157°C; 1H-NMR (DMSO-d6): 9.5 (s, 1H, NH), 8.00 (S, 1H, NH), 7.62 –6.95 (m, 6H, arom, H-imidazole), 5.54 (3H, CH2N, C-H quinazoline), 2.93 - 2.54 (m, 7H, SCH3, 6,8,CH2 quinazoline) 1.11-0.83 (m, 6H, CH3 quinazoline). Anal. Calcd for C21H24N4OS2: C, 63.13; H, 5.86; N, 13.58. Found: C, 61.27; H, 5.88; N, 13.63.
4-[1-(2-chlorobenzyl)-2-(methylthio)-1H-imidazol-5-yl]-3,4,7,8- tetrahydro- 7,7 dimethyl quinazoline-2,5-(1H,6H)-diones (4c)
This compound was obtained in 61% yield; mp 149°C; 1H-NMR (DMSO-d6): 9.5(s, 1H, NH), 8.1(s, 1H, NH), 7.43-6.02 (m, 5H, arom, H-imdazole), 5.53 (3H, CH2N, C-H quinazoline), 2.82 -2.53 (m, 7H, SCH3, 6,8- CH2 quinazoline), 1.11-0.74 (m, 6H, CH3 quinazoline). Anal. Calcd for C21H23ClN4O2S: C, 58.53; H, 5.38; N, 13.00. Found: C, 58.68; H, 5.40; N, 12.94.
4-[1-(2-chlorobenyl)-2-methylthio-1H-imidazol-5-yl]-1,2,3,4,7,8- hexahydro-7,7 dimethyl-2-thio oxoquinazoline-5-(6H)-one (4d)
This compound was obtained in 58.1% yield; mp 142°C; 1H-NMR (DMSO-d6): 9.5 (s, 1H,NH), 8.13 (s, 1H, NH) 7.63-6.41 (m, 5H, arom, H-imdazole) 5.52 (3H, CH2N, C-H quinazoline), 2.83 -2.45 (m, 7H, SCH3, 6,8- CH2 quinazoline), 1.23-1.02 (m, 6H, CH3 quinazoline). Anal. Calcd for C21H23ClN4OS2: C, 56.42; H, 5.19; N, 12.53. Found: C, 56.31; H, 5.21; N, 12.45.
4-[1-(4-fluorobenzyl)-2-(methylthio)-1H-imidazol-5-yl]-3,4,7,8 – tetrahydro-7,7 dimethyl quinazoline-2,5-(1H,6H)-diones (4e)
This compound was obtained in 68.7% yield; mp 82°C; 1H-NMR (DMSO-d6): 9.52 (s, 1H, NH), 7.91 (s, 1H, NH), 7.14- 7.11 (m, 5H, arom, H-imdazole), 5.41-5.23 (3H, CH2N, C-H quinazoline), 2.81- 2.05 (m, 7H, SCH3, 6,8- CH2 quinazoline), 1.22 -0.92 (m, 6H, CH3 quinazoline). Anal. Calcd for C21H23FN4O2S: C, 60.85; H, 5.59; N, 13.52. Found: C, 60.80; H, 5.61; N, 13.46.
4-[1-(4-fluorobenzyl)-2-methylthio-1H-imidazol-5-yl]-1,2,3,4,7,8 -hexahydro-7,7-dimethyl-2-thiooxoquinazoline-5-(6H)-one (4f)
This compound was obtained in 58% yield; mp 149°C; 1H-NMR (DMSO-d6): 9.5 (s, 1H, NH), 8.1 (s, 1H, NH), 7.33-7.10 (m, 5H, arom, H-imdazole), 5.43 (3H, CH2N, C-H quinazoline), 2.85- 2.12 (m, 7H, SCH3, 6,8-CH2 quinazoline), 2.81 -2.43 (m, 3H, SCH3), 1.22-0.94 (m, 6H, CH3 quinazoline). Anal. Calcd for C21H23FN4OS2: C, 58.58; H, 5.38; N, 13.01. Found: C, 58.61; H, 5.39; N, 13.06.
Docking
The X-ray crystal structure of Eg5-enastron complex (Protein Data Bank ID: 2X7C) was obtained from the Protein Data Bank. The three-dimensional structures of the derivatives were constructed using molecular mechanic force field (MM+), pre-optimization and AM1 semiemperical calculation in Hyperchem 7 software. The final corrected PDB file of the protein and synthesized analogs were submitted to AutoDock tools in order to run docking process. Docking studies were performed by AutoDock software Version 4. Searching was conducted within a specified 3D docking box (40 angstrom in all aspects) around enastron and the number of GA runs adjusted to 20 using Lamarckian genetic algorithm and all other parameters set as default. At the final stage through the docked structures of all analogs, best conformation was selected and saved as PDB file.
PDB files of best docked analogs along with Eg5 protein were submitted to MOE 2007.11 (License purchased from Chemical Computing Group by Mashhad University Medical Sciences, http://www.chemcomp.com/ for preparing figures and running protein ligand interaction fingerprint (PLIF).
The virtual physicochemical parameters of the synthesized compounds were also determined using MOE 2007.11 software.
Cell culture
HeLa cell line was obtained from National Cell Bank of Iran, Pasteur Institute of Iran. Cells were cultured in DMEM medium (Gibco, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco, USA), 100 U/ml penicillin (Biosera, UK), and 100 mg/ml streptomycin (Biosera, UK) at 37°C in a humidified atmosphere (95%) containing 5% CO2.
Cell viability
Cytotoxic effects of synthesized compounds were determined using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay [18-20]. Briefly, 5000 HeLa cells/well were seeded in a 96-well plate and cultured overnight. Different concentrations of synthesized compounds were added to each well. After incubation for 24 h, cells were treated with MTT solution (final concentration 0.5 mg/ml; Sigma, USA) for 4 h at 37°C. Then, the medium was removed, and the purple formazan crystals were dissolved in 150 μl dimethylsulfoxide (Merck, Germany). Absorbance was measured at 545 nm (630 nm as a reference) in Synergy H4 Hybrid Multi-Mode Microplate Reader (Biotek, Model: H4MLFPTAD). IC50 was calculated using CalcuSyn (BioSoft).
Kinesins activity assay
Briefly MT ELIPA master mix was prepared using 2 ml of reaction buffer containing 15 mM PIPES pH 7, 5 mM MgCl2, 160 μl of 1 μg/μl tubuline solution in reaction buffer, 480 μl of ELIPA reagent 1 containing 1 mM 2-amino-6-mercapto-7-methylpurine riboside (MESG) and 24 μl of ELIPA reagent 2 containing 0.1 units/μl of purine nucleoside phosphorylase (PNP). Final reaction mixture was prepared by adding 75 μl of ELIPA master mix, 1 μl of 2.5 mg/ml kinesin heavy chain Motor (Cytoskeleton, Cat # KR01-A) and 20 μl of different concentrations of synthesized inhibitors and Taxol. Reaction was started by adding 8 μl of 100 mM ATP stock solution (Sigma, Cat # A3377). Absorbance of each well was recorded on a kinetic mode at λ360 nm wavelength using Synergy H4 Hybrid Multi-Mode Microplate Reader (Biotek, Model: H4MLFPTAD). Phosphate standard curve was constructed by adding 0–25 μl of 0.5 mM phosphate stock, to ELIPA master mix (Without tubulin). Activity of kinesin was reported as nmole Pi/min/2.5 μg kinesin.
Statistical analysis
Data are expressed as mean ± SD. Statistical analyses were performed with ANOVA followed by Tukey–Kramer test to compare the differences between means. Differences were considered statistically significant when P < 0.05.
Results and discussion
Chemistry
In this study the new dimethylenastron derivatives (4a-f) were produced by substitution of 3-hydroxyphenyl in dimethylenastron with substituted benzyl imidazolyl under Biginelli condition.
The purity of the compounds was checked by TLC and melting points. The structure of the compounds was confirmed on the basis of its 1H NMR spectral data and elemental analyses. All spectral data are in accordance with assigned structures. In IR spectra, N-H and C-O stretching bands were observed at spectra expected values. In the 1H NMR spectra, methyl protons were seen at 0.90-1.00 ppm as separated singlets. Aromatic, methylene, methine and NH protons were found at expected values.
Docking analysis
Accuracy of docking protocol was examined by docking enastron in active site of Eg5 enzyme. Figure 3 shows docked enastron and co-crystallized one in almost same position among the receptor (RMSD = 1.24Ǻ) that confirmed validation of docking protocol. All dimethylenastron derivatives were docked into active site of Eg5.
Figure 3.
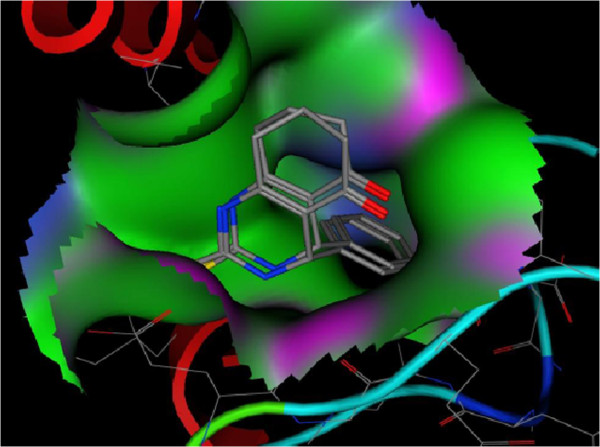
Docked and co-crystalized enasteron in Eg5 enzyme.
Table 1 shows estimated free energy of binding and calculated ki of synthesized compounds extracted from docking studies, these data in addition to Figure 4 which indicates synthetic ligands were in suitable position between active site of enzyme approve suitable interaction between ligands and protein.
Table 1.
Cytotoxicity on HeLa cell line (n = 5), inhibition of Kinesin activity (n = 2) and docking results of synthesized analogues 4a-f
| Compound | X* | Y* | IC 50 (μg/ml) ± SD on HeLa cell line | IC 50 (μg/ml) for Kinesin inhibition | Estimated** free energy of binding | Calculated*** Ki (nM) for Kinesin inhibition |
|---|---|---|---|---|---|---|
|
4a |
H |
O |
210 ± 21 |
72 |
-8.94 |
281.5 nM |
|
4b |
H |
S |
301 ± 22 |
77 |
-8.67 |
438.63 nM |
|
4c |
2-Cl |
O |
98 ± 19 |
79 |
-9.39 |
130.76 nM |
|
4d |
2-Cl |
S |
ND** |
ND** |
-8.83 |
334.27 nM |
|
4e |
4-F |
O |
110 ± 28 |
96 |
-8.55 |
536.29 nM |
|
4f |
4-F |
S |
339 ± 23 |
198 |
-8.52 |
573.33 nM |
| dimethylenastrone |
- |
- |
338 ± 26 |
210 |
-8.72 |
409.07 nM |
| Taxol | - | - | ND** | 7 | ND | ND |
Figure 4.
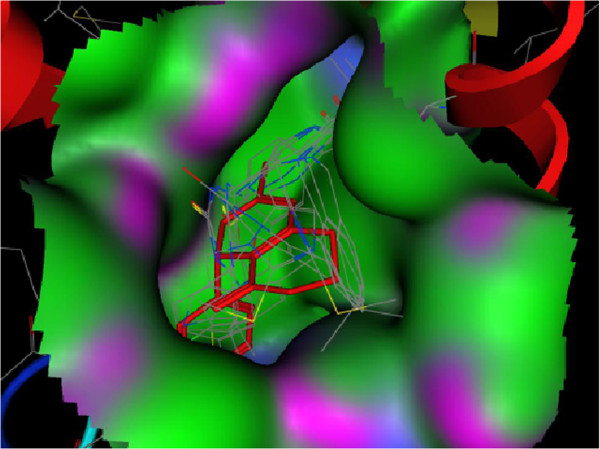
Map surface of docked analogs in active site of enzyme (Green: hydrophobic; Violet: H bonding; Blue: mild polar). Crystallized enasteron is indicated by red and stick lines.
According to PLIF, the most consistence interaction is H-bound between side chain of Glu116 and nitrogen atom on quinazoline ring. Compounds 4a, 4b, 4c and 4d have shown mentioned interaction and a backbone H-bound donor interaction was also detected between Glu118 and nitrogen atom in imidazolic ring of compound 4a. On the other hand by calculating ligand-protein interactions using LigX module in MOE software, except these H-bound interactions, surface contact interactions (arene-cation) between imidazolic ring of ligands and Arg 221 were identified. (Figure 5) Figure 5 represents 2D graph of interactions between synthesized compound 4b and Eg5 protein calculated by LigX module.
Figure 5.
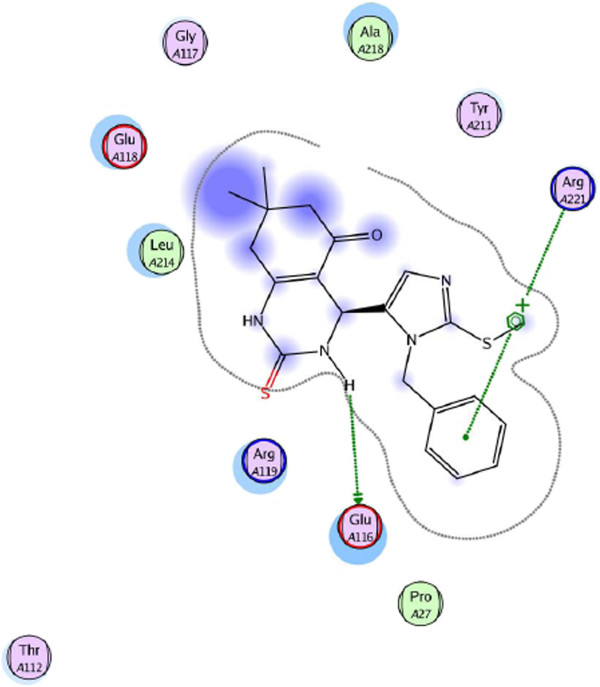
2D graph of interactions between synthesized compound 4b and protein made by LigX module of MOE software. In the 2D graphs hydrophobic/aromatic residues are colored in green, whereas polar amino acids are shown in magenta. H-bonds and all π-stacking interactions are represented as green dotted lines. The active site contour is also shown.
Docking analysis revealed that the all compounds interacted with Eg5 in good manner and confirms the importance of H-bound donor group in quinazoline ring and the role of imidazole ring as well as benzene ring in surface contact interaction on antiproliferative effects of synthesized compounds.
Effects of synthesized compounds on cell viability
Cytotoxicity of synthesized compounds was evaluated using MTT assay (Table 1). HeLa cells were incubated with different concentrations of newly synthesized compounds for 24 h and cytotoxicities were evaluated using MTT assay. Our data showed that analogue 4c (X = 2-Cl Y = O, IC50: 98 ± 19 μg/ml) was the most toxic compound among the newly synthesized compounds. Analogue 4f (X = 4-F Y = S, IC50: 339 ± 23 μg/ml), 4b (X = H Y = S, IC50: 301 ± 22 μg/ml) and dimethylenasteron (IC50: 338 ± 26 μg/ml) showed less cytotoxicities.
The significant difference between cytotoxicity of analogues 4f (X = 4-F Y = S, IC50: 339 ± 23 μg/ml) and 4e (X = 4-F Y = O, IC50: 110 ± 28 μg/ml) or analogue 4b (X = H Y = S, IC50: 301 ± 22 μg/ml) and 4a (X = H Y = O, IC50: 210 ± 21 μg/ml)demonstrated the essential role of carbonyl group at position C2 of tetrahydro-quinazoline on antiproliferative effect of synthesized compounds.
Meanwhile there was no big difference between cytotoxicity of compound 4c (X = 2-Cl Y = O, IC50: 98 ± 19 μg/ml) and 4e (X = 4-F Y = O, IC50: 110 ± 28 μg/ml); they had so much better cytotoxic effect in comparison with compounds 4b (X = H Y = S, IC50: 301 ± 22 μg/ml) and 4a (X = H Y = O, IC50: 210 ± 21 μg/ml). The obtained results illustrated the presence of an electron withdrawing groups (F or Cl) on the benzylimidazolyl substituent at position C4 of tetrahydro-quinazoline structure could increase the cytotoxicity of prepared compounds.
Effects of synthesized compounds on Kinesin activity
Kinesins operate by utilizing the energy of ATP hydrolysis, generating Pi, to move along microtubule (MT) substrates. To determine activity of kinesins, rate of Pi production was measured using ELIPA (Enzyme Linked Inorganic Phosphate Assay) Biochem kit (Cytoskeleton, cat # BK060). The assay was based on an absorbance shift (330–360 nm) that occurred when 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG) was catalytically converted to 2-amino-6-mercapto-7-methylpurine in the presence of inorganic phosphate (Pi). The reaction was catalyzed by purine nucleoside phosphorylase (PNP). One molecule of Pi produces one molecule of 2-amino-6-mercapto-7-methylpurine in an essentially irreversible reaction. Thus, the absorbance at 360 nm is directly proportional to the amount of Pi. Our data (Table 1) showed that analogue 4a (X = H Y = O, IC50: 72 μg/ml), 4b (X = H Y = S, IC50: 77 μg/ml), 4c (X = 2-Cl Y = O, IC50: 79 μg/ml) and 4e (X = 4-F Y = O, IC50: 96 μg/ml) were stronger Kinesin inhibitor as compared with analogue 4f (X = 4-F Y = S, IC50: 198 μg/ml) and dimethylenasteron (IC50: 210 μg/ml. None of our compounds were able to inhibit kinesin as much as Taxol (IC50: 7 μg/ml).
According to kinesin inhibitory effect of analogues 4f and 4e, the carbonyl group at C2 position of tetrahydro-quinozoline was crucial in kinesin inhibitory activities of synthesized compounds. On the other hand, our results suggested that the presence of an electron withdrawing group in benzene ring of benzylimidazolyl at position C4 of tetrahydro-quinazoline structure had no significant effect either positive or negative on kinesin inhibitory activity of synthesized compounds.
Conclusion
In this study a series of six novel dimethylenastron analogs based on Biginelli reaction were synthesized with IC50 in the range of 98 to 210 μg/mL against HeLa cell line. Compared with dimethylenastron, these new series were found to have stronger antiproliferative activity against HeLa cell line. Structural-activity relationship study highlighted the important role of carbonyl substitution at C2 position of tetrahydro-quinazoline structure in antiproliferative and kinesin inhibitory effect of newly synthesized compounds. The reported results could be helpful in developing potential formulation of new anticancer drugs.
Abbreviations
DMF: Dimethylformamide; ELIPA: Enzyme linked inorganic phosphate assay; IR: Infrared; MESG: 2-amino-6-mercapto-7-methylpurine ribonucleoside; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MT: Microtubule; NMR: Nuclear magnetic resonance; PLIF: Protein ligand interaction fingerprint; PNP: Purine nucleoside phosphorylase; RMSD: Root-mean-square deviation; TLC: Thin layer chromatography; TMSCL: Trimethylsilyl chloride.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KA: Supervision of biological studies including cytotoxicity (MTT) and Kinesin inhibition assay. BB: performed synthesis of the target compounds. SM: Collaboration in biological studies, MRMF: Performed the cytotoxic tests and Kinesin Assay. MA: manuscript draft preparation and Collaboration in biological studies. FM: performed synthesis and identifying of the target compounds. MG: performed molecular modeling studies. FH: supervision of design and synthesis of target compounds and manuscript preparation. All authors read and approved the final manuscript.
Contributor Information
Khalil Abnous, Email: abnouskh@mums.ac.ir.
Batoul Barati, Email: batoulbarati@yahoo.com.
Soghra Mehri, Email: mehris@mums.ac.ir.
Mohammad Reza Masboghi Farimani, Email: masboughimr@yahoo.com.
Mona Alibolandi, Email: alibolandim901@mums.ac.ir.
Fatemeh Mohammadpour, Email: Mohammadpf911@mums.ac.ir.
Morteza Ghandadi, Email: ghandadim891@mums.ac.ir.
Farzin Hadizadeh, Email: hadizadehf@mums.ac.ir.
Acknowledgements
The authors are thankful for financial support from the Research Council of Mashhad University of Medical Sciences. This work was part of Pharm.D thesis.
References
- Jun Z, Paraskevi G. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anti Canc Agents. 2005;21:65–71. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]
- Verweij J, Clavel M, Chevalier B. Paclitaxel (Taxol) and docetaxel (Taxotere): not simply two of a kind. Ann Oncol. 1994;21:495–505. doi: 10.1093/oxfordjournals.annonc.a058903. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;21:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Crews CM, Mohan R. Small-molecule inhibitors of the cell cycle. Curr Opin Chem Biol. 2000;21:47–53. doi: 10.1016/S1367-5931(99)00050-2. [DOI] [PubMed] [Google Scholar]
- Liu X, Gong H, Huang K. Oncogenic role of kinesin proteins and targeting kinesin therapy. Cancer Sci. 2013;21:651–656. doi: 10.1111/cas.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;21:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBonis S, Simorre J-P, Crevel I, Lebeau L, Skoufias DA, Blangy A, Ebel C, Gans P, Cross R, Hackney DD. et al. Interaction of the mitotic inhibitor monastrol with human kinesin Eg5. Biochemistry. 2002;21:338–349. doi: 10.1021/bi026716j. [DOI] [PubMed] [Google Scholar]
- Cochran JC, Gilbert SP. ATPase mechanism of Eg5 in the absence of microtubules: insight into microtubule activation and allosteric inhibition by monastrol. Biochemistry. 2005;21:16633–16648. doi: 10.1021/bi051724w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque SA, Hasaka TP, Brooks AD, Lobanov PV, Baas PW. Monastrol, a prototype anti-cancer drug that inhibits a mitotic kinesin, induces rapid bursts of axonal outgrowth from cultured postmitotic neurons. Cell Motil Cytoskeleton. 2004;21:10–16. doi: 10.1002/cm.10176. [DOI] [PubMed] [Google Scholar]
- Maliga Z, Kapoor TM, Mitchison TJ. Evidence that monastrol is an allosteric inhibitor of the mitotic kinesin Eg5. Chem Biol. 2002;21:989–996. doi: 10.1016/S1074-5521(02)00212-0. [DOI] [PubMed] [Google Scholar]
- Muller C, Gross D, Sarli V, Gartner M, Giannis A, Bernhardt G, Buschauer A. Inhibitors of kinesin Eg5: antiproliferative activity of monastrol analogues against human glioblastoma cells. Cancer Chemother Pharmacol. 2007;21:157–164. doi: 10.1007/s00280-006-0254-1. [DOI] [PubMed] [Google Scholar]
- Leizerman I, Avunie-Masala R, Elkabets M, Fich A, Gheber L. Differential effects of monastrol in two human cell lines. Cell Molecular Life Sci. 2004;21:2060–2070. doi: 10.1007/s00018-004-4074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russowsky D, Canto RFS, Sanches SAA, DOca MGM, de Fatima A, Pilli RA, Kohn LK, Antonio MA, de Carvalho JE. Synthesis and differential antiproliferative activity of biginelli compounds against cancer cell lines: monastrol, oxo-monastrol and oxygenated analogues. Bioorg Chem. 2006;21:173–182. doi: 10.1016/j.bioorg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Gartner M, Sunder-Plassmann N, Seiler J, Utz M, Vernos I, Surrey T, Giannis A. Development and biological evaluation of potent and specific inhibitors of mitotic kinesin Eg5. Chem Bio Chem. 2005;21:1173–1177. doi: 10.1002/cbic.200500005. [DOI] [PubMed] [Google Scholar]
- Sunder-Plassmann N, Sarli V, Gartner M, Utz M, Seiler J, Huemmer S, Mayer TU, Surrey T, Giannis A. Synthesis and biological evaluation of new tetrahydro-β-carbolines as inhibitors of the mitotic kinesin Eg5. Bioor Med Chem. 2005;21:6094–6111. doi: 10.1016/j.bmc.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Klein E, DeBonis S, Thiede B, Skoufias DA, Kozielski F, Lebeau L. New chemical tools for investigating human mitotic kinesin Eg5. Bioor Med Chem. 2007;21:6474–6488. doi: 10.1016/j.bmc.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Hadizadeh F, Mohajeri S, Hosseinzadeh H, Salami S, Motamedshariat F. Synthesis of novel 4-[1-(4-fluorobenzyl)-5-imidazolyl] dihydropyridines and study of their effects on rat blood pressure. Iranian J. Basic Medical Sci. 2011;21(5):213–218. [PMC free article] [PubMed] [Google Scholar]
- Hadizadeh F, Moallem SA, Jaafari MR, Shahab M, Alahyari M, Rameshrad M, Samiei A. Synthesis and immunomodulation of human lymphocyte proliferation and cytokine (interferon-gamma) production of four novel malonitrilamides. Chem Biol Drug Des. 2009;21(6):668–673. doi: 10.1111/j.1747-0285.2009.00812.x. [DOI] [PubMed] [Google Scholar]
- Vosooghi M, Yahyavi H, Divsalar K, Shamsa H, Kheirollahi A, Safavi M, Ardestani SK, Sadeghi-Neshat S, Mohammadhosseini N, Edraki N, Khoshneviszadeh M, Shafiee A, Foroumadi A. Synthesis and in vitro cytotoxic activity evaluation of (E)-16-(substituted benzylidene) derivatives of dehydroepiandrosterone. Daru J Pharmaceut Sci. 2013;21:34. doi: 10.1186/2008-2231-21-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushini S, Alipour E, Emami S, Safavi M, Ardestani SK, Gohari AR, Shafiee A, Foroumadi A. Synthesis and cytotoxic properties of novel (E)-3-benzylidene-7-methoxychroman-4-one derivatives. Daru J Pharmaceut Sci. 2013;21:31. doi: 10.1186/2008-2231-21-31. [DOI] [PMC free article] [PubMed] [Google Scholar]