

Abstract
The time-dependent integration of excitatory and inhibitory synaptic currents is an important process for shaping the input–output profiles of individual excitable cells, and therefore the activity of neuronal networks. Here, we show that the decay time course of GABAergic inhibitory synaptic currents is considerably faster when recorded with physiological internal Cl− concentrations than with symmetrical Cl− solutions. This effect of intracellular Cl− is due to a direct modulation of the GABAA receptor that is independent of the net direction of current flow through the ion channel. As a consequence, the time window during which GABAergic inhibition can counteract coincident excitatory inputs is much shorter, under physiological conditions, than that previously measured using high internal Cl−. This is expected to have implications for neuronal network excitability and neurodevelopment, and for our understanding of pathological conditions, such as epilepsy and chronic pain, where intracellular Cl− concentrations can be altered.
Introduction
The time course of synaptic GABAA receptor-mediated IPSC will profoundly influence the excitability of individual neurons and networks, by setting the duration of a time window during which an inhibitory input can most effectively counteract a coincident excitatory input.
The IPSC time course is determined by both presynaptic and postsynaptic factors at inhibitory synapses. These include the concentration–time profile of GABA in the synaptic cleft, which is itself affected by the density of GABA transporters and the degree of synchrony for presynaptic GABA release (Overstreet and Westbrook, 2003; Hefft and Jonas, 2005; Keros and Hablitz, 2005), as well as several postsynaptic factors, such as the following: the presence of perisynaptic GABAA receptors, which prolong the IPSC decay phase (Wei et al., 2003), the subunit composition of synaptic receptors (Barberis et al., 2007), and the phosphorylation state of GABAA receptor subunits (Jones and Westbrook, 1997; Houston et al., 2008). Another factor, which has received less consideration and may also influence IPSC decays, is the nature and concentration of permeant ions through the ligand-gated ion channel. These are known to affect the kinetics of voltage-gated ion channels (Stanfield et al., 1981; Swenson and Armstrong, 1981), but there are very few reports of such an effect on synaptic ligand-gated channels (Marchais and Marty, 1979; Onodera and Takeuchi, 1979; Schneggenburger and Ascher, 1997). Indeed, for GABA ion channels, such a modulation by Cl− would not have been readily observed before, as most patch-clamp studies of synaptic inhibition routinely use matching high internal and external Cl− concentrations to set the Cl− reversal potential near to zero. This is convenient because it increases the amplitude of the IPSC at the negative holding potentials that are suitable for whole-cell recording.
Here, we report that intracellular Cl−, by acting directly on the GABAA receptor, critically affects the GABAergic IPSC decay phase at interneuron–Purkinje cell inhibitory synapses in the cerebellum. At low Cl− concentrations, typically approaching those found in neurons (∼5–10 mm), the IPSC decay phase is rapid, and the duration of the IPSC shorter. We found that by incrementally raising the internal Cl− concentration, the IPSC decay time was proportionately increased. Thus, variations of the intracellular Cl− concentration, following physiological or pathophysiological processes, will have a crucial impact on the IPSC profile with important implications for the dynamics of inhibitory transmission throughout the CNS.
Materials and Methods
Preparation of slices.
Thin sagittal slices (250 μm) were cut from the cerebellum of postnatal day 12 Sprague Dawley rats using a Leica VT1200s vibroslicer, in accordance with the UK Animals (Scientific Procedures) Act of 1986. Slices were cut in an artificial CSF (aCSF) solution containing (in mm) 125 NaCl (or 85 NaCl, 75 sucrose), 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 CaCl2, 5 MgCl2, and 11 glucose, gassed with 95% O2/5% CO2 at 4°C. The slices were then incubated at 35°C for 45 min before being allowed to cool to room temperature (23°C) before use. Over this time the solution was slowly changed to standard aCSF and slices were perfused in the recording bath at room temperature containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 11 glucose, gassed with 95% O2/5% CO2 (Duguid and Smart, 2004; Bright et al., 2007). For the bicarbonate-free experiments, the ACSF contained (in mm) 135 NaCl, 2.5 KCl, 1.25 NaH2PO4, 20 HEPES, 2 CaCl2, 2 MgCl2, and 11 glucose (Kaila et al., 1997), gassed with 100% O2. To block NMDA and AMPA receptor activation, 20 μm AP-5 and 10 μm CNQX were added to the standard aCSF.
Expression of recombinant receptors in HEK293 cells and fast concentration jumps.
HEK293 cells were transfected 6 h after plating using the calcium phosphate precipitation method with 4 μg of DNA of αβγ GABAA receptor subunits and EGFP in equimolar ratios per 35 mm dish. Cells were bathed in a solution composed of (in mm) 102.7 NaCl, 20 Na gluconate, 2 KCl, 2 CaCl2, 1.2 MgCl2, 10 HEPES, 14 glucose, 15 sucrose, and 20 TEA-Cl, pH adjusted to 7.4 with NaOH (osmolarity ∼320 mOsm). Recordings were obtained in the outside-out patch configuration using either a high-Cl− pipette solution (107.1 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 11 EGTA, 20 TEA-Cl, and 2 MgATP) or a low-Cl− solution (121.1 K gluconate, 1 CaCl2, 1 MgCl2, 10 HEPES, 11 EGTA, 6 TEA-Cl, and 2 MgATP). Concentration jumps were performed using a theta tube (outer diameter 2 mm, inner diameter 1.7, septum 0.117, 14-072-01, Hilgenberg) whose tip was cut with a diamond pen to a final diameter of ∼150 μm. The theta tubes were filled with control solution, and 3 mm GABA was added to one of the two barrels. Fast drug applications were performed by delivering a short (1–2 ms) voltage pulse to a piezo-stepper (Burleigh Instruments). The exchange time was measured by applying a 20% diluted solution to the open tip of the recording pipette after rupture of the patch and measuring the resulting junction potentials. Experiments were included in the analysis if the exchange time was faster than 150 μs. A minimum of 10 individual responses at five different voltages (from −100 to +60 mV in 40 mV steps) were averaged and fitted with a mixture of two or three exponentials. Weighted time constants are reported throughout the text.
Electrophysiology.
Whole-cell patch-clamp recordings were made from single cells identified by differential interference contrast microscopy (Nikon Eclipse E 600FN) using a MultiClamp 700A amplifier in the voltage-clamp configuration (Molecular Devices). Patch pipettes (1.5–2 MΩ) were filled with solutions containing the following (in mm): 140 CsCl (150 mm Cl−), 140 Cs-gluconate (10 mm Cl−), or 20 CsCl and 120 Cs-gluconate (30 mm Cl−); and 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, 2 Mg2ATP, and 5 QX-314-Cl, pH adjusted to 7.3 with CsOH. The osmolarity was adjusted to 310 mOsm with sucrose. To make a 5 mm Cl− internal solution, 4 NaCl was replaced with 4 Na-gluconate and 4 QX-314-Cl.
Currents were filtered at 3 kHz (Bessel filter), digitized at 20 kHz and analyzed using Clampex 8.2 (Molecular Devices). Series resistance after compensation was typically between 2 and 3 MΩ (<1 MΩ for experiments at 37°C) and was constantly monitored throughout recording. Cells were not used for analysis if their series resistance changed by >10–15%. The capacitance of a typical Purkinje cell at this age was 355 ± 37 pA (n = 7), and cells that deviated from this by >20% were not used for analysis. Cells were continuously superfused with CNQX (10 μm) and AP-5 (20 μm) to block glutamate-mediated fast synaptic transmission.
The stimulating electrode was a patch electrode filled with aCSF positioned within the lower third of the molecular layer, ∼150 μm lateral to the Purkinje cell body (Mittmann and Hausser, 2007) to preferentially stimulate basket cells that form a GABAergic synapse close to the soma at the axon initial segment (Ango et al., 2004). Stimulation was applied through a constant current stimulator (DS3, Digitimer) and gradually increased until an evoked IPSC (eIPSC) could be detected in the recorded Purkinje cell. This stimulation was increased until the amplitude of the eIPSC increased substantially presumably due to recruitment of more basket cells. The stimulation intensity was then set at ∼1.5 times the threshold, and only cells where there was a clear separation between the stimulation transient and the rising phase of the eIPSC were used.
Data analysis.
The decay times of IPSCs were determined in Clampfit 8.2 by fitting a single exponential to the (90–10%) decay phase. At each holding potential, individual IPSCs were examined and those with any deflection in the rising or decaying phases were deleted, as were any failures, before fitting the mean IPSC. Averaged IPSCs included ∼30–50 individual sweeps. In cumulative distribution plots the individual decay times of each IPSC were plotted and normalized between cells. Spontaneous (s)IPSCs were analyzed using MiniAnalysis (version 6.0.1, Synaptosoft) and were also fitted with a single exponential (Houston et al., 2008). IPSC events were selected for analysis if there were no deflections in their rising or decaying phases and they decayed back to the baseline holding current. Events of low amplitude (<10 pA) and slow rise times (10–90% > 3 ms) were discarded from this analysis as they were thought to represent events occurring at more distant synapses and therefore more likely to be affected by dendritic cabling (Llano et al., 2000).
Dynamic clamp experiments.
Current-clamp recordings from Purkinje neurons used a standard intracellular solution composed of (in mm) 130 KCl, 10 HEPES, 1 CaCl2, 4 NaCl, 5 EGTA, and 4 MgATP, pH 7.3 with KOH. A GABAergic conductance was simulated with a function of the following form:
where t 0 represents the start of the waveform, τrise was 1.8 ms (determined from our eIPSC recordings), and τdecay was either 8.5 ms (simulating low Cl− conditions) or 12.5 ms (replicating our decays in high Cl− at −60 mV, data not shown). This waveform was injected through a dynamic clamp amplifier (SM-1, Cambridge Conductance) setting the reversal potential to −85 mV (Chavas and Marty, 2003) and a conductance of 15 nS. Trains of spikes were evoked by constant current injection and superimposed with either a fast or a slow conductance. Excitatory (AMPA-like) conductances had the same functional profile as those used for simulating inhibition, but with a rise time of 0.3 ms and a decay time of 3 ms. The value of the conductance was adjusted to be 10–20% above the threshold for firing (typically 12–18 nS). The inhibitory conductance (either fast or slow) was injected with a variable latency within a 120 ms window and repeated at 4 ms intervals.
Statistics.
For the statistical comparison of two means, an unpaired, two-tailed Student's t test was used, or the nonparametric alternative, Mann–Whitney test, if there was a significant difference in the group SD as determined by an F test. With all statistical tests, two means were considered significantly different if p < 0.05. Statistical tests were performed using GraphPad Instat version 3.01.
Results
Evoked, spontaneous, and miniature IPSC decay times are affected by internal Cl− concentration
To determine whether the intracellular Cl− concentration affects the kinetics of IPSCs, we recorded evoked and spontaneous IPSCs in cerebellar Purkinje cells, which receive inhibitory afferents from basket and stellate cells in the molecular layer. We used several different internal Cl− concentrations, from the high symmetrical Cl− frequently used in most electrophysiological experiments, to the low intracellular Cl− normally found in mature Purkinje neurons (Chavas and Marty, 2003); IPSCs were evoked by applying focal stimulation from a patch pipette filled with aCSF positioned in the lower third of the molecular layer. At a holding potential of −100 mV, the eIPSC decay was fit with a single exponential (Fig. 1 A) (Puia et al., 1994), with a time constant, τ, that visibly increased with the internal Cl− concentration (Fig. 1 A,C). This consistent change in the decay times with the internal Cl− concentrations was clearly evident from the lateral shifts in the cumulative distribution for IPSC decay time constants (Fig. 1 B). By reducing internal Cl− from 150 to 10 mm, the median τ was reduced from 10.2 to 6.5 ms at −100 mV (150 mm: mean τ = 10 ± 0.5 ms; 10 mm: τ = 6.6 ± 0.2 ms, p = 0.0001, Mann–Whitney test, n = 13), and from 20.5 to 14 ms at 20 mV (150 mm: mean τ = 20.7 ± 1.0 ms; 10 mm: τ = 14.5 ± 1.2 ms, p = 0.004, Mann–Whitney test, n = 6) (Fig. 1 C). At −100 mV, increasing internal Cl− over a physiologically relevant range from 5 to 30 mm, caused τ to increase by ∼59%.
Figure 1.

Reducing intracellular Cl− concentration decreases the decay time constant of evoked IPSCs. A, Consecutively evoked (gray) and averaged (black) eIPSCs recorded at −100 mV from Purkinje cells in an acute cerebellar slice using: 5, 10, 30, or 150 mm internal Cl− solutions. B, Cumulative distributions for eIPSC decay times measured at −100 mV in Purkinje cells recorded with 5, 10, 30, or 150 mm internal Cl−. C, Bar chart of the decay-time constants for eIPSCs at −100 (n = 13) and 20 mV (n = 6) holding potentials with high (black, 150 mm) or low (gray, 10 mm) internal Cl− concentrations (means ± SEM, *p < 0.05). D, Consecutive (gray) and averaged (black) eIPSCs, recorded with either a 10 or 150 mm internal Cl− solutions at 20 mV.
By comparing the eIPSC decay time constants at different holding potentials, it was apparent that the IPSC decays were also voltage dependent (Fig. 1 C,D) [cf. Collingridge et al. (1984) and Otis and Mody (1992)]. The ratio of τ at +20 mV and −100 mV (τ20/τ100) was 2.10 ± 0.19 with symmetrical Cl− (mean ± SEM; n = 6) and 2.35 ± 0.04 (n = 5) with low internal Cl−, suggesting that the voltage dependence of the IPSC decay was unaltered by the Cl− concentration. In marked contrast, at −100 mV, τ was consistently reduced by lowering the internal Cl− concentration [−100 mV: 9.7 ms (150 mm Cl−), 8.9 ms (30 mm), 6.3 ms (10 mm Cl−), 5.6 ms (5 mm)], a feature also noted at 20 mV [20.4 ms (150 mm), 13.9 ms (10 mm); n = 5–6]. Given that internal Cl− affected the speed of the eIPSC decay at −100 and 20 mV to a similar extent, it is clear that this regulation must be independent of the net direction of current flow (Fig. 1 D).
We also examined whether the internal Cl− concentration similarly affected spontaneous IPSCs (sIPSCs). In accord with the eIPSC data, the decay profiles of sIPSCs were described by single exponentials and displayed a similar voltage sensitivity from −100 mV to +20 mV, with τ increasing with membrane depolarization (Fig. 2 A–D). Recording from Purkinje cells with 150 mm internal Cl− concentrations the τ20/τ100 ratio was ∼2.33 ± 0.14 (n = 6) (Fig. 2 E), and remained constant at 10 mm Cl− concentration (2.64 ± 0.13, n = 6) (Fig. 2 E). As observed from experiments with eIPSCs, a low internal Cl− concentration (10 mm), reduced the mean decay time constant for sIPSCs at all holding potentials examined [−100 mV: 9.2 ± 0.6 ms (150 mm Cl−), 6.4 ± 0.5 ms (10 mm Cl−); 20 mV: 21.0 ± 1.1 ms (150 mm Cl−), 16.6 ± 0.6 ms (10 mm Cl−), p = 0.009 at −100 and 20 mV, Mann–Whitney test, n = 6] (Fig. 2 E). The displacements in the cumulative distributions for sIPSC decay times also revealed a consistent effect of the low Cl− concentration in reducing the median decay time constant [−100 mV, 9.1 ms (150 mm Cl−), 6.2 ms (10 mm Cl−); 20 mV: 21.9 ms (150 mm Cl−), 15.9 ms (10 mm Cl−); n = 6] (Fig. 2 F).
Figure 2.
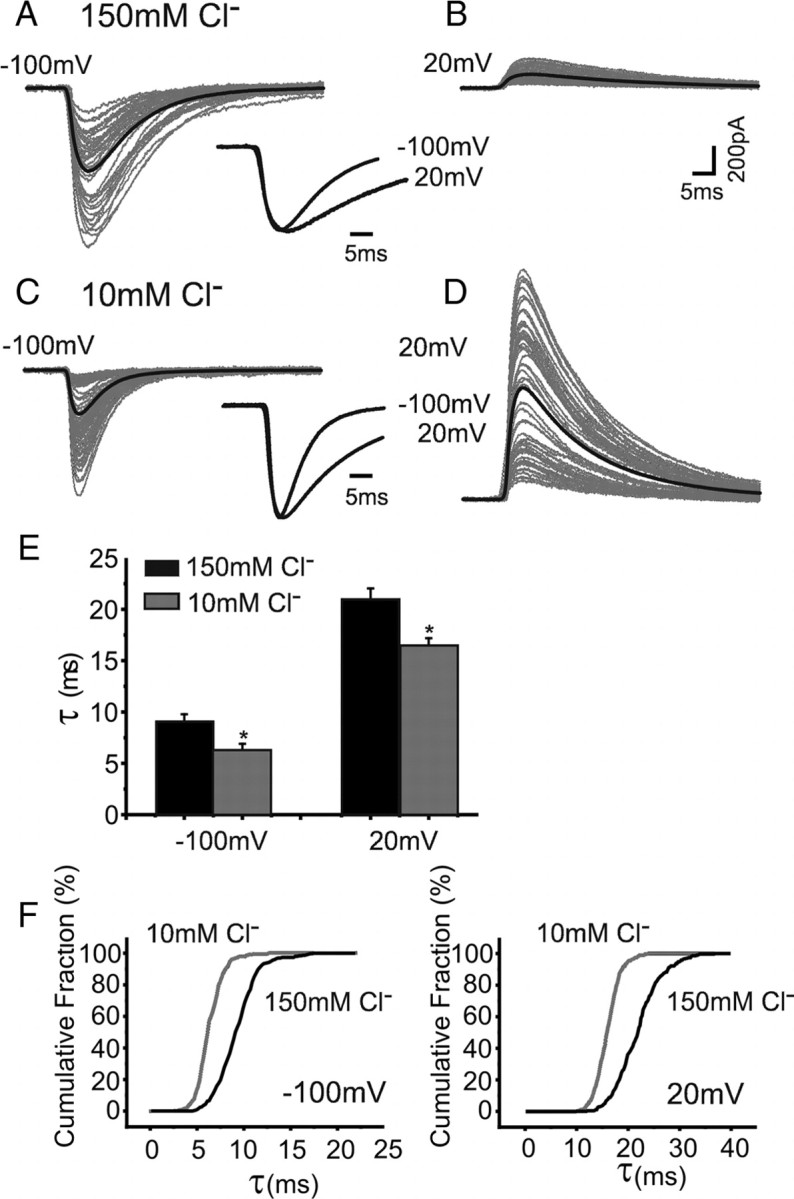
Intracellular Cl− regulates the decay phase of spontaneous IPSCs. A, sIPSCs (gray) and averaged sIPSC (black) recorded from Purkinje cells with 150 mm Cl− internal solution at −100 mV. The averaged and peak-scaled sIPSCs recorded at −100 and 20 mV (sIPSCs are inverted) are overlaid (inset). B, The same protocol as in A for individual and averaged sIPSCs recorded at 20 mV. C, sIPSCs recorded using 10 mm Cl− in the patch pipette (gray lines), at −100 mV. Averaged and peak-scaled sIPSCs at −100 and 20 mV are overlaid (inset). D, sIPSCs clamped at 20 mV (10 mm Cl−). The scale bar in B applies to all. E, Bar chart of mean sIPSC decay-time constants at different holding potentials with high (black) or low (gray) internal Cl−. F, Cumulative probability plots for sIPSC decay times at −100 mV and +20 mV with high (black) and low (gray) Cl−.
In accord with our results with eIPSCs and sIPSCs, internal Cl− also regulated the decay times of miniature IPSCs (mIPSCs). By reducing internal Cl−, the mIPSC decay time constant measured at −100 mV was reduced from τ (high Cl−) = 9.6 ± 0.8 ms to τ (low Cl−) = 6.8 ± 0.4 ms (n = 6; p = 0.018). Similarly, at a holding potential of +20 mV, τ (high Cl−) was reduced from 23.1 ± 0.6 ms and τ (low Cl−) to 16.5 ± 0.7 ms (n = 6; p = 0.0003; unpaired t test).
The increased decay rate for IPSCs recorded with low internal Cl− is expected to increase further when the recording temperature is raised from 23°C to 37°C. When eIPSCs were recorded at physiological temperatures from Purkinje neurons held at −80 mV, reducing the internal Cl− from 150 to 10 mm also reduced their median decay time constant from 2.7 ms to 1.9 ms (p = 0.01; Mann–Whitney; n = 6–8) (Fig. 3 A,B). The cumulative distribution for the decay times (Fig. 3 C) indicated that the median τ for 10 mm Cl− at 37°C approached the limit we can effectively resolve with the level of low-pass filtering imposed by the combination of whole-cell capacitance and series resistance. It is for this reason that most of the subsequent experiments were conducted at room temperature.
Figure 3.

Effect of low internal Cl− on IPSC decays at 37°C. Consecutively evoked (gray) and averaged eIPSCs (black) recorded at 37°C from cerebellar Purkinje cells held at −80 mV with a 150 mm (A) or 10 mm (B) internal Cl− solution. C, Cumulative probability relationships for eIPSC decay time constants at −80 mV and 37°C with 150 mm (black) and 10 mm (gray) internal Cl− (n = 6–8).
IPSC decay times are unaffected by HCO3 − permeation through GABA channels
Although Cl− is the major permeating anion through GABA channels, it is known that HCO 3 − ions also have an appreciable permeability through synaptic GABA channels in mammalian neurons (Bormann et al., 1987; Kaila et al., 1993), with a P HCO3/P Cl ratio of ∼0.2 (Kaila, 1994). Whether the internal HCO 3 − concentration was also important for the regulation of the IPSC decay time was examined by comparing eIPSCs recorded from Purkinje neurons bathed in aCSF or in an O2-gassed/HEPES-based external solution to create a nominally HCO 3 −-free solution (Fig. 4 A–D). In the HCO 3 −-free, 10 mm Cl− solution, the amplitudes of the eIPSCs were reduced to 65.8 ± 7.5% of their value compared in normal aCSF (p = 0.05, unpaired two-tailed t test, n = 6–8).
Figure 4.

Bicarbonate ions do not affect the IPSC decay time constant. A, In the presence of bicarbonate ions, contained in normal aCSF, consecutively evoked (gray) and averaged eIPSCs (black) were recorded from cerebellar Purkinje cells with 150 mm internal Cl− solution at −100 mV. B, Evoked IPSCs recorded from the same cell after exchanging aCSF with HCO3 −-free solution (100% O2/HEPES buffered). C, Consecutive (gray) and averaged (black) eIPSCs with 10 mm internal Cl− solution at −100 mV. D, Evoked IPSCs recorded from the same cell after exchange with HCO3 −-free aCSF. E, Cumulative distribution for eIPSC decay time constants at −100 mV with 150 mm (black) and 10 mm (gray) internal Cl− recorded in normal aCSF (dashed lines) and HCO3 −-free aCSF (solid lines).
However, by using an 150 mm internal Cl− concentration, eIPSC amplitudes remained unaffected by switching to an HCO3 −-free aCSF (95.2 ± 9.8%; p = 0.5, n = 6–8). This reflected the relative contributions of HCO3 − and Cl− ions to the IPSC and the lower permeability of HCO3 − through the GABA channel. The decay time constant was again reduced, as expected, by low internal Cl− concentrations in normal aCSF (Fig. 4 E) (150 mm Cl−, median = 8.9 ms; 10 mm Cl− median = 6.3 ms). The extent of the reduction in eIPSC decay times by low internal Cl− remained unaffected by the HCO3 −-free solution (150 mm Cl−, median = 9.5 ms; 10 mm Cl− median = 6.3 ms) (Fig. 4 E), indicating that as a permeant ion, only Cl− are necessary and sufficient to regulate the decay phase of the eIPSC.
Low Cl− regulates IPSC decay times by directly affecting GABAA receptor function
To determine whether the Cl− modulation is intrinsic to the synaptic GABAA receptor, rather than mediated by receptor-associated molecules located at inhibitory synapses (Lüscher and Keller, 2004), we expressed α1β2γ2L subunits in human embryonic kidney (HEK) cells. This combination of subunits was chosen to replicate the likely composition of synaptic GABAA receptors in Purkinje cells (Laurie et al., 1992; Fritschy and Mohler, 1995). Outside-out patches were briefly exposed to saturating concentrations of GABA (3 mm, 1 ms) to emulate the GABA concentration transient in the synaptic cleft (Jones and Westbrook, 1995). With either 150 mm (Fig. 5 A,B) or 10 mm (Fig. 5 C,D) internal Cl− concentrations, the GABA-activated current (I GABA) decays were best fit by two or three exponential components. Over a patch potential range from −100 mV to +60 mV, the weighted τdecay for I GABA increased monotonically in a similar manner to the decay time constants for evoked and spontaneous IPSCs. The weighted τdecay for inward I GABA at −100 mV was significantly reduced by 10 mm intracellular Cl− from 54.5 ± 5.8 (n = 10) to 21.1 ± 2.9 ms (n = 10, p = 0.0002, two-tailed t test) (Fig. 5 E). A similar reduction in τdecay was observed (from 75.4 ± 13.6 to 40.9 ± 8.3 ms, p = 0.04) when the same patches were held at 60 mV causing the direction of the GABA-activated currents to become net outward. The consistent effects of internal Cl− concentration on the current decays mediated by both synaptic and recombinant GABAA receptors, with their similar subunit composition, strongly indicates that Cl− is modulating the receptor directly, rather than acting via receptor-associated molecules. However, while elements of the postsynaptic density will not be present in HEK cells, it is conceivable that some remnants of receptor-associated molecules, such as protein kinases, may be present in outside-out patches and these might be affected by Cl− concentration thereby affecting macroscopic GABA current decays. To address this, we included the broad spectrum serine/threonine kinase inhibitor, staurosporine (500 nm), in the patch pipette and repeated the rapid GABA applications in the presence of low and high Cl− concentrations. The weighted τdecay for inward I GABA at −100 mV was again significantly reduced by 10 mm internal Cl− from 45.1 ± 2.7 (n = 6) to 20.1 ± 3.3 ms (n = 5, p = 0.0004, unpaired t test), yielding a τ(high Cl)/τ(low Cl) ratio of 2.3, which is very similar to that measured in the absence of staurosporine (2.5). Thus internal Cl− is most likely modulating the receptor by a direct action rather than through receptor-associated signaling molecules.
Figure 5.

Recombinant α1β2γ2L GABAA receptor kinetics are modulated by intracellular Cl−. Shown are currents activated by 3 mm GABA (1 ms pulse) recorded from outside-out patches of HEK cells expressing GABAA receptors. A, GABA currents recorded at −100 to 60 mV (40 mV steps, average of 10 sweeps per voltage) with 150 mm Cl− in the pipette solution decay more slowly at positive potentials. Inset, An I–V plot for GABA-activated currents (E rev is 7 mV). B, Peak-scaled, superimposed currents at −100 and 60 mV. Note the currents recorded at −100 mV have been flipped to match those at 60 mV. C, The same experiment repeated with 10 mm intracellular Cl−. Currents reverse at −56 mV (inset). The relative voltage dependence of the weighted decay time constant is maintained (compare peak-scaled currents in D), but at all membrane voltages, the decay time is 2–3 times faster in low Cl− (average of 12 sweeps). E, Summary of weighted decay times in 150 mm (black) and 10 mm (gray) Cl− at all the voltages tested. The calibrations in A and B apply to the bars in C and D, respectively.
Implications of fast-decaying IPSCs for synaptic inhibition
Given that the physiological Cl− concentration in an adult neuron is lower than we routinely use in our symmetrical Cl− solutions, it is likely that the real profile of an IPSC in an intact cell is much shorter than we previously thought. Therefore, does the speed of the IPSC decay, and the corresponding effect this has on the time window for inhibition, reduce the effectiveness of synaptic inhibition in controlling neuronal excitability? This question was addressed by recording from Purkinje cells under current-clamp conditions and then injecting inhibitory conductances using a dynamic clamp amplifier. We evoked a continuous spike train in the Purkinje cell with a constant current pulse (∼200 pA) before introducing an artificial inhibitory conductance to inhibit the train. To reproduce the IPSCs expected in a Purkinje cell with either high (150 mm) or low (10 mm) internal Cl− concentrations, the conductance decay time constants were set to 12.5 and 8.5 ms, respectively (Fig. 6 A). Action potentials ceased immediately after the onset of the artificial inhibitory conductance, and the latency to the first spike was significantly increased when the injected conductance had a longer decay time (average first latency changed from 41 ± 2 to 54 ± 4 ms, n = 9) (Fig. 6 A,B).
Figure 6.

Decay times of inhibitory synaptic conductances profoundly affect Purkinje neuron excitability. A, Constant current pulse (1 s, 200 pA) evoked spike firing in a Purkinje neuron with slow (12.5 ms decay time, black) and fast (8.5 ms decay time, blue) inhibitory conductances (E rev = −82 mV) (Chavas and Marty, 2003) injected through a conductance clamp amplifier during the train. The fast conductance is reflected by the shorter inhibition time window [see expanded timescale, right panel; 99 (fast conductance) and 102 (slow) overlaid sweeps at −65 mV]. B, The cumulative distribution for first spike latencies after conductance injection is significantly displaced to the right for longer-lasting conductances (n = 9, time course of the conductance injection is shown below). C, Injecting an excitatory conductance (E rev = 0 mV, τrise = 0.3 ms, τdecay = 3 ms, conductance fixed at 20% above threshold, typically 12–18 nS) to induce a single spike with a concurrent sliding inhibitory conductance (fixed at 15 nS) with either slow (left, black) or fast (right, blue) kinetics. Sweeps are separated by 4 ms, and those shown in the top panel correspond to conductances depicted as thick lines in the lower panel. The cell is prevented from firing by the inhibition time window, which is reduced from ∼50 to ∼20 ms for the fast conductance injection (measured over 50–60 trains of injections, alternating between fast and slow conductance injection). D, Summary of the duration of the inhibition windows for fast (blue) and slow (black) conductance injections.
As an alternative approach to assess the impact of the kinetics of inhibition on cell excitability, we injected a brief excitatory conductance into the soma of the Purkinje cell under current clamp to emulate the activation of synaptic AMPA-type glutamate receptors at climbing fiber excitatory synapses and initiate action potential firing (Traynelis et al., 1993). An inhibitory conductance with either slow or fast kinetics (based on the internal Cl− concentration) was also introduced and its latency varied so that the inhibitory conductance was active over a time window that encompassed the excitation. A slow inhibitory conductance, which preceded an excitatory conductance, inhibited spike firing within a time window of ∼37 ms (Fig. 6 C,D). For the faster inhibitory conductance, typical of neurons exposed to low internal Cl−, this window is significantly reduced by over half to 15 ms.
Discussion
Internal Cl− regulates IPSC decay by a direct effect on the GABAA receptor
We have demonstrated here, for the first time, that the duration of GABAergic synaptic inhibition is strongly influenced by the intracellular concentration of Cl−, the GABAA receptor's major permeating anion. Indeed, by studying a range of internal Cl− concentrations, we observed that the IPSC decay time constant was clearly affected by changes to the Cl− concentration over a range that is considered to be physiologically relevant (e.g., between 5 and 30 mm).
It is important to note that GABAA receptor channels are also permeable to HCO3 − ions (Kaila et al., 1993), and it is conceivable that these might have emulated or supplemented the effect of internal Cl− on the IPSC decay times, by competing with Cl− ions for the same binding site(s) on the receptor. Under our recording conditions, using aCSF with a partial pressure of CO2 of ∼37.5 mmHg, the intracellular HCO3 − concentration lies between 8 and 13 mm, at an intracellular pH of 7.2–7.4. This implies the equilibrium potential for HCO3 − is between −18 and −31 mV. Therefore, the maximum contribution of HCO3 − to the eIPSC will occur when we are recording with low internal Cl− at −100 mV. However, even under these conditions, the removal of HCO3 − ions did not affect the decay time constant, indicating that Cl−, in the context of permeant ions, is the major determinant of the IPSCs decay kinetics.
A link between the effectiveness of inhibition and internal Cl− has been noted previously from early intracellular current-clamp recordings of CA1 pyramidal neurons using 3 m KCl and 2 m KCH3SO4-filled electrodes (Alger and Nicoll, 1979). A prolonged time course of the depolarizing IPSP was observed in neurons where Cl− leakage had occurred from the KCl pipette. Although the duration of synaptic potentials is also dependent on the membrane time constant, most likely this prolongation reflected the phenomenon of internal Cl− affecting IPSC decays that we report in the present study.
There are several mechanisms by which Cl− could regulate the decay phase kinetics of the IPSC. One possibility is that high internal Cl− prolongs the IPSC decay by slowing the uptake of GABA by the principal neuronal GABA transporter subtype, GAT-1 (Guastella et al., 1990), which is thought to have a stoichiometry of 2 Na+:1 GABA:1 Cl− (Cammack et al., 1994). This is unlikely to account for our findings because GABA current kinetics also became slower with high internal Cl− in excised outside-out patches where GABA is applied by a rapid application system and GABA transport plays no role in the current decay process. This result also indicated that the regulation by internal Cl− must be an intrinsic property of the GABAA receptor and does not depend on intracellular signaling mechanisms and on receptor-associated molecules, many of which, such as gephyrin, are not expressed in HEK cells (Thomas and Smart, 2005).
The results obtained with the recombinant GABAA receptors suggest that one or more sites must exist on the receptors that are capable of binding Cl−. There appear to be no clear structural consensus sequences for Cl− binding sites (Plested and Mayer, 2007), but as internal Cl− regulates the IPSC decay, such sites are most likely to be found within the intracellular domains between M1 and M2, or between M3 and M4, or conceivably within the ion channel and its associated internal vestibule. Some support for the latter structure as a potential site(s) comes from prior studies of acetylcholine receptors in Aplysia neurons, where the concentration of the permeating ion was directly related to the mean channel open time, and occupancy of such a site(s) in the ion channel was postulated to prolong the channel open duration (Ascher et al., 1978; Marchais and Marty, 1979).
Physiological impact of regulating the speed of synaptic inhibition
We predict that the regulation of GABAA receptor function by internal Cl− is likely to have significant consequences for neuronal excitability, because the duration over which an inhibitory input can effectively counteract coincident neuronal excitation is appreciably shorter with low physiological levels of Cl− compared with that observed with symmetrical high Cl−. This is fundamentally important, as intracellular Cl− concentrations are subject to alteration by the density and activity of Cl− transporters (DeFazio et al., 2000), which will influence IPSC time profiles and thereby signal integration.
Our finding that the kinetics of GABAA receptor currents are modulated by internal Cl− ions has many implications for our understanding of synaptic inhibition. It is clear that we have been underestimating the speed of inhibitory transmission. This has occurred because of the routine use of symmetrical Cl− recording solutions to depolarize E Cl close to 0 mV, to ensure that the IPSCs are of large amplitudes for ease of measurement when holding cells near to their resting membrane potentials. By using physiological concentrations of internal Cl−, the shorter time window now available for synaptic inhibition will reduce its effect on cell excitability with consequences for synaptic plasticity and presumably the modeling of neuronal network behavior. Indeed, at physiological body temperatures, the real durations of IPSCs are so short that they now approach 2–3 ms, and therefore become difficult to resolve accurately with the level of filtering imposed by the recording apparatus.
Furthermore, the kinetics of I GABA for recombinant GABAA α1β2γ2L receptors expressed in HEK cells exhibited a similar sensitivity to the internal Cl− concentration. This combination of GABAA receptor subunits is considered to account for at least 30% of inhibitory synaptic GABAA receptors in the CNS (Whiting et al., 1995), indicating that their regulation by internal Cl−, may well be a universal feature of many inhibitory synapses across the CNS. Indeed, a similar phenomenon is also apparent for the glycine receptor (Pitt et al., 2008), indicating that regulation by internal Cl− may be an intrinsic property of all ligand-gated anion channels in the Cys-loop superfamily.
The regulation of the speed of IPSC decays may also be relevant to neurons during postnatal development. The higher internal Cl− concentrations that characterize this stage of neurodevelopment (Ben-Ari et al., 2007) would allow GABAA receptor activation to initiate more prolonged depolarization, eventually exceeding the threshold for voltage-gated calcium channel activation (Cherubini et al., 1991). Even for more mature neurons with lower basal Cl− concentrations, the prolonged activation of GABAA receptors in structurally small dendrites can eventually lead to Cl− loading and membrane depolarization, which is exacerbated by HCO 3 − efflux (Staley and Proctor, 1999). Under these conditions, the time course of the synaptic GABA currents would again be prolonged with consequences for neuronal excitability.
It has also been reported that E Cl can vary between the dendrites and the soma, affecting synaptic inhibition in the same neuron (Duebel et al., 2006; cf. Glickfeld et al., 2009). We would predict that the time course of, and therefore the time window for, synaptic inhibition will be shorter, where the Cl− concentration is lowest and thus IPSCs with dendritic and somatic origins could have different time courses in the same cell. This newly discovered role for Cl− will have important implications not only for how synaptic inhibition regulates the activity of neuronal networks, but also for understanding how alterations in internal Cl− affect early neuronal development (Ben-Ari, 2002) and the progression of pathophysiological conditions, such as epilepsy (Huberfeld et al., 2007) and chronic pain (Coull et al., 2003).
Footnotes
This work was supported by the Medical Research Council (UK) to T.G.S. and L.G.S., and by a Royal Society University Research Fellowship and Wellcome Trust support to M.B. We thank Angus Silver and Jason Rothman for the loan of the dynamic clamp amplifier. After completion of our study, we learned that unpublished observations similar to those described in the present paper were independently made by Joel Charvas, Christophe Pouzat, and Alain Marty.
References
- Alger BE, Nicoll RA. GABA-mediated biphasic inhibitory responses in hippocampus. Nature. 1979;281:315–317. doi: 10.1038/281315a0. [DOI] [PubMed] [Google Scholar]
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at Purkinje axon initial segment. Cell. 2004;119:257–272. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Ascher P, Marty A, Neild TO. Life time and elementary conductance of the channels mediating the excitatory effects of acetylcholine in Aplysia neurones. J Physiol. 1978;278:177–206. doi: 10.1113/jphysiol.1978.sp012299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis A, Mozrzymas JW, Ortinski PI, Vicini S. Desensitization and binding properties determine distinct α1β2γ2 and α3β2γ2 GABAA receptor-channel kinetic behavior. Eur J Neurosci. 2007;25:2726–2740. doi: 10.1111/j.1460-9568.2007.05530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI, Brickley SG. Synaptic release generates a tonic GABAA receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammack JN, Rakhilin SV, Schwartz EA. A GABA transporter operates asymmetrically and with variable stoichiometry. Neuron. 1994;13:949–960. doi: 10.1016/0896-6273(94)90260-7. [DOI] [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Gage PW, Robertson B. Inhibitory post-synaptic currents in rat hippocampal CA1 neurones. J Physiol. 1984;356:551–564. doi: 10.1113/jphysiol.1984.sp015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci. 2000;20:8069–8076. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron. 2006;49:81–94. doi: 10.1016/j.neuron.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- Fritschy J-M, Mohler H. GABAA receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Houston CM, Hosie AM, Smart TG. Distinct regulation of β2 and β3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2008;28:7574–7584. doi: 10.1523/JNEUROSCI.5531-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Shaping of IPSCs by endogenous calcineurin activity. J Neurosci. 1997;17:7626–7633. doi: 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, González J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci. 2000;3:1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- Lüscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Marchais D, Marty A. Interaction of permeant ions with channels activated by acetylcholine in Aplysia neurones. J Physiol. 1979;297:9–45. doi: 10.1113/jphysiol.1979.sp013025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Häusser M. Linking synaptic plasticity and spike output at excitatory and inhibitory synapses onto cerebellar Purkinje cells. J Neurosci. 2007;27:5559–5570. doi: 10.1523/JNEUROSCI.5117-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera K, Takeuchi A. An analysis of the inhibitory post-synaptic current in the voltage-clamped crayfish muscle. J Physiol. 1979;286:265–282. doi: 10.1113/jphysiol.1979.sp012618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience. 1992;49:13–32. doi: 10.1016/0306-4522(92)90073-b. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Westbrook GL. Synapse density regulates independence at unitary inhibitory synapses. J Neurosci. 2003;23:2618–2626. doi: 10.1523/JNEUROSCI.23-07-02618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Sivilotti LG, Beato M. High intracellular chloride slows the decay of glycinergic currents. J Neurosci. 2008;28:11454–11467. doi: 10.1523/JNEUROSCI.3890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJR, Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Puia G, Costa E, Vicini S. Functional diversity of GABA-activated Cl− currents in Purkinje versus granule neurons in rat cerebellar slices. Neuron. 1994;12:117–126. doi: 10.1016/0896-6273(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Ascher P. Coupling of permeation and gating in an NMDA-channel pore mutant. Neuron. 1997;18:167–177. doi: 10.1016/s0896-6273(01)80055-6. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3 − transport. J Physiol. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Ashcroft FM, Plant TD. Gating of a muscle K+ channel and its dependence on the permeating ion species. Nature. 1981;289:509–511. doi: 10.1038/289509a0. [DOI] [PubMed] [Google Scholar]
- Swenson RP, Jr, Armstrong CM. K+ channels close more slowly in the presence of external K+ and Rb+ Nature. 1981;291:427–429. doi: 10.1038/291427a0. [DOI] [PubMed] [Google Scholar]
- Thomas P, Smart TG. HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 2005;51:187–200. doi: 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-s. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ, McKernan RM, Wafford KA. Structure and pharmacology of vertebrate GABAA receptor subtypes. Int Rev Neurobiol. 1995;38:95–138. doi: 10.1016/s0074-7742(08)60525-5. [DOI] [PubMed] [Google Scholar]