

Abstract
We report a suite of enzyme redesign protocol based on the surface charge–charge interaction calculation, which is potentially applied to improve the stability of an enzyme without compromising its catalytic activity. Together with the experimental validation, we have released a suite of enzyme redesign algorithm Enzyme Thermal Stability System, written based on our model, for open access to meet the needs in wet labs. Lipk107, a lipase of a versatile industrial use, was chosen to test our software. Our calculation determined that four residues, D113, D149, D213, and D253, located on the surface of LipK107 were critical to the stability of the enzyme. The model was validated with mutagenesis at these four residues followed by stability and activity tests. LipK107 mutants D113A and D149K were more resistant to thermal inactivation with ∼10°C higher half-inactivation temperature than wild-type LipK107. Moreover, mutant D149K exhibited significant retention in residual activity under constant heat, showing a 14-fold increase in the half-inactivation time at 50°C. Activity tests showed that these mutants retained the equal or higher specific activity, among which noteworthy was the mutant D253A with as much as 20% higher activity. We suggest that our protocol could be used as a general guideline to redesign protein enzymes with increased stabilities and enhanced activities.
Keywords: designed protein stabilization, electrostatic stabilization, lipase stabilization, surface charge-charge interaction, Lipk107
Introduction
Enhanced stability of an enzyme without compromising the enzyme activity can be achieved by rational protein engineering.1 Although the mode of approach to that goal remains to be established, economic interests have recently grown over its potential application in diagnostics, pharmaceutical and food processing, and fine-chemical synthesis.2 Many approaches have been proposed. To name a few, hydrogen bond redesign, Van der Waals redesign, and salt bridge redesign have been employed to improve the stability of enzymes in laboratory conditions. However, they all seem technically complicated to write a simple computational program for rational design in industrial scales,3–8 and hence have failed to gain popular acceptance.
Recently, a suite of protocol based on the Tanford–Kirkwood solvent accessibility (TK-SA) model and surface charge–charge interaction analysis has been released, and subsequent applications proved its mettle.1,9–13 TK model, formulated based on the charge–charge interaction by Tanford and Kirkwood,14,15 treats a protein as a sphere and the ionizable groups on protein as point charges that occupy fixed positions (Supporting Information Fig. S1). The interactions between point charges are appraised to delineate the electrostatic properties on a protein.16–18 Additionally, SA was introduced to refine TK-SA model.19,20 Bashford and Karplus21 included the effects of partition function and Gibbs free energy in their improved TK-SA model. Then, different studies have made further improvements with the model. Taking the solvent effect into account,22–24 TK-SA model was shown to approach experimental outcomes.25,26 Subsequent efforts on TK-SA model calculation yielded successful cases of modifying characteristics of enzymes.1,9,10,27–31 Unfortunately, no computational tool using TK-SA model has been publicly released to facilitate the applicability of this protocol among bench scientists.
In our previous study, the lipase LipK107, which belongs to the Proteus/psychrophilic subfamily of I.1 lipases, was cloned from Proteus sp., and was successfully expressed in Escherichia coli BL21, and its structure was solved by X-ray crystallography (PDB accession: 3W9U). LipK107 has garnered interest as a biocatalyst by its high enzyme activity, tolerance to short-chain alcohols, and ability to be expressed soluble in E. coli in a chaperone-independent manner.32–34 Although the hydrolytic activity of LipK107 was as high as 1200 U/mg at ambient temperature, its thermal stability was not very satisfactory.35 In this study, we refined the protocol of TK-SA model calculation and surface charge–charge interactions analysis. We also released a suite of computational program: Enzyme Thermal Stability System (ETSS) intended to assist researchers engaged in enzyme redesign field. To our knowledge, this is the first program freely accessible to the public for protein redesign purpose. Using ETSS, here we report a novel protocol of enzyme redesign and the development of several LipK107 variants with enhanced thermal stability.
Results and Discussion
Engineering of Lipk107 mutants with increased thermal stability in silico
Source code was compiled by gcc 4.0 in the Linux system of Centos 6.0 (the source code is available upon request). The parameters of Lipk107 were calculated by the program which found 79 charged amino acids in LipK107 monomer. Total Eij is shown in Figure 2. Positive energies represented the overall unfavorable interactions of a given residue with all other ionizable residues in the protein, whereas negative values reflected the overall favorable interactions. Hence, we posited that alteration of the electrical property at the residues with positive values would shift the protein's stability character. Although Figure 2 shows dozens of residues fitting this criterion, the best candidate is also needed to meet the following rules. The residues far away from the active center and with high positive value of Eij should be considered first because the introduction of these mutations would have little impacts on enzyme activity. Residues located at the secondary structure should be deliberated carefully because the introduction of these mutations may break the secondary structure, destroying the whole protein or unexpectedly improve the stability of the secondary structure making a rigid protein. Finally, introduction of mutations on the injurious residue to opposite electrical property would be better than introduction of mutations on unprotonable one. However, such mutations could trigger potential allosteric effects and change the whole structure. Therefore, four residues far away from active site, three locating at the loop region, and one locating at the α-helix were selected and mutated for thermostability test. Among them, three residues were mutated to unprotonable Ala and one residue was mutated to Lys (which has opposite electrical property with Asp). With the above issues taken into account, four residues, D113 (total Eij = 33.60 KJ/mol), D149 (total Eij = 34.08 KJ/mol), D213 (total Eij = 32.28 KJ/mol), and D253 (total Eij = 34.85 KJ/mol), were selected for mutation. Figure 3 shows that each mutational site was located on the protein surface spaced sufficiently from the catalytic center. Further precaution was taken regarding to which amino acids the chosen residues should be mutated to. Theoretically, mutation of a negatively charged residue to a positive one may cause a significant alteration of surface potential and thus of binding property. In this case, possible outcome is uncertain and unfavorable to our study. To circumvent the uncertainty, D113, D213, and D253 were mutated to a simple neutral amino acid alanine, and only D149 was mutated to positively charged residue lysine.
Figure 2.
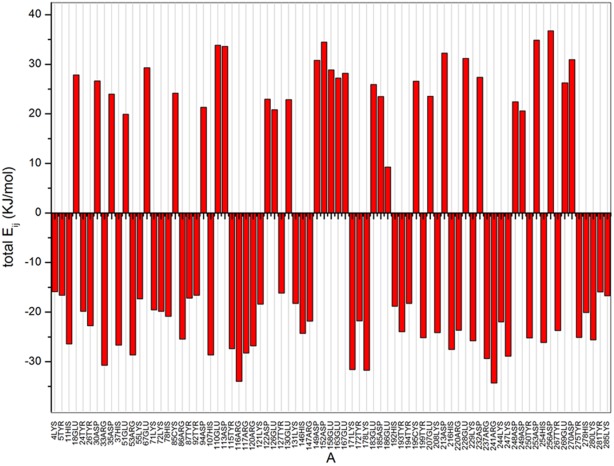
Total energy calculation of each chargeable residue. Positive value represents the overall contributions from repulsive force, which is pernicious to the thermostability of the protein structure, whereas negative value represents the overall contributions from gravitation, which is beneficial to the thermostability of the protein structure.
Figure 3.
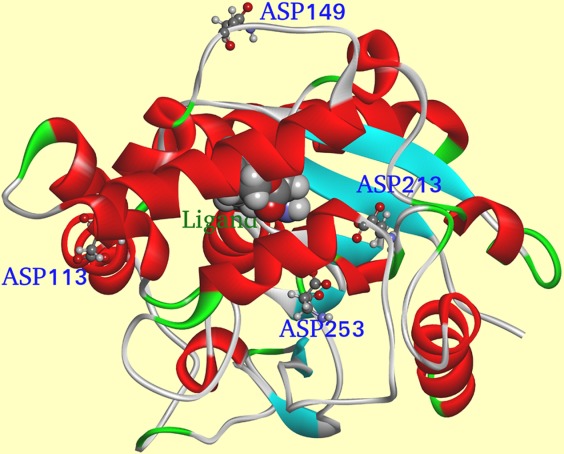
Cartoon representation of LipK107 structure. Residues selected by our ETSS software were shown in stick model and labeled. The ligand is shown in ball model and labeled. α-Helices are colored in red, β-strands are colored in blue, and random coils are colored in green.
Engineered Lipk107 mutants display increased thermal stability
Mutants were constructed by site-directed mutagenesis as described in Materials and Methods section. As an industrial application of an enzyme demands its robustness in a temperature higher than ambience, the effect of the mutation on resistance to heat stress was assayed by monitoring the residual lipase activity after heat treatment. Theoretically, the percentage of activity lost in heat treatment is in accordance with the number of protein molecules denatured by high temperature. Therefore, if the interaction between point charges is enhanced with mutation, the substrate binding pocket will evade a collapse under the heat. To test the reliability of our rational design, we introduced a set of standards and different conditions. First, the half-IT1/2 was determined by measuring the activity of WT LipK107 and its variants on hydrolysis of pNPP at various temperatures. As shown in Figure 4(A), point mutations of D213A and D253A shifted IT1/2 upward by 5°C, from 50 to 55°C, and similarly D113A and D149K by 10°C, from 50 to 60°C, indicating that the mutation significantly stabilized LipK107. Moreover, the extent of temperature inactivation was evaluated on time-course among the WT and variants, based on their residual activity during incubation of the enzymes at constant 50°C (Fig. 4B). Point mutations of D213A and D253A lengthened the inactivation half-life by ∼4.5-fold (2.25 h) and 6-fold (3 h), respectively, compared to that of the WT (<30 min). Notably, mutants D113A and D149K showed a further improvement over D213A and D253A with an inactivation half-life of ∼6 and ∼7 h, which was more than 12- to 14–folds of improvement over WT, suggesting that point mutation had a positive effect on the thermal stability. D113A is located at α-helix, whereas D213A and D253A located at the loop area. For a secondary structure, reducing the negativity of D113 may improve the whole stability of the α-helix. Hence, D113A showed a better effect than D213A and D253A. The experiment results showed that D149K was the best mutant. To explain the reason, virtual mutation has been done by Discovery studio 3.0, and we recalculated the mutated protein by ETSS. When D149 was mutated to K149, the Eij was decreased from +34.08 KJ/mol (D149) to −59.73 KJ/mol (K149), indicating a significant improvement of thermostability. In contrast, the other three residues were mutated to unprotonable Ala, and hence their impacts on thermostability were smaller than that introduced by D149K.
Figure 4.

Thermostability assays and the relative specific activity of LipK107 and its variants. (A) Inactivation as a function of temperature. Residual activity was measured at 37°C after incubation of LipK107 and its variants for 30 min at various temperatures. (B) Time-course of thermal inactivation at 50°C. Results are the average of three independent experiments. (█) LipK107 WT, (•) LipK107 D113A, (▴) LipK107 D149K, (▾) LipK107 D213A, and (◆) LipK107 D253A. (C) The relative specific activity of LipK107 and its mutants. Lipase activities were determined at 37°C, and the specific activities of mutants were expressed as a percentage of the LipK107. The values shown are the means of three independent experiments.
Engineered Lipk107 mutants maintain the high enzyme activities
As our main concern in this study is to maintain the enzyme activity while enhancing thermal stability, the specific activity of each mutant was examined and compared to that of WT. Figure 4C shows that those point mutations did not compromise the enzyme activity, and that they retained at least 80% of WT enzyme activity. Notably, D253A rather improved the enzyme activity by 20%.
In this study, ETSS enabled us to identify four LipK107 mutants with significantly improved thermal stability. Admittedly, our search for stability-enhanced mutants has not exhausted the pool of possible redesigns. Further sophistication of our redesign suite will entail overcoming computational ambiguity in such parameters as the impacts of ionic strength on short-range and long-range charge–charge interactions, roles of surface charges in those interactions, and their synergy with water molecules.22,24,45,46 Still, our prediction of key residues by ETSS turned out to be precise enough to reduce the candidates to as few as four single-point mutants. Directed evolution with error-prone PCR could be an alternative method to find out a mutant with as much thermal stability as in our hands. However, assays concerning enzyme activity and stability require a highly purified protein instead of lysate, which renders high-throughput mutagenesis infeasible.
Conclusion and Remarks
Our rational redesign strategy was proven promising to pinpoint mutant candidates with enhanced thermal stability. It will have broad applications for the rational design of thermal stability-enhanced proteins. A clear advantage of this study is that the source code of the program has been introduced here and supplied in the Material on Line. Thus, researchers can optimize the parameters to suit their experimental systems freely, which, we hope, will cater to thermal stability engineering field greatly. Any feedback on Open Source Principle will be appreciated.
Materials and Methods
Computational theory
The workflow of the program consists of three parts (Fig. 1). The first part is to retrieve the amino acid sequence of the protein from Protein Data Bank (http://www.rcsb.org/pdb) and to calculate the theoretical parameters of atoms in the protein, including the number of charges, pKa value, and Van der Waals radius, the whole volume of the protein and the Cartesian coordinate of each chargeable atom. It should be noted that theoretical values of Van der Waals radius vary and affect end-state calculation as in Gerstein's, Rowland's, and Li's.36–39 In view of SA calculation, small instead of large size of Van der Waals radius should be chosen to improve the accessibility of the residues surrounded by big atoms. Therefore, parameters of Van der Waals radius have been set changeable here for users to choose the best one for their system. Herein, the BL+ of Jerry's40 was chosen to do the calculation.
Figure 1.
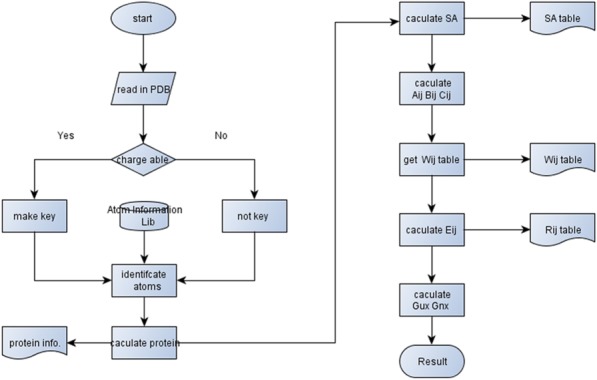
Workflow chart of end-state calculation. All steps shown in the workflow chart are evaluated and performed automatically.
The second part of the program is the calculation of SA for each amino acid. We consider the plausible conditions where protonation happens only to the charged atoms, not to the one whole amino acid. Thus, the SA of a charged atom is not viewed as replacing the SA of the whole ammonic acid. Source code for SA calculation follows the typical Rich method,41 an ingenious method balancing between accuracy and efficiency.42
The third part is to figure out the charge–charge interaction of each amino acid, including long-range and short-range interactions. The TK-SA model is applied to determine the charge–charge interactions inside and outside the protein. The numerical processing is recapitulated in Eq. (1).10 In TK-SA model, the interaction energy between the surface charges of point i and j is calculated as follows (Supporting Information Fig. S1): b is the radius of the sphere representing the protein, a is the radius of the sphere from which solvent ions are excluded. SAij is the average SA of group i and j. ε is the unit charge whose value is 1.602 × 10−19C. Aij, Bij, and Cij are three coefficients to revise the value of the charge–charge interaction. The details of the equations are described in Supporting Information material.
| (1) |
Construction and preparation of mutant enzymes
The construction of wild–type (WT) and mutant LipK107 followed our previous study.35 Briefly, lipK107 (GenBank accession no. EU600201) was cloned from the reverse transcripts of total RNA from Proteus sp. into pET28a in frame with hexahistidine tag. The plasmid pET28a-lipK107 was used as a template for mutagenic PCR using KOD-Plus Mutagenesis Kit (Toyobo, Japan). The respective point mutations introduced were D113A, D149K, D213A, and D253A, and these four clones were separately transformed with E. coli strain BL21. The protein expression was induced with 0.8 mM of isopropyl-β-d-thiogalactopyranoside, 30°C for 10 h. The bacterial cell was harvested, sonicated, cleared by centrifugation, and loaded onto Ni-NTA affinity column (Qiagen). The chromatographic method was described previously.35 Similarly, the purification of LipK107 mutants was performed. All the proteins were dialyzed into 50 mM of Tris–HCl, pH 7.5, and subjected to catalytic assay.
Thermal stability assay of LipK107 and its mutants
The method of activity assay was described previously.43 The activity was determined by measuring the release of para-nitrophenol (pNP) from pNP-derivative substrates. For colorimetric assays, pNP-myristate was dissolved in isopropanol and 6.25 mM of stock solution was prepared. The reaction mixture contained 100 µL of substrate stock solution, 900 µL of Tris–HCl buffer (100 mM, pH 8.0), and 500 µL of purified enzyme. This mixture was incubated at 37°C for 5 min, the reaction was terminated by addition of 100 µL of Na2CO3 (100 mM), and the release of pNP was determined by measuring the absorbance at 410 nm. One unit of activity was defined as the amount of enzyme needed to release 1 µmol of pNP per minute under the assay conditions. The activity assay of LipK107 and its mutants were carried out to determine the apparent half-inactivation temperature (IT1/2). Residual activity was measured at 37°C after incubation of LipK107 and its mutants at various temperatures for 30 min. For thermal stability assay, the residual activity of LipK107 and its variants as a function of time at 50 or 60°C was determined by hydrolysis of para-nitrophenyl phosphate (pNPP) at 37°C. All the values were based on the average of triplicate measurements. The protein concentrations were determined by Bradford method44 using BSA as a standard.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Schweiker KL, Makhatadze GI. Protein stabilization by the rational design of surface charge-charge interactions. Methods Mol Biol. 2009;490:261–283. doi: 10.1007/978-1-59745-367-7_11. [DOI] [PubMed] [Google Scholar]
- 2.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serrano L, Kellis JT, Jr, Cann P, Matouschek A, Fersht AR. The folding of an enzyme: II. Substructure of barnase and the contribution of different interactions to protein stability. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 4.Desjarlais JR, Handel TM. De novo design of the hydrophobic cores of proteins. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazar GA, Desjarlais JR, Handel TM. De novo design of the hydrophobic core of ubiquitin. Protein Sci. 1997;6:1167–1178. doi: 10.1002/pro.5560060605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loladze VV, Ermolenko DN, Makhatadze GI. Thermodynamic consequences of burial of polar and non-polar amino acid residues in the protein interior. J Mol Biol. 2002;320:343–357. doi: 10.1016/S0022-2836(02)00465-5. [DOI] [PubMed] [Google Scholar]
- 7.Pace CN, Shirley BA, McNutt M, Gajiwala K. Forces contributing to the conformational stability of proteins. FASEB J. 1996;10:75–83. doi: 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- 8.Griko YV, Makhatadze GI, Privalov PL, Hartley RW. Thermodynamics of barnase unfolding. Protein Sci. 1994;3:669–676. doi: 10.1002/pro.5560030414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strickler SS, Gribenko AV, Keiffer TR, Tomlinson J, Reihle T, Loladze VV, Makhatadze GI. Protein stability and surface electrostatics: a charged relationship. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- 10.Gribenko AV, Patel MM, Liu J, McCallum SA, Wang CY, Makhatadze GI. Rational stabilization of enzymes by computational redesign of surface charge-charge interactions. Proc Natl Acad Sci USA. 2009;106:2601–2606. doi: 10.1073/pnas.0808220106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gribenko AV, Makhatadze GI. Role of the charge–charge interactions in defining stability and halophilicity of the CspB proteins. J Mol Biol. 2007;366:842–856. doi: 10.1016/j.jmb.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 12.Permyakov SE, Makhatadze GI, Owenius R, Uversky VN, Brooks CL, Permyakov EA, Berliner LJ. How to improve nature: study of the electrostatic properties of the surface of α-lactalbumin. Protein Eng Des Sel. 2005;18:425–433. doi: 10.1093/protein/gzi051. [DOI] [PubMed] [Google Scholar]
- 13.Schweiker KL, Zarrine-Afsar A, Davidson AR, Makhatadze GI. Computational design of the Fyn SH3 domain with increased stability through optimization of surface charge-charge interactions. Protein Sci. 2007;16:2694–2702. doi: 10.1110/ps.073091607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkwood JG. Theory of solutions of molecules containing widely separated charges with special application to zwitterions. J Chem Phys. 1934;2:351. [Google Scholar]
- 15.Tanford C, Kirkwood JG. Theory of protein titration curves. I. General equations for impenetrable spheres. J Am Chem Soc. 1957;79:5333–5339. [Google Scholar]
- 16.Matthew JB, Gurd F. Calculation of electrostatic interactions in proteins. Methods Enzymol. 1986;130:413–436. doi: 10.1016/0076-6879(86)30019-3. [DOI] [PubMed] [Google Scholar]
- 17.Matthew JB, Gurd F. Stabilization and destabilization of protein structure by charge interactions. Methods Enzymol. 1986;130:437–453. doi: 10.1016/0076-6879(86)30020-x. [DOI] [PubMed] [Google Scholar]
- 18.Matthew JB, Gurd FR, Garcia-Moreno BE, Flanagan MA, March KL, Shire SJ. pH-dependent processes in protein. Crit Rev Biochem Mol Biol. 1985;18:91–197. doi: 10.3109/10409238509085133. [DOI] [PubMed] [Google Scholar]
- 19.Matthew J, Hanania G, Gurd F. Electrostatic effects in hemoglobin: hydrogen ion equilibria in human deoxy-and oxyhemoglobin A. Biochemistry. 1979;18:1919. doi: 10.1021/bi00577a011. [DOI] [PubMed] [Google Scholar]
- 20.Richmond TJ. Solvent accessible surface area and excluded volume in proteins: analytical equations for overlapping spheres and implications for the hydrophobic effect. J Mol Biol. 1984;178:63–89. doi: 10.1016/0022-2836(84)90231-6. [DOI] [PubMed] [Google Scholar]
- 21.Bashford D, Karplus M. Multiple-site titration curves of proteins: an analysis of exact and approximate methods for their calculation. J Phys Chem. 1991;95:9556–9561. [Google Scholar]
- 22.García-Moreno EB, Fitch CA. Structural interpretation of pH and salt-dependent processes in proteins with computational methods. Methods Enzymol. 2004;380:20–51. doi: 10.1016/S0076-6879(04)80002-8. [DOI] [PubMed] [Google Scholar]
- 23.Warshel A. Computer simulations of enzyme catalysis: methods, progress, and insights. Ann Rev Biophys Biomol Struct. 2003;32:425–443. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- 24.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 25.Havranek JJ, Harbury PB. Tanford-Kirkwood electrostatics for protein modeling. Proc Natl Acad Sci USA. 1999;96:11145–11150. doi: 10.1073/pnas.96.20.11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Ruiz JM, Ibarra-Molero B, Loladze VV, Makhatadze GI. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 1999;38:8138–8149. doi: 10.1021/bi9905819. [DOI] [PubMed] [Google Scholar]
- 27.Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol. 2001;312:885–896. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez-Ruiz JM, Makhatadze GI. To charge or not to charge? Trends Biotechnol. 2001;19:132–135. doi: 10.1016/s0167-7799(00)01548-1. [DOI] [PubMed] [Google Scholar]
- 29.Ibarra-Molero B, Sanchez-Ruiz JM. Genetic algorithm to design stabilizing surface-charge distributions in proteins. J Phys Chem B. 2002;106:6609–6613. [Google Scholar]
- 30.Makhatadze GI, Loladze VV, Ermolenko DN, Chen XF, Thomas ST. Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J Mol Biol. 2003;327:1135–1148. doi: 10.1016/s0022-2836(03)00233-x. [DOI] [PubMed] [Google Scholar]
- 31.Makhatadze GI, Loladze VV, Gribenko AV, Lopez MM. Mechanism of thermostabilization in a designed cold shock protein with optimized surface electrostatic interactions. J Mol Biol. 2004;336:929–942. doi: 10.1016/j.jmb.2003.12.058. [DOI] [PubMed] [Google Scholar]
- 32.Xu T, Zhang L, Su E, Cui D, Wang X, Wei D. Disparity in productive binding mode of the slow-reacting enantiomer determines the novel catalytic behavior of Candida antarctica lipase B. J Mol Catal B Enzym. 2010;62:288–296. [Google Scholar]
- 33.Xu T, Gao B, Zhang L, Lin J, Wang X, Wei D. Template-based modeling of a psychrophilic lipase: conformational changes, novel structural features and its application in predicting the enantioselectivity of lipase catalyzed transesterification of secondary alcohols. Biochim Biophys Acta. 2010;1804:2183–2190. doi: 10.1016/j.bbapap.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Xu T, Zhang L, Wang X, Wei D, Li T. Structure-based substrate screening for an enzyme. BMC Bioinformat. 2009;10:257. doi: 10.1186/1471-2105-10-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao B, Su E, Lin J, Jiang Z, Ma Y, Wei D. Development of recombinant Escherichia coli whole-cell biocatalyst expressing a novel alkaline lipase-coding gene from Proteus sp. for biodiesel production. J Biotechnol. 2009;139:169–175. doi: 10.1016/j.jbiotec.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Harpaz Y, Gerstein M, Chothia C. Volume changes on protein folding. Structure. 1994;2:641–649. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 37.Gerstein M, Tsai J, Levitt M. The volume of atoms on the protein surface: calculated from simulation, using Voronoi polyhedra. J Mol Biol. 1995;249:955–966. doi: 10.1006/jmbi.1995.0351. [DOI] [PubMed] [Google Scholar]
- 38.Rowland RS, Taylor R. Intermolecular nonbonded contact distances in organic crystal structures: comparison with distances expected from van der Waals radii. J Phys Chem. 1996;100:7384–7391. [Google Scholar]
- 39.Li AJ, Nussinov R. A set of van der Waals and coulombic radii of protein atoms for molecular and solvent-accessible surface calculation, packing evaluation, and docking. Proteins. 1998;32:111–127. [PubMed] [Google Scholar]
- 40.Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: standard radii and volumes. J Mol Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 41.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 42.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 43.Prim N, Blanco A, Martínez J, Pastor FI, Diaz P. estA, a gene coding for a cell-bound esterase from Paenibacillus sp. BP-23, is a new member of the bacterial subclass of type B carboxylesterases. Res Microbiol. 2000;151:303–312. doi: 10.1016/s0923-2508(00)00150-9. [DOI] [PubMed] [Google Scholar]
- 44.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 45.Kumar S, Nussinov R. Close-range electrostatic interactions in proteins. ChemBioChem. 2002;3:604–617. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 46.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.