

Summary
Hyperphosphorylation of the microtubule associated protein, Tau, is the hallmark of a group of neurodegenerative disorders known as the tauopathies which includes Alzheimer's disease. Precisely how and why Tau phosphorylation is increased in disease is not fully understood, nor how individual sites modify Tau function. Several groups have used the Drosophila visual system as an in vivo model to examine how the toxicity of Tau varies with phosphorylation status. This system relies on overexpression of Tau from transgenes but is susceptible to position effects altering expression and activity of the transgenes. We have refined the system by eliminating position effects through the use of site-specific integration. By standardising Tau expression levels we have been able to compare directly the toxicity of different isoforms of Tau and Tau point mutants that abolish important phosphorylation events. We have also examined the importance of human kinases in modulating Tau toxicity in vivo. We were able to confirm that human GSK3β phosphorylates Tau and increases toxicity but, unexpectedly, we identified that preventing phosphorylation of Ser404 is a protective event. When phosphorylation at this site is prevented, Tau toxicity in the Drosophila visual system is increased in the presence of GSK3β. Our data suggest that not all phosphorylation events on Tau are associated with toxicity.
Keywords: Tau, Phosphorylation, GSK3β, Drosophila
Introduction
The tauopathies are a group of neurodegenerative diseases characterised by the accumulation of intra-neural aggregates of the microtubule-associated protein, Tau. They include Alzheimer's disease (AD), fronto-temporal lobar degeneration (FTLD-Tau), Pick's disease, progressive supranuclear palsy and corticobasal degeneration. The microtubule-binding properties of Tau were identified more than 30 years ago but, and despite intense research, its true physiological role remains unclear, thus the cellular mechanisms underpinning the tauopathies are also poorly understood. More recently, additional properties of Tau have been identified, including an ability to bundle actin filaments, and multiple Tau-binding partners have been identified, including tyrosine kinases. Together, these have led to the suggestion that Tau may act as a signalling scaffold (reviewed by Morris et al., 2011).
The functions of Tau may be regulated both by alternative splicing (six isoforms are expressed in the human CNS) and through a multitude of post-translational modifications. These include multiple phosphorylation and acetylation events, glycosylation, ubiquitylation and sumolyation amongst others (reviewed by Martin et al., 2011). The phosphorylation of Tau has received the most attention because Tau within neuropathological aggregates is found primarily in a highly phosphorylated form (Buée et al., 2000; Lee et al., 2001; Ballatore et al., 2007; Hanger et al., 2009; Iqbal et al., 2009). In vitro, hyperphosphorylation of Tau reduces its microtubule binding affinity (Lindwall and Cole, 1984). This leads to an increase of hyperphosphorylated Tau in the cytosol and a destabilization of the microtubule network (Cowan et al., 2010; Feuillette et al., 2010). Both the accumulation of highly phosphorylated cytosolic Tau and destabilization of the microtubules are suggested to lead to neurodegeneration (Cowan et al., 2010; Wu et al., 2011). Yet, increased phosphorylation of Tau is not necessarily toxic to neurons in all circumstances as it occurs during fetal development (Yu et al., 2009) and transiently in hibernating mammals (Arendt et al., 2003). An alternative view sees elevated Tau phosphorylation as part of a protective response to oxidative stress rather than a direct cause of pathology (Castellani et al., 2008; Bonda et al., 2011). The complex relationship between Tau phosphorylation and neurodegeneration is also highlighted by observations that phosphorylation resistant Tau constructs either lose or maintain their toxicity, suggesting a non-equivalent role for groups of phosphorylation sites. In Drosophila both TauAP and TauS11A forms, with 14 or 11 Ser/Thr sites mutated to alanine respectively, retain microtubule binding function yet TauAP loses toxicity while TauS11A retains toxicity (Steinhilb et al., 2007b; Feuillette et al., 2010; Talmat-Amar et al., 2011). Other studies have shown alternatively that phosophorylation of Tau at specific sites can promote microtubule binding or reduce Tau toxicity (Wada et al., 1998; Feijoo et al., 2005; Thies and Mandelkow, 2007) while increased binding of Tau to microtubules may also be deleterious to neurons through interference with axonal trafficking (Talmat-Amar et al., 2011). Thus, a precise balance of differential Tau phosphorylation at individual sites may be required to appropriately regulate levels of cytosolic or microtubule bound Tau essential for microtubule dynamics and axon transport.
Since Tau phosphorylation is likely to contribute in some way to pathology, one therapeutic strategy being followed is to reduce the phosphorylation load on Tau by targeting Tau kinases (Churcher, 2006; Mazanetz and Fischer, 2007; Brunden et al., 2009). For this approach to be effective, it is important to identify which of the many Tau phosphorylation events that have been identified in vitro are critical for toxicity in vivo and to establish which kinases phosphorylate Tau in disease states and whether Tau forms resistant to phosphorylation show reduced toxicity. The scale of this task is significant because recent studies have identified 45 distinct sites that are phosphorylated on Tau from AD brains compared with only 17 from healthy brains, with many different kinases capable of phosphorylating Tau in vitro (Morishima-Kawashima et al., 1995; Hanger et al., 1998; Hanger et al., 2007; Hanger et al., 2009; Lebouvier et al., 2009). Moreover, some phosphorylation events will modify Tau in vitro but not necessarily in a manner that is physiologically relevant. There is a need, therefore, for a model system in which individual kinases can be tested for their ability to alter Tau toxicity in vivo.
Tau-mediated degeneration of photoreceptor neurons in the Drosophila visual system is a commonly used in vivo model to study the cell biology of the tauopathies (Wittmann et al., 2001; Jackson et al., 2002; Muqit and Feany, 2002; Nishimura et al., 2004; Karsten et al., 2006; Steinhilb et al., 2007a; Khurana, 2008; Chatterjee et al., 2009). Typically, human Tau is expressed ectopically in the developing Drosophila brain or visual system, resulting in neurodegeneration that bears several hallmarks of the tauopathies, including age dependency, abnormally phosphorylated Tau and, in some cases, Tau aggregates (e.g. Wittmann et al., 2001; Jackson et al., 2002; reviewed by Gistelinck et al., 2012). The powerful genetics of Drosophila can be employed to identify endogenous genes that are required for tau-mediated degeneration or can modify the degree of degeneration mediated by human Tau. Using this approach, several Drosophila kinases, including the homologues of GSK3β, MARK, cdk5, JNK and PKA, have been implicated in Tau toxicity (Wittmann et al., 2001; Jackson et al., 2002; Shulman and Feany, 2003; Chau et al., 2006; Steinhilb et al., 2007a; Steinhilb et al., 2007b; Chatterjee et al., 2009).
Following our studies of Tau phosphorylation in AD post-mortem brain (Hanger et al., 1998; Hanger et al., 2007), we were interested to determine whether the Drosophila photoreceptor model could be used to assess the roles of human kinases in mediating neurodegeneration in vivo and to identify particular phosphorylation events on Tau that are important for toxicity. Transgene expression in Drosophila is affected by positional effects on transgene activity which complicate comparisons of the toxicity mediated by different isoforms or mutant forms of human Tau. To overcome this, we used an alternative methodology to express Tau where Tau transgenes are targeted to pre-determined sites in the Drosophila genome to control for any positional effects and permit direct comparisons of toxicity. We sought to confirm the importance of GSK3β as a pathological kinase in vivo, both individually and in combination with CK1δ or DYRK1A, both of which could act as priming kinases for GSK3β by phosphorylating Tau residues adjacent to GSK3β target sites (reviewed by Doble and Woodgett, 2003). CK1δ is highly overexpressed in AD brain (Ghoshal et al., 1999) and is tightly associated with PHF Tau (Kuret et al., 1997). DYRK1A is both overexpressed in AD brain material and duplicated in the critical region of chromosome 21 in Down's syndrome which is associated with early onset AD (Kimura et al., 2007). We were able to confirm the importance of GSK3β for toxicity in vivo, but found little role for priming kinases in this system. Surprisingly, we identified a GSK3β-mediated phosphorylation event that seems to be protective. We also found that, in contrast to previous work, the R406W mutation in Tau associated with FTLD-Tau has no effect on toxicity in this system.
Results
Tau-mediated degeneration in transgenic flies is modified by position effects
We sought to use Drosophila as a model to assess the roles of human kinases to generate toxic forms of Tau and to identify the particular phosphorylation events on Tau responsible for toxicity. Previous studies of human Tau toxicity in Drosophila have used transgenic lines generated by P-element mediated transgenesis (Wittmann et al., 2001; Jackson et al., 2002; Nishimura et al., 2004; Steinhilb et al., 2007a; Steinhilb et al., 2007b; Chatterjee et al., 2009) where constructs containing human Tau cDNA are inserted at random into the Drosophila genome (Spradling and Rubin, 1982). Expression of Tau is achieved using the UAS/GAL4 system or by use of a tissue specific promotor. However variable levels of expression results from the same Tau construct inserted at different genomic locations. This position-dependent expression is a well-characterised phenomenon of P-element mediated transgenesis in Drosophila and, whilst this can be exploited to obtain a range of expression levels (Wilson et al., 1990), it may limit the ability to directly compare the toxicity of variant forms of the transgene. We investigated the extent to which Tau toxicity can vary due to positional effects resulting from random insertion of the same transgene. We generated five different random insertions of UAS-2N4R Tau and used GMR-GAL4 to drive Tau expression in the developing visual system from one copy of the transgene. The disruption of the regular, crystalline array of ommatidia in the adult compound eye ranged from mild (e.g. line 4) to severe (e.g. line 3) (Fig. 1A).
Fig. 1. Position effect causes variations in Tau-mediated toxicity in the Drosophila eye.
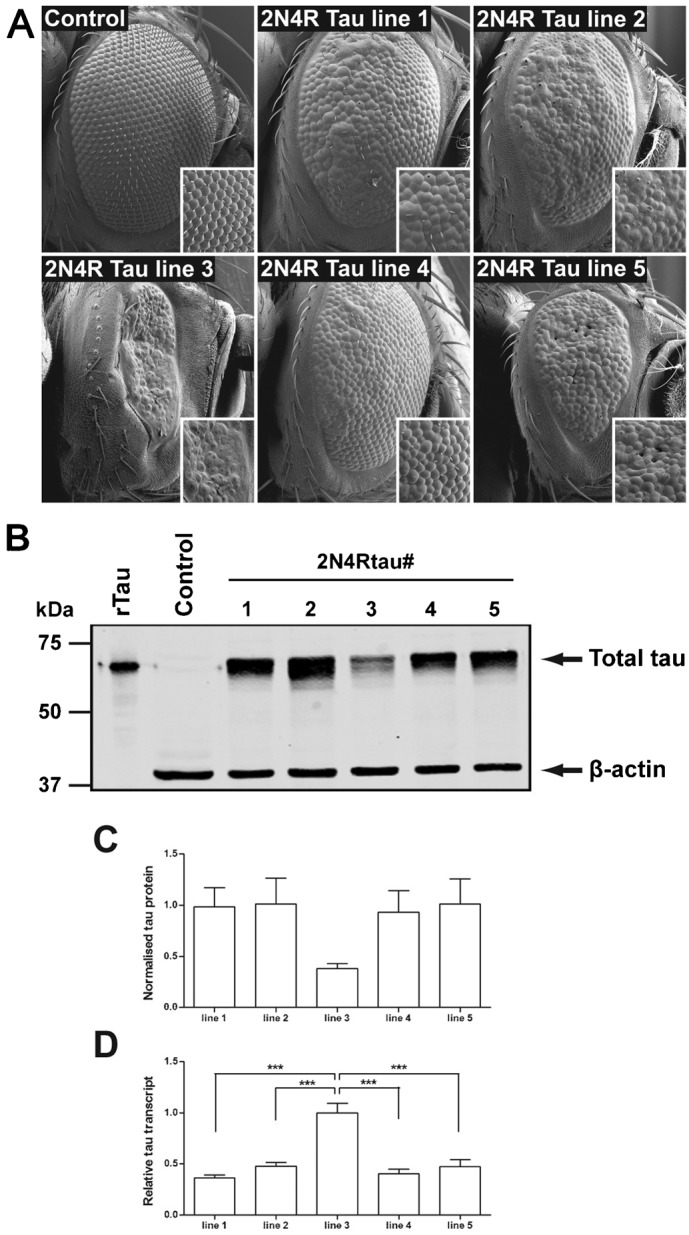
(A) Scanning electron micrographs of Drosophila eyes. The regular array of ommatidia of the compound eye is disrupted by overexpression of Tau to a variable degree. The GMR-gal4 driver was mated to wild-type (control) or UAS-2N4R Tau flies. Five different randomly inserted UAS-lines were assayed. (B, C) Quantitative Western blotting of Tau levels in adult Drosophila heads from GMR-gal4 × UAS-2N4R Tau flies. B. Blot probed with anti-total Tau and anti-β-actin. (C) Quantification of Tau protein levels normalised to actin. Recombinant Tau was included to permit normalisation between blots. (D) Tau transcript levels in brain and eye discs from third instar larvae before degeneration occurs. Tau transcripts were normalised to membrane GFP expressed from the GMR-gal4 chromosome after recombination (see Materials and Methods).
We identified the genomic insertion site of UAS-Tau for each line (supplementary material Table S1). Four of the five lines (lines 1, 2, 4, and 5) were homozygous viable insertions and none of these showed an eye phenotype in the absence of the GMR-GAL4 driver. Line 3 exhibited the most severe degeneration (Fig. 1). This line contained Tau inserted into the l(3)87Df gene, and was lethal when homozygous. To exclude the possibility that the severe degeneration apparent in line 3 was due to a genetic interaction between Tau and l(3)87Df gene, which encodes a probable chaperone, we used GMR-GAL4 to express Tau from the mildly disruptive line 1 locus in a genetic background heterozygous for an allele of l(3)87Df. No increase in degeneration resulted, excluding the possibility that l(3)87Df plays a role in Tau-mediated degeneration (supplementary material Fig. S1). We concluded that the Tau-mediated degeneration is highly dependent on positional effects and we would need to control for these if we were going to compare the toxicity of different forms of Tau.
The amount of Tau protein does not correlate with degeneration
To determine the relationship between the level of disruption to the eye in the differing transgenic lines and Tau protein expression, we quantified the amount of Tau expressed in each line in Drosophila heads normalised to β-actin (Fig. 1B,C). We detected significantly less Tau protein in line 3, compared to the other lines, despite this line displaying the most severe degeneration. The amounts of Tau protein present in lines 1, 2, 4, and 5 were equivalent to each other, despite differences in the degree of disruption to the eye (Fig. 1C). The reduced level of Tau protein in the lines with greater levels of disruption may potentially be due to increased numbers of dying or dead cells. To circumvent this we used qPCR to measure transcript levels of each Tau transgene earlier in the development of the visual system, within late third instar larval eye imaginal discs, prior to onset of degeneration. We controlled for the number of cells expressing GMR-GAL4 (and therefore Tau) by recombining UAS-GFP with GMR-Gal4 to drive the expression of GFP. Normalisation of the Tau transcript levels to that of GFP for each line revealed significantly increased Tau transcription in the highly degenerate line 3 compared to all of the other Tau insertion lines (Fig. 1D). Lines 1, 2, 4, and 5 each express approximately equivalent amounts of Tau mRNA and protein, despite variation in the levels of Tau toxicity apparent in each line. Line 3 expresses significantly more Tau mRNA which correlates with the more severe degenerate phenotype, suggesting the lower levels of Tau protein in adults is a consequence of increased cell loss. Thus, the level of toxicity mediated by 2N4R-Tau driven from differing transgenes varies significantly and is influenced by positional effects. We could not be confident that protein or mRNA levels would predict toxicity sufficiently accurately to be used as a reliable method for selecting comparable insertion lines.
Site-specific integration of transgenes eliminates the effects of positional variation
To overcome the problem of positional effects on Tau expression, we turned to the φC31 system that allows site specific integration of Drosophila transgenes at specific “landing” sites in the genome (Groth et al., 2004). We integrated UAS-2N4R Tau at five different landing sites spread across chromosomes II and III (insertion sites at 51C, 68A, 68C, 86F and 96E). After crossing to GMR-GAL4 driver flies, toxicity in the eyes was assessed (Fig. 2A). In two cases (51C and 68E; Fig. 2A, upper row) there was very little disruption of the crystalline array of ommatidia; in three others (68A, 86F and 96E; Fig. 2A, lower row) disruption was moderate. In none of the lines was disruption seen to the level of any of our randomly integrated lines (cf. Fig. 1). We quantified Tau transcript expression in eye discs from two of the five site-specific lines, 68A and 86F, relative to GFP, as before. As expected, Tau is expressed at significantly lower levels in both of these lines. Tau expression levels in the targeted insertion lines was approximately half of the level in the random insertion lines 1 and 2 and considerably lower than the highly degenerate line 3 (Fig. 2B). To check reproducibility of the landing sites, we generated an additional independent line with UAS-2N4R Tau inserted at the 68A locus. This second insertion generated an identical eye phenotype to the initial 68A transgenic when expression was driven with GMR-GAL4 (data not shown).
Fig. 2. Site-specific insertions of UAS-2N4R Tau mediate lower levels of toxicity.
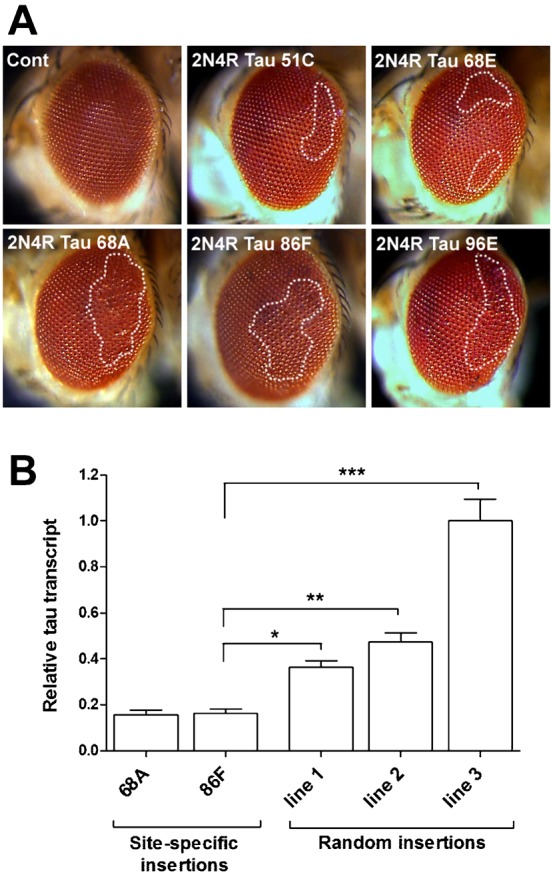
UAS-2N4R Tau was inserted into five specific genomic landing sites by φC31-mediated integration. (A) Light micrographs of adult eyes from GMR-gal4 × wild-type (control) or UAS-2N4R Tau crosses. Areas of the eye displaying significant disruption are highlighted with dashed lines. (B) Tau transcript levels driven by GMR-gal4 from two site-specific UAS insertions normalised to our random insertion line 3.
Using site-specific insertion lines, R406W mutation does not enhance the toxicity of Tau
We used the φC31-mediated site-specific insertion to test the contribution of specific residues within Tau to cause toxicity in vivo. The R406W mutation in Tau is associated with some cases of autosomal dominant FTLD-Tau (Hutton et al., 1998) and has been reported to be more toxic than wild-type Tau when overexpressed in Drosophila (Wittmann et al., 2001; Jackson et al., 2002; Khurana et al., 2006). The R406W mutation affects the ability of Tau to regulate microtubule dynamics in vitro (Bunker et al., 2006) and impairs axonal transport in mice when overexpressed at high levels (Zhang et al., 2004) and may also affect the phosphorylation of Tau by specific kinases (Mack et al., 2001). We used GMR-GAL4 to drive expression of the 2N4R and 0N4R wild-type and R406W Tau isoforms from constructs inserted into the 68A locus. We found that there were no differences in toxicity between unmodified 0N4R or 2N4R Tau and those containing the R406W point mutant when expressed from the same insertion site, in contrast to the severe increase in toxicity in the eye reported previously for the R406W mutation (Wittmann et al., 2001; Jackson et al., 2002; Nishimura et al., 2004) (Fig. 3A, upper panels). In parallel, we examined the eye phenotype of flies overexpressing 0N4R Tau R406W from the randomly inserted construct generated by the Feany lab and used in several previous studies (Wittmann et al., 2001; Jackson et al., 2002; Nishimura et al., 2004) and confirmed this line generated a much stronger phenotype in the eye (Fig. 3B). We quantified transcript expression in larval eye discs and normalised expression to that of the strongest 2N4R Tau random insertion, line no. 3 (Fig. 3C). This revealed that each of the four Tau isoforms were expressed from the 68A site at similar levels, as expected (no significant differences were observed; Fig. 3C), but notably, expression from the 68A insertion site was approximately half that from four of the five randomly integrated lines we generated. The 0N4R Tau R406W line was expressed at a level twice that of the lines inserted at 68A and comparable to the level seen for most of our random insertions (Fig. 3C). We wondered whether Tau expression from the 68A insertions was below a threshold needed to observe any differential toxicity. We increased expression by combining GMR-GAL4 with double the copy number of the 68A UAS-Tau insertion in homozygous animals. As expected, the eye phenotype worsened in each case (Fig. 3A, lower panels) but the R406W variants of both 2N4R and 0N4R Tau continued to show similar toxicity to the non-mutated forms.
Fig. 3. No increase in Tau-mediated toxicity caused by the R406W mutation.
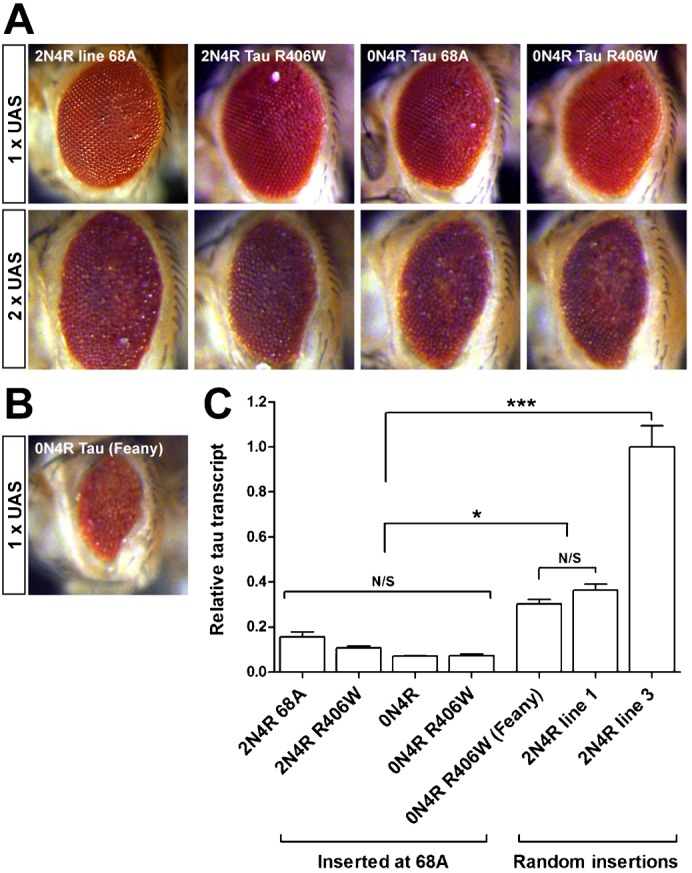
(A) GMR-gal4 driving expression from the 68A integration site of 2N4R or 0N4R Tau with or without the R406W mutations. Upper row: one copy of the UAS-insert. Lower row: two copies of the UAS-insert. GMR-gal4 is heterozygote in all cases. The R406W mutation does not increase toxicity. (B) GMR-gal4 driving 0N4R Tau R406W from a randomly inserted UAS-line generated by the Feany lab (Wittmann et al., 2001). (C) Tau transcripts levels driven by GMR-gal4 normalised to our random insertion line 3.
Assay of human GSK3β activity for Tau toxicity
The low level of Tau overexpression from our 68A insertions results in moderate toxicity and provides a baseline to investigate factors that increase toxicity. This level of expression may be representative of human disease where Tau expression is not elevated.
Several groups have previously investigated phosphorylation of exogenous human Tau by endogenous Drosophila kinases (Nishimura et al., 2004; Steinhilb et al., 2007a; Steinhilb et al., 2007b). A key role has been reported for Shaggy, the Drosophila homologue of GSK3β, consistent with the suggestion that GSK3β is a major pathological kinase in human tauopathies (Hanger et al., 1992; Lovestone et al., 1994; Lucas et al., 2001; Huang and Klein, 2006). We investigated whether Shaggy overexpression could similarly increase Tau toxicity in our targeted expression system. We used the GMR-GAL4 driver to express Shaggy from a UAS-shaggy transgene in the presence or absence of 2N4R Tau inserted at the 68A locus. We found that, whereas expression of Shaggy alone caused mild disruption of the ommatidia, co-expression with 2N4R Tau resulted in increased eye disruption (Fig. 4A, dashed area highlights severe disruption and glazing of the eye). The ommatida were more severely disrupted and patches of pigment loss and glazing were visible in the eyes of the Shaggy and Tau expressing flies. Thus, Tau-mediated toxicity can be increased by co-expression of human Tau with Drosophila GSK3β, even at low levels of Tau expression.
Fig. 4. Co-expression of human GSK3β increases Tau-mediated toxicity.

(A) Light micrographs of adult fly eyes. Drosophila (Shaggy) or hGSK3β was driven in the visual system under the control of GMR-gal4 either alone (upper row) or alongside 2N4R Tau from the 68A insertion site. Shaggy expression alone causes disruption of eye development but hGSK3β does not. Co-expression of GSK3β increases the toxicity of Tau. Areas of the eye displaying significant disruption including substantial areas of glazing are highlighted with dashed lines. (B) Relative expression of hGSK3β driven from four random UAS-insertions. Higher transcript levels correspond to higher Tau-mediated toxicity. (C) Increased toxicity does not correlate with a change in solubility of Tau. Tau in sarcosyl-soluble and -insoluble fractions was quantified by western blotting with or without co-expression of hGSK3β.
In order to examine whether human GSK3β (hGSK3β) could also enhance Tau-mediated toxicity in Drosophila, we first confirmed that hGSK3β was capable of phosphorylating human Tau in Drosophila cells. When hGSK3β was co-expressed with human 2N4R Tau in Drosophila S2 cells, we identified hGSK3β-mediated phosphorylation events using PHF1, AT8, AT270 and AT100 antibodies which recognise specific Tau phospho-epitopes (supplementary material Fig. S2). We next generated four UAS-hGSK3β transgenic lines by random insertion and found each line was expressed to varying amounts (Fig. 4B) yet expression of hGSK3β alone did not disrupt the ommatidial structure (Fig. 4A, upper panels). When hGSK3β expression was driven together with 2N4R Tau inserted in the 68A locus an enhanced disruption of the eye occurred (Fig. 4A, lower panels). The degree of enhancement was dependent on the level of GSK3β expression from each transgene (Fig. 4A,B). hGSK3β line 1 has the highest expression and generated the greatest toxicity when expressed with Tau, all subsequent experiments were performed using this hGSK3β line 1. We examined whether the increased Tau toxicity caused by hGSK3β was a consequence of changes to Tau solubility since altered solubility of Tau is associated with neurodegenerative tauopathies (Lewis et al., 2000; Ishihara et al., 2001; Zhukareva et al., 2002; Hirata-Fukae et al., 2009). Sarcosyl fractionation is a commonly used method to assay Tau solubility (Lewis et al., 2000; Zhukareva et al., 2002). We extracted sarcosyl-soluble and sarcosyl-insoluble fractions from Drosophila heads and found no change in the ratio of soluble to insoluble Tau in these fractions when hGSK3β was co-expressed together with Tau in Drosophila (Fig. 4C).
GSK3β-mediated phosphorylation events on Tau
We used antibodies to four phospho-epitopes, in addition to total Tau to examine the effects of hGSK3β expression on phosphorylation of Sarcosyl-soluble and Sarcosyl-insoluble extracts in the Tau transgenic flies. Three of the antibodies (AT270, PHF1, and AT8) recognise known GSK3β target sites in Tau (Thr181, Ser396/404, and Ser202/Thr205, respectively), whereas labelling by antibody AT100 requires phosphorylation of both Thr212 and Ser214 by GSK3β in combination with another kinase (Sato et al., 2002). Soluble and insoluble Tau was readily detected in the heads from Drosophila expressing the 2N4RTau transgene inserted into the 68A landing site (Fig. 5A–D). We found at the levels of Tau expression seen in our system neither soluble or insoluble Tau were phosphorylated appreciably on Ser396/Ser404 by endogenous Drosophila kinases but both fractions were phosphorylated when hGSK3β was co-expressed (Fig. 5A,E,F; PHF1). Residue Thr181 on Tau was phosphorylated in both soluble and insoluble fractions by endogenous kinases and this phosphorylation was significantly increased by hGSK3β (Fig. 5B,E,F; AT270). In contrast, Ser202/Thr205 was phosphorylated by endogenous kinases but phosphorylation at this epitope was not increased by hGSK3β (Fig. 5C,E,F; AT8). Phosphorylation of Thr212 and Ser214 was barely detectable in soluble Tau and was absent from insoluble Tau. Furthermore, this epitope was not generated by co-expression of hGSK3β (Fig. 5D; AT100). Thus, Tau residues Ser202/Thr205 and Thr212/Ser214 are not targets for hGSK3β in vivo, at least in Drosophila, but phosphorylation of Thr231 and Ser396/Ser404 is substantially increased in this system.
Fig. 5. Tau phospho-epitopes phosphorylated by human GSK3β in the Drosophila visual system.

Sarcosyl-soluble and -insoluble Tau fractions were purified from fly heads overexpressing 2N4R Tau from the 68A locus with or without hGSK3β. Four phospho-specific antibodies were used to identify specific phosphorylation events mediated by hGSK3β in vitro.
No role for GSK3-priming kinases in mediating Tau toxicity in the Drosophila eye
Our failure to detect phosphorylation of Thr212/Ser214 by hGSK3β to generate the AT100 epitope may indicate a requirement for priming at adjacent upstream Ser residues by an additional kinase (Doble and Woodgett, 2003). One candidate is casein kinase 1δ (CK1δ) which acts co-operatively withGSK3β on some substrates (Zeng et al., 2005). CK1δ is highly overexpressed in AD brain (Ghoshal et al., 1999) and our previous work has indicated a potential role for this kinase in aberrant phosphorylation of Tau (Hanger et al., 2007). We also wished to see if dual-specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A) would act similarly. DYRK1A phosphorylates Tau in vitro and acts as a priming kinase for GSK3β (Woods et al., 2001). The DYRK1A gene lies within the critical region on chromosome 21 and is duplicated and overexpressed in Down Syndrome (DS). DS patients have a high incidence of early onset AD and one possible cause is hyperphosphorylation of Tau by the overexpressed DYRK1A (Ryoo et al., 2007; Liu et al., 2008). We generated transgenic UAS-human CK1δ and UAS-rat DYRK1A lines by random insertion. CK1δ was truncated after amino acid 371 because the shorter form is constitutively active. The kinases were expressed alongside Tau either with or without hGSK3β but the toxicity was not enhanced in any situation (supplementary material Fig. S3).
GSK3β-mediated S404 phosphorylation is protective
To determine which, if any, of the hGSK3β sites on Tau that we confirmed as phosphorylated in vivo are essential for increased toxicity, we replaced the individual residues, Ser202, Ser205, Thr212 or Ser404 in 2N4R Tau with non-phosphorylatable alanine. In a fifth mutant, all four of these residues were substituted by alanine (termed Tau4xA). We examined the effect of these mutations on the ability of hGSK3β to phosphorylate 2N4R Tau in Drosophila cells. When 2N4R Tau is expressed in cells, multiple Tau bands were detected using a phospho-independent antibody. These likely correspond to different phosphorylated forms mediated by endogenous kinases. Co-expression of hGSK3β resulted in a retardation of the Tau bands in the gel, indicating increased phosphorylation (Fig. 6A; see 2N4R wt). Mutating Tau at S202, S205 or T212 had no effect on the banding pattern of Tau, either with or without co-expression of hGSK3β (Fig. 6A). In contrast, introducing the S404A mutation abolished the slowest migrating Tau bands, suggesting that this site is an important target of hGSK3β. The 4xA form of Tau carrying each of the four point mutations showed an identical reduction in phosphorylation and, as expected, the PHF1 antibody, which recognises the pS396/pS404 epitope on Tau, detected phosphorylation by hGSK3β on WT, S202A, S205A and T212A forms of Tau not in the S404A and 4xA mutant forms (Fig. 6B).
Fig. 6. Phosphorylation of S404 by human GSK3β is protective in the Drosophila visual system.

(A,B) Wild-type or point mutated forms of human 2N4R Tau were expressed in Drosophila S2 cells with or without myc-tagged hGSK3β. (A) Multiple bands representing differentially phosphorylated forms of Tau were detected between 60 and 70 kDa with a phospho-independent antibody. Co-expression of hGSK3β retards Tau mobility, indicative of increased phosphorylation load but this is absent in the Tau S404A or 4xA mutants. Recombinant Tau was included as a positive control for anti-Tau. (B) The pS396/pS404 epitope recognised by PHF1 is phosphorylated by hGSK3β but not by endogenous kinases. Phosphorylation is absent in the Tau S404A and the 4xA mutants. (C) Light photomicrographs of adult fly eyes. GMR-gal4 was used to express wild-type 2N4R Tau or S202A, T205A, T212A or S404A or Tau4xA Tau point mutants (top row). hGSK3β was co-expressed with each Tau isoform (bottom row). Areas displaying severe disruption to eye development including glazing and loss of pigmentation are highlighted by dashed lines. The S404A and 4xA mutant display increased toxicity when co-expressed with hGSK3β.
We made transgenic flies by inserting the mutant Tau constructs into the same 68A locus as before to examine the resistance of each of these mutations to hGSK3β-induced toxicity. When each of the single point mutations, Tau4xA, or wild-type Tau, was expressed in the Drosophila visual system, toxicity was equivalent. Each resulted in mild disruption of ommatidia (Fig. 6C, also cf. Fig. 2). Co-expression of three of the Tau mutants, Tau S202A, T205A or T212A, with hGSK3β, resulted in no changes in toxicity (Fig. 6). However, hGSK3β expression unexpectedly increased the amount of degeneration observed with either TauS404A or Tau4xA. The eyes had a greater degree of external disorganisation and extensive patches of depigmentation indicative of increased toxicity (Fig. 6; depigmented areas marked by dashed lines). This result indicates that phosphorylation of Ser404 in Tau by hGSK3β modulates the toxicity of Tau in vivo but in this system, phosphorylation may not increase Tau toxicity but instead may be protective against degeneration.
Discussion
Hyperphosphorylated Tau is associated with a number of neurodegenerative diseases, strongly suggesting that disregulated phosphorylation of Tau leads to neural pathology. In order to progress towards effective therapies it is key to identify which of the many Tau phosphorylation events are critical for toxicity in vivo and to establish which kinases phosphorylate Tau to cause disease. Previous studies have used Drosophila to identify endogenous kinases that increase Tau toxicity (Jackson et al., 2002; Mudher et al., 2004; Nishimura et al., 2004; Chatterjee et al., 2009). We were interested to determine whether the Drosophila photoreceptor model could be used to assess the roles of human kinases in mediating neurodegeneration in vivo and to identify particular phosphorylation events on Tau that are important for toxicity. Use of this model requires that the individual transgenic lines generated to test manipulated forms of Tau be expressed identically. However, we and others have demonstrated that Tau transgenes randomly integrated within the Drosophila genome produced variable levels of expression. This presented a significant challenge in seeking to identify the contribution to in vivo toxicity of Tau phosphorylation events mediated by human kinases. In previous studies, transgenic flies were selected for comparable Tau expression based on western blot analysis (Wittmann et al., 2001; Steinhilb et al., 2007a; Steinhilb et al., 2007b). However, we have found a highly degenerate eye phenotype was associated with reduced Tau protein, presumably due to cell death. Rather, the toxicity correlated with the level of RNA expression at earlier developmental stages, before cell death was apparent. We demonstrated that the toxicity mediated by different forms of Tau was difficult or impossible to compare even after selecting lines with comparable protein or mRNA expression levels because even small changes in Tau transcript level resulted in quite variable degrees of toxicity. We concluded that, using the standard method of generating transgenic flies that is susceptible to position effects, it would be difficult to be certain that changes in toxicity mediated by different forms of Tau were due to the properties of the Tau protein itself rather than subtle changes in expression level.
Controlling for integration site is necessary to assess Tau toxicity
To remove any possible confounding positional effects generated by random integration of Tau transgenes, we adopted φC31-mediated site-specific integration to standardise the genomic integration locus (Groth et al., 2004). We found that, as expected, Tau transgenes inserted into the same locus resulted in the equivalent expression of tau transcript. We selected Drosophila landing sites that expressed Tau at levels low enough to produce only a mild disruption of the structure of the ommatidia of the eye. This has a major advantage because, although the ratio of 4R:3R Tau varies between tauopathies (Ingelsson et al., 2007), Tau is not thought to be significantly overexpressed in AD. Thus, our model system, in which Tau is not highly overexpressed, may be more useful for uncovering mechanisms underlying Tau toxicity in disease.
Using site-specific integration, we examined the toxicity of two Tau isoforms (0N4R and 2N4R), with or without a point mutation, R406W, which is found in some autosomal dominant forms of FTLD-Tau (Hutton et al., 1998). Previous studies by others have reported a strong, highly disrupted eye phenotype when 0N4R R406W Tau is overexpressed in the Drosophila visual system, indicating enhanced toxicity (Wittmann et al., 2001; Jackson et al., 2002; Nishimura et al., 2004). We confirmed the increased toxicity of 0N4R Tau R406W in vivo using a previously generated strain with a randomly integrated Tau transgene (Wittmann et al., 2001). However, we found that, when we controlled for the integration site and reduced Tau overexpression using φC31-mediated site-specific integration, we failed to see any increase in toxicity caused by this mutation. We were also unable to detect any difference in toxicity generated by expression of the 0N4R and 2N4R Tau isoforms. Doubling the copy number of each of the UAS-transgenes increased the amount of toxicity observed, as expected from the increased expression of Tau. However, despite two copies of UAS-Tau increasing Tau expression to a level similar to that of the 0N4R Tau R406W line developed previously (Wittmann et al., 2001), the R406W mutation still had no effect on the organisation of the Drosophila eye. When we controlled for positional effects, our results suggest that the R406W mutation does not have a significant effect on Tau-mediated toxicity. Interestingly, this conclusion is in agreement with previous studies assaying the effect of FTLD-Tau-associated point mutations on the microtubule-binding properties of Tau (Delobel et al., 2002; Bunker et al., 2006). In an in vitro study using purified microtubules (Bunker et al., 2006) and an in vivo assay in Xenopus oocytes (Delobel et al., 2002), Tau R406W displayed only subtle differences in microtubule-binding compared to wild-type Tau. Taken together, these findings are consistent with the late onset of symptoms and slow disease progression observed in FTLD-Tau patients carrying the R406W Tau mutation (Heutink, 2000).
GSK3β-mediated Tau toxicity is enhanced by S404A
GSK3β is a key candidate pathological Tau kinase in AD (Hanger et al., 1992; Lovestone et al., 1994; Lucas et al., 2001) to the extent that lithium and other GSK3β inhibitors have been trialled clinically for AD (reviewed by Mangialasche et al., 2010). GSK3β can phosphorylate many residues on Tau in vitro but it is not yet clear how each phosphorylation event contributes to Tau toxicity (Hanger et al., 2007) or whether all sites increase toxicity. We examined the role of priming kinases as a possible level of regulation. However, we were unable to detect any significant role for CK1δ or DYRK1A on Tau toxicity in this model system. Although hGSK3β did increase Tau toxicity, in our study it was not possible to identify a specific phosphorylation event that is responsible for this increased toxicity, suggesting that phosphorylation at multiple residues generate toxicity confirming previous observations investigating endogenous kinases (Steinhilb et al., 2007a; Steinhilb et al., 2007b; Chatterjee et al., 2009). Unexpectedly we found that phosphorylation of S404 in Tau appeared to be protective when co-expressed with hGSK3β, and substitution of S404 with alanine resulted in an enhanced toxicity compared to expressing either S404A or hGSK3β alone. A previous study examining the role of phosphorylation for Tau-mediated toxicity in the Drosophila eye identified that the double mutant S396A/S404A did not affect Tau toxicity (Steinhilb et al., 2007a) produced from endogenous kinases. We also found that S404A did not affect toxicity when acted on by endogenous kinases but see an enhancement of toxicity when S404A Tau was expressed together with hGSK3β together. We did not observe any phosphorylation of Ser396/Ser404 by endogenous Drosophila kinases but this epitope was phosphorylated when hGSK3β was co-expressed with Tau. Our results contrast therefore with the previous study, in which phosphorylation by endogenous kinases was detected at this site (Steinhilb et al., 2007a). This difference may be due to the increased toxicity of Tau in the previous study due to elevated Tau expression, which could potentially affect the recognition of Tau by endogenous kinases.
Importantly, although increased Tau phosphorylation is considered by many to be pathological, our observations indicate that some phosphorylation events may have a protective effect. Previous studies have also indicated that phosphorylation of Tau may reduce toxicity (Thies and Mandelkow, 2007). The PHF1 epitope (comprising pSer396 and pSer404) appears to be dynamically regulated by stress. Phosphorylation of these Tau residues is affected in various stress situations in vivo, including post-ischemic injury (Wen et al., 2004; Gordon-Krajcer et al., 2007) and following anaesthesia-induced hypothermia (Planel et al., 2007). In vitro, both phosphorylation (Gómez-Ramos et al., 2003) and dephosphorylation (Zambrano et al., 2004) of Ser396/Ser404 have been reported under conditions of oxidative stress. These data presumably reflect dynamic regulation: in ischaemic injury, Ser396/Ser404 becomes almost completely dephosphorylated, followed by a rapid increase in phosphorylation (Gordon-Krajcer et al., 2007). The phosphorylation of Ser396/Ser404 in neuroblastoma cells treated with lipid peroxidising agents can be blocked by inhibitors of either GSK3β or the stress-activated kinase p38, but not by cyclin-dependent kinase-5, supporting the view that phosphorylation of this epitope could be part of a co-ordinated neuronal stress response. It has been argued that increased Tau phosphorylation is a protective response to stress, rather than a primary cause of neuronal toxicity (Castellani et al., 2008; Buée et al., 2010), a view supported by evidence that expression of hypophosphorylated forms of Tau can also cause neurotoxicity (Chatterjee et al., 2009; Talmat-Amar et al., 2011). The inability to phosphorylate Ser404 combined with increased GSK3β activity may lead to a pattern of Tau phosphorylation that modifies its microtubule binding properties in such a way that toxicity is increased, potentially by generating a form that irreversibly binds microtubules. Alternatively GSK3β may modify additional components that contribute to Tau toxicity via a mechanism that is sensitive to the phosphorylation load on Tau. GSK3β expression can increase the toxicity of some but not all phosphorylation resistant forms of Tau, suggesting an interplay between Tau phosphorylation status and further kinase dependent components. Our modification of the Drosophila in vivo toxicity model may help to decipher the different roles of specific Tau phosphorylation events leading to changes in Tau toxicity.
Materials and Methods
Drosophila husbandry and stocks
Flies were maintained on standard cornmeal agar medium. Crosses were maintained at 18°C or 25°C on semi-defined rich “German food”. Medium recipes are available from the Bloomington Drosophila Stock Centre website. Transgenic flies were generated by standard P-element mediated transgenesis or φC31-mediated transgenesis by BestGene Inc. (Chino Hills, CA, USA) or GenetiVision (Houston, TX, USA). All transgenic flies produced were confirmed by sequencing and Splinkerette PCR was used to identify genomic DNA flanking regions (Potter and Luo, 2010). The control strain used for all experiments was an isogenic w1118 line (Vienna Drosophila RNAi Center). Details of genetic markers and balancer chromosome are described at Flybase (http://flybase.org). GMR-GAL4 was used to drive expression of UAS-Tau and UAS-kinases in the visual system as previously described (Tuxworth et al., 2009).
Molecular biology
Generation of UAS constructs
Full length open reading frames (ORFs) of cDNAs for Tau and each kinase were amplified with proof-reading Taq polymerase, cloned into pENTR (Invitrogen) and verified by sequencing. Constructs were recombined into the Murphy collection of Destination vectors supplied by the Drosophila Genomic Resource Centre (Bloomington, IN). The kinases were epitope-tagged with Myc or Flag sequences, while Tau was not tagged.
Site-directed mutagenesis of Tau
The ORF of 2N4R human Tau cloned into pTW was mutated using the QuikChange Multi kit (Stratagene) and confirmed by sequencing.
cDNA synthesis
RNA was extracted from 5–10 adult fly heads using Tri Reagent (Sigma) and used immediately for cDNA synthesis with the ImProm-IITM Reverse Transcription System (Promega) following the manufacturer's instructions. 500 ng RNA was used per reaction.
qPCR
Quantitative PCR was performed by QStandard (http://www.qstandard.co.uk). Transcript levels for the following genes were quantified: Drosophila actin5c (CG4027), Drosophila GAPDH2 (CG8893), Drosophila EIF-4a (CG9075), mouse CD8a (geneID: 12525), human Tau (geneID: 4137).
Imaging of fly eyes
Whole flies were processed for scanning electron microscopy or light microscopy and imaged as described (Tuxworth et al., 2009).
Biochemistry
Homogenisation of Drosophila heads
Fly heads were used as material for qPCR and biochemistry. Whole flies were snap-frozen in liquid nitrogen and shaken at 6.5 m/s for 20 secs in a FastPrep24 homogeniser (MP Biomedical) to decapitate. Fly heads were separated from thoraces and abdomens by shaking through a fine sieve.
A two-stage, neutral-alkaline extraction procedure was used to maximise recovery of Tau protein. Fly heads were homogenised (20 heads/100 µl) in 100 µl ice-cold homogenisation buffer (50 mM Tris-HCl; 300 mM NaCl; 1% v/v β-mercaptoethanol, protease and phosphatase inhibitor cocktails [Calbiochem]), pH 6.8, and garnet beads in a FastPrep24 homogeniser (MP Biomedicals). Heads were homogenised twice at 6.5 m/s for 20 s and centrifuged at 20,000 g for 10 min at 4°C. The supernatant was removed and stored on ice. A second homogenisation was then performed by adding 100 µl of ice-cold homogenisation buffer at pH 9.2 to the pellet and centrifuging as above. The supernatants were combined, the pH adjusted to 8.0 and then centrifuged at 20,000 g for 30 min at 4°C. The final supernatant was stored at −80°C.
Sarcosyl-solubility of Tau
Ice-cold Tris buffer (10 mM Tris-HCl, pH 7.5, 800 mM NaCl, 1 mM EGTA, pH 8.0, 10% w/v sucrose, supplemented with protease and phosphatase inhibitor cocktails) was added to the fly heads (30 heads/60 µl buffer) and garnet beads. Fly heads were homogenised three times at 6.5 m/s for 20 s as above, then centrifuged at 1,000 g for 5 min at 4°C. The supernatant was removed and stored at −80°C and referred to as the sarcosyl-soluble fraction. N-lauroylsarcosinate (Sigma) was added to the pellet to give a final concentration of 1% (w/v) and the suspension was incubated for 1 h at ambient room temperature with gentle shaking. The suspension was centrifuged at 100,000 g for 1 h at 4°C. The insoluble pellet was resuspended in 50 mM Tris-HCl, pH 7.5, stored at −80°C and referred to as the sarcosyl-insoluble fraction.
Western blotting
SDS-PAGE, Western blotting, membrane blocking and probing were all performed by standard protocols. The membrane used was supported nitrocellulose (BioRad) which was blocked for 1 h in 5% milk in Tris-buffered saline (TBS) without Tween-20. Antibodies were diluted in 5% milk in TBS, 0.1% Tween-20 (TBST). Blots were washed with TBST, followed by two washes in TBS without Tween prior to imaging.
Antibodies used
Primary antibodies raised against: total human Tau (phosphoinsensitive), used at 1/10,000 (Dako); PHF1 human Tau pS396/pS404, used at 1/5,000 (gift of P. Davies, Albert Einstein College of Medicine, NY); AT8 human Tau pS202/pT205 at 1/2,000 (Innogenetics); Tau1 human Tau 197–208 at 1/1,000 (Chemicon); AT100 human Tau pT212/pS214 at 1/1,000 (Innogenetics); AT270 human Tau pT181 at 1/1,000 (Innogenetics); 9E10 c-Myc at 1/1,000 (Santa Cruz); M2 Flag-tag at 1/1,000 (Sigma); β-actin at 1/5,000 (Calbiochem). Secondary antibodies: Alexa-680 mouse anti-IgG at 1/15,000 (Invitrogen); Alexa-800 mouse anti-IgM at 1/15,000 (Rockland); Alexa-800 rabbit anti-IgG at 1/15,000 (Rockland).
Signal quantification
Membranes were scanned and quantified using an Odyssey infra-red scanner (Licor Biosciences) and Prism 5 software (GraphPad).
Normalisation of Tau levels between membranes
A constant amount of recombinant Tau was included in an individual lane on each Tau protein gel to act as a normalisation control.
Statistical analysis
Statistical analysis was performed using Prism 5 (GraphPad). Transcript and protein quantification data were analysed for significance by one-way ANOVA with Bonferroni correction. Resulting p values lower than 0.05 were deemed significant.
Supplementary Material
Acknowledgments
The authors would like to thank Megan O'Hare for her support with this project, Dr Darren Williams for suggesting we use the φC31 integrase system and Dr Koen Venken for sharing information and materials on the system prior to publication. We thank Dr Mel Feany for providing the UAS-Tau 0N3R R406W line, Dr Peter Davies (Albert Einstein College of Medicine) for PHF1 antibody, Dr David Meek (University of Dundee) for CK1δ cDNA, and Prof. Dr Walter Becker (RWTH Aachen University) for DYRK1A cDNA.
Footnotes
Author Contributions: G.P. and R.I.T. performed all of the experiments. R.I.T., G.T. and D.P.H. conceived the project initially. Each of the authors contributed to the drafting of the manuscript.
Competing interests: The authors have no competing interests to declare.
Funding
This work was supported by Alzheimer's Research UK [ART-PhD2008-4 to G.T.] and the Medical Research Council [G0300408 to D.P.H. and G0500261 to G.T.].
References
- Arendt T., Stieler J., Strijkstra A. M., Hut R. A., Rüdiger J., Van der Zee E. A., Harkany T., Holzer M., Härtig W. (2003). Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 23, 6972–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C., Lee V. M., Trojanowski J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- Bonda D. J., Castellani R. J., Zhu X., Nunomura A., Lee H. G., Perry G., Smith M. A. (2011). A novel perspective on tau in Alzheimer's disease. Curr. Alzheimer Res. 8, 639–642 10.2174/156720511796717131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden K. R., Trojanowski J. Q., Lee V. M. (2009). Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat. Rev. Drug Discov. 8, 783–793 10.1038/nrd2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130 10.1016/S0165-0173(00)00019-9 [DOI] [PubMed] [Google Scholar]
- Buée L., Troquier L., Burnouf S., Belarbi K., Van der Jeugd A., Ahmed T., Fernandez-Gomez F., Caillierez R., Grosjean M. E., Begard S. et al. (2010). From tau phosphorylation to tau aggregation: what about neuronal death? Biochem. Soc. Trans. 38, 967–972 10.1042/BST0380967 [DOI] [PubMed] [Google Scholar]
- Bunker J. M., Kamath K., Wilson L., Jordan M. A., Feinstein S. C. (2006). FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J. Biol. Chem. 281, 11856–11863 10.1074/jbc.M509420200 [DOI] [PubMed] [Google Scholar]
- Castellani R. J., Nunomura A., Lee H. G., Perry G., Smith M. A. (2008). Phosphorylated tau: toxic, protective, or none of the above. J. Alzheimers Dis. 14, 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Sang T. K., Lawless G. M., Jackson G. R. (2009). Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 18, 164–177 10.1093/hmg/ddn326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau K. W., Chan W. Y., Shaw P. C., Chan H. Y. (2006). Biochemical investigation of Tau protein phosphorylation status and its solubility properties in Drosophila. Biochem. Biophys. Res. Commun. 346, 150–159 10.1016/j.bbrc.2006.05.112 [DOI] [PubMed] [Google Scholar]
- Churcher I. (2006). Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr. Top. Med. Chem. 6, 579–595 10.2174/156802606776743057 [DOI] [PubMed] [Google Scholar]
- Cowan C. M., Bossing T., Page A., Shepherd D., Mudher A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 120, 593–604 10.1007/s00401-010-0716-8 [DOI] [PubMed] [Google Scholar]
- Delobel P., Flament S., Hamdane M., Jakes R., Rousseau A., Delacourte A., Vilain J. P., Goedert M., Buée L. (2002). Functional characterization of FTDP-17 tau gene mutations through their effects on Xenopus oocyte maturation. J. Biol. Chem. 277, 9199–9205 10.1074/jbc.M107716200 [DOI] [PubMed] [Google Scholar]
- Doble B. W., Woodgett J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 10.1242/jcs.00384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C., Campbell D. G., Jakes R., Goedert M., Cuenda A. (2005). Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci. 118, 397–408 10.1242/jcs.01655 [DOI] [PubMed] [Google Scholar]
- Feuillette S., Miguel L., Frébourg T., Campion D., Lecourtois M. (2010). Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 113, 895–903 10.1111/j.1471-4159.2010.06663.x [DOI] [PubMed] [Google Scholar]
- Ghoshal N., Smiley J. F., DeMaggio A. J., Hoekstra M. F., Cochran E. J., Binder L. I., Kuret J. (1999). A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer's disease. Am. J. Pathol. 155, 1163–1172 10.1016/S0002-9440(10)65219-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gistelinck M., Lambert J. C., Callaerts P., Dermaut B., Dourlen P. (2012). Drosophila models of tauopathies: what have we learned? Int. J. Alzheimers Dis. 2012, 970980 10.1155/2012/970980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Ramos A., Díaz-Nido J., Smith M. A., Perry G., Avila J. (2003). Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 71, 863–870 10.1002/jnr.10525 [DOI] [PubMed] [Google Scholar]
- Gordon-Krajcer W., Kozniewska E., Lazarewicz J. W., Ksiezak-Reding H. (2007). Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem. Res. 32, 729–737 10.1007/s11064-006-9199-3 [DOI] [PubMed] [Google Scholar]
- Groth A. C., Fish M., Nusse R., Calos M. P. (2004). Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166, 1775–1782 10.1534/genetics.166.4.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger D. P., Hughes K., Woodgett J. R., Brion J. P., Anderton B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62 10.1016/0304-3940(92)90774-2 [DOI] [PubMed] [Google Scholar]
- Hanger D. P., Betts J. C., Loviny T. L., Blackstock W. P., Anderton B. H. (1998). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 71, 2465–2476 10.1046/j.1471-4159.1998.71062465.x [DOI] [PubMed] [Google Scholar]
- Hanger D. P., Byers H. L., Wray S., Leung K. Y., Saxton M. J., Seereeram A., Reynolds C. H., Ward M. A., Anderton B. H. (2007). Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 282, 23645–23654 10.1074/jbc.M703269200 [DOI] [PubMed] [Google Scholar]
- Hanger D. P., Anderton B. H., Noble W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119 10.1016/j.molmed.2009.01.003 [DOI] [PubMed] [Google Scholar]
- Heutink P. (2000). Untangling tau-related dementia. Hum. Mol. Genet. 9, 979–986 10.1093/hmg/9.6.979 [DOI] [PubMed] [Google Scholar]
- Hirata-Fukae C., Li H. F., Ma L., Hoe H. S., Rebeck G. W., Aisen P. S., Matsuoka Y. (2009). Levels of soluble and insoluble tau reflect overall status of tau phosphorylation in vivo. Neurosci. Lett. 450, 51–55 10.1016/j.neulet.2008.11.023 [DOI] [PubMed] [Google Scholar]
- Huang H. C., Klein P. S. (2006). Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer's disease. Curr. Drug Targets 7, 1389–1397 10.2174/1389450110607011389 [DOI] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- Ingelsson M., Ramasamy K., Russ C., Freeman S. H., Orne J., Raju S., Matsui T., Growdon J. H., Frosch M. P., Ghetti B. et al. (2007). Increase in the relative expression of tau with four microtubule binding repeat regions in frontotemporal lobar degeneration and progressive supranuclear palsy brains. Acta Neuropathol. 114, 471–479 10.1007/s00401-007-0280-z [DOI] [PubMed] [Google Scholar]
- Iqbal K., Liu F., Gong C. X., Alonso A. C., Grundke-Iqbal I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69 10.1007/s00401-009-0486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T., Zhang B., Higuchi M., Yoshiyama Y., Trojanowski J. Q., Lee V. M. (2001). Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am. J. Pathol. 158, 555–562 10.1016/S0002-9440(10)63997-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson G. R., Wiedau-Pazos M., Sang T. K., Wagle N., Brown C. A., Massachi S., Geschwind D. H. (2002). Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34, 509–519 10.1016/S0896-6273(02)00706-7 [DOI] [PubMed] [Google Scholar]
- Karsten S. L., Sang T. K., Gehman L. T., Chatterjee S., Liu J., Lawless G. M., Sengupta S., Berry R. W., Pomakian J., Oh H. S. et al. (2006). A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 51, 549–560 10.1016/j.neuron.2006.07.019 [DOI] [PubMed] [Google Scholar]
- Khurana V. (2008). Modeling tauopathy in the fruit fly Drosophila melanogaster. J. Alzheimers Dis. 15, 541–553. [DOI] [PubMed] [Google Scholar]
- Khurana V., Lu Y., Steinhilb M. L., Oldham S., Shulman J. M., Feany M. B. (2006). TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 16, 230–241 10.1016/j.cub.2005.12.042 [DOI] [PubMed] [Google Scholar]
- Kimura R., Kamino K., Yamamoto M., Nuripa A., Kida T., Kazui H., Hashimoto R., Tanaka T., Kudo T., Yamagata H. et al. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23 10.1093/hmg/ddl437 [DOI] [PubMed] [Google Scholar]
- Kuret J., Johnson G. S., Cha D., Christenson E. R., DeMaggio A. J., Hoekstra M. F. (1997). Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer's disease brain. J. Neurochem. 69, 2506–2515 10.1046/j.1471-4159.1997.69062506.x [DOI] [PubMed] [Google Scholar]
- Lebouvier T., Scales T. M., Williamson R., Noble W., Duyckaerts C., Hanger D. P., Reynolds C. H., Anderton B. H., Derkinderen P. (2009). The microtubule-associated protein tau is also phosphorylated on tyrosine. J. Alzheimers Dis. 18, 1–9 10.3233/JAD-2009-1116 [DOI] [PubMed] [Google Scholar]
- Lee V. M., Goedert M., Trojanowski J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X. et al. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405 10.1038/78078 [DOI] [PubMed] [Google Scholar]
- Lindwall G., Cole R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305. [PubMed] [Google Scholar]
- Liu F., Liang Z., Wegiel J., Hwang Y. W., Iqbal K., Grundke-Iqbal I., Ramakrishna N., Gong C. X. (2008). Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233 10.1096/fj.07-104539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovestone S., Reynolds C. H., Latimer D., Davis D. R., Anderton B. H., Gallo J. M., Hanger D., Mulot S., Marquardt B., Stabel S. et al. (1994). Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 4, 1077–1086 10.1016/S0960-9822(00)00246-3 [DOI] [PubMed] [Google Scholar]
- Lucas J. J., Hernández F., Gómez-Ramos P., Morán M. A., Hen R., Avila J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39 10.1093/emboj/20.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack T. G., Dayanandan R., Van Slegtenhorst M., Whone A., Hutton M., Lovestone S., Anderton B. H. (2001). Tau proteins with frontotemporal dementia-17 mutations have both altered expression levels and phosphorylation profiles in differentiated neuroblastoma cells. Neuroscience 108, 701–712 10.1016/S0306-4522(01)00434-1 [DOI] [PubMed] [Google Scholar]
- Mangialasche F., Solomon A., Winblad B., Mecocci P., Kivipelto M. (2010). Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716 10.1016/S1474-4422(10)70119-8 [DOI] [PubMed] [Google Scholar]
- Martin L., Latypova X., Terro F. (2011). Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem. Int. 58, 458–471 10.1016/j.neuint.2010.12.023 [DOI] [PubMed] [Google Scholar]
- Mazanetz M. P., Fischer P. M. (2007). Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 6, 464–479 10.1038/nrd2111 [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Watanabe A., Titani K., Ihara Y. (1995). Hyperphosphorylation of tau in PHF. Neurobiol. Aging 16, 365-371, discussion 371-380 10.1016/0197-4580(95)00027-C [DOI] [PubMed] [Google Scholar]
- Morris M., Maeda S., Vossel K., Mucke L. (2011). The many faces of tau. Neuron 70, 410–426 10.1016/j.neuron.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A., Shepherd D., Newman T. A., Mildren P., Jukes J. P., Squire A., Mears A., Drummond J. A., Berg S., MacKay D. et al. (2004). GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 9, 522–530 10.1038/sj.mp.4001483 [DOI] [PubMed] [Google Scholar]
- Muqit M. M., Feany M. B. (2002). Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat. Rev. Neurosci. 3, 237–243 10.1038/nrn751 [DOI] [PubMed] [Google Scholar]
- Nishimura I., Yang Y., Lu B. (2004). PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116, 671–682 10.1016/S0092-8674(04)00170-9 [DOI] [PubMed] [Google Scholar]
- Planel E., Richter K. E., Nolan C. E., Finley J. E., Liu L., Wen Y., Krishnamurthy P., Herman M., Wang L., Schachter J. B. et al. (2007). Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J. Neurosci. 27, 3090–3097 10.1523/JNEUROSCI.4854-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter C. J., Luo L. (2010). Splinkerette PCR for mapping transposable elements in Drosophila. PLoS ONE 5, e10168 10.1371/journal.pone.0010168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo S. R., Jeong H. K., Radnaabazar C., Yoo J. J., Cho H. J., Lee H. W., Kim I. S., Cheon Y. H., Ahn Y. S., Chung S. H. et al. (2007). DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 282, 34850–34857 10.1074/jbc.M707358200 [DOI] [PubMed] [Google Scholar]
- Sato S., Tatebayashi Y., Akagi T., Chui D. H., Murayama M., Miyasaka T., Planel E., Tanemura K., Sun X., Hashikawa T. et al. (2002). Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 277, 42060–42065 10.1074/jbc.M202241200 [DOI] [PubMed] [Google Scholar]
- Shulman J. M., Feany M. B. (2003). Genetic modifiers of tauopathy in Drosophila. Genetics 165, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A. C., Rubin G. M. (1982). Transposition of cloned P elements into Drosophila germ line chromosomes. Science 218, 341–347 10.1126/science.6289435 [DOI] [PubMed] [Google Scholar]
- Steinhilb M. L., Dias-Santagata D., Fulga T. A., Felch D. L., Feany M. B. (2007a). Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 18, 5060–5068 10.1091/mbc.E07-04-0327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb M. L., Dias-Santagata D., Mulkearns E. E., Shulman J. M., Biernat J., Mandelkow E. M., Feany M. B. (2007b). S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J. Neurosci. Res. 85, 1271–1278 10.1002/jnr.21232 [DOI] [PubMed] [Google Scholar]
- Talmat-Amar Y., Arribat Y., Redt-Clouet C., Feuillette S., Bougé A. L., Lecourtois M., Parmentier M. L. (2011). Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum. Mol. Genet. 20, 3738–3745 10.1093/hmg/ddr290 [DOI] [PubMed] [Google Scholar]
- Thies E., Mandelkow E. M. (2007). Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896–2907 10.1523/JNEUROSCI.4674-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuxworth R. I., Vivancos V., O'Hare M. B., Tear G. (2009). Interactions between the juvenile Batten disease gene, CLN3, and the Notch and JNK signalling pathways. Hum. Mol. Genet. 18, 667–678 10.1093/hmg/ddn396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y., Ishiguro K., Itoh T. J., Uchida T., Hotani H., Saito T., Kishimoto T., Hisanaga S. (1998). Microtubule-stimulated phosphorylation of tau at Ser202 and Thr205 by cdk5 decreases its microtubule nucleation activity. J. Biochem. 124, 738–746 10.1093/oxfordjournals.jbchem.a022174 [DOI] [PubMed] [Google Scholar]
- Wen Y., Yang S., Liu R., Simpkins J. W. (2004). Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 1022, 30–38 10.1016/j.brainres.2004.05.106 [DOI] [PubMed] [Google Scholar]
- Wilson C., Bellen H. J., Gehring W. J. (1990). Position effects on eukaryotic gene expression. Annu. Rev. Cell Biol. 6, 679–714 10.1146/annurev.cb.06.110190.003335 [DOI] [PubMed] [Google Scholar]
- Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., Feany M. B. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Woods Y. L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T. G., Cohen P. (2001). The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P. R., Tsai P. I., Chen G. C., Chou H. J., Huang Y. P., Chen Y. H., Lin M. Y., Kimchi A., Chien C. T., Chen R. H. (2011). DAPK activates MARK1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ. 18, 1507–1520 10.1038/cdd.2011.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Run X., Liang Z., Li Y., Liu F., Liu Y., Iqbal K., Grundke-Iqbal I., Gong C. X. (2009). Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 108, 1480–1494 10.1111/j.1471-4159.2009.05882.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano C. A., Egaña J. T., Núñez M. T., Maccioni R. B., González-Billault C. (2004). Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radic. Biol. Med. 36, 1393–1402 10.1016/j.freeradbiomed.2004.03.007 [DOI] [PubMed] [Google Scholar]
- Zeng X., Tamai K., Doble B., Li S., Huang H., Habas R., Okamura H., Woodgett J., He X. (2005). A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 438, 873–877 10.1038/nature04185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Higuchi M., Yoshiyama Y., Ishihara T., Forman M. S., Martinez D., Joyce S., Trojanowski J. Q., Lee V. M. (2004). Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J. Neurosci. 24, 4657–4667 10.1523/JNEUROSCI.0797-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V., Shah K., Uryu K., Braak H., Del Tredici K., Sundarraj S., Clark C., Trojanowski J. Q., Lee V. M. (2002). Biochemical analysis of tau proteins in argyrophilic grain disease, Alzheimer's disease, and Pick's disease: a comparative study. Am. J. Pathol. 161, 1135–1141 10.1016/S0002-9440(10)64390-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.