

Abstract
Purpose:
We have utilized a genome-wide approach to identify novel differentially methylated CpG dinucleotides that are seen in different anatomic sites of head and neck squamous cell carcinoma (HNSCC), as well as those that might be related to HPV status in the oropharynx.
Experimental Design:
We performed genome-wide DNA methylation profiling of primary tumor samples and corresponding adjacent mucosa from 118 HNSCC patients undergoing treatment at Montefiore Medical Center, Bronx, NY using the Illumina HumanMethylation27 beadchip. For each matched tissue set, we measured differentially methylated CpG loci using a change in methylation level (M-value).
Results:
When datasets were individually analyzed by anatomic site of the primary tumor, we identified 293 differentially methylated CpG loci in oral cavity SCC, 219 differentially methylated CpG loci in laryngeal SCC, and 460 differentially methylated in oropharyngeal SCC. A subset of these differentially methylated CpG loci was common across all anatomic sites of HNSCC. Stratification by HPV status revealed a significantly higher number of differentially methylated CpG loci in HPV-positive patients.
Conclusion:
Novel epigenetic biomarkers derived from clinical HNSCC specimens can be used molecular classifiers of this disease, revealing many new avenues of investigation for this disease.
Keywords: methylation, prognosis, head and neck, squamous, carcinoma
Introduction
Head and neck squamous cell carcinoma (HNSCC), an anatomically heterogeneous group arising most often from the oral cavity, oropharynx, hypopharynx, larynx and nasopharynx, is the fifth most common malignancy in men worldwide, representing a major international health problem (1). Tumors originating from these different locations can exhibit varying behavior that is not predictable by histopathology of the primary tumor but is discernible by other methods such as gene profiling (2). Conventional treatments employ surgery, radiation therapy and chemotherapy either alone or in combination with any, or all, of the other modalities. Any of these therapies can, and do, produce morbidities that affect speech, swallowing and overall quality of life. Despite treatment intervention, recurrence of the disease is observed in about 50% of patients, either locally, regionally or at a distant site with high rates of associated mortality (3). As a result, the 5-year survival rate has improved only marginally over the past decade; it is estimated that over 45,000 new cases and 11,000 deaths will occur each year in the United States from HNSCC (4).
The search for biomarkers predictive of patient prognosis and treatment response has now progressed to include epigenetic changes associated with tumor initiation and progression. By far, the most common epigenetic event in the human genome is the addition of a methyl group to the carbon-5 position of cytosine nucleotides. This covalent DNA modification occurs predominantly in cytosines immediately preceding guanine nucleotides (the CpG dinucleotide). In HNSCC, promoter methylation of tumor suppressor genes appears to be a common mechanism of transcriptional silencing, as discussed by Ha and Califano (5), Shaw (6) and Pérez-Sayáns (7). In many cases, epigenetic events associated with individual genes have been of prognostic value. For example, methylation of the promoter region of MGMT is associated with increased tumor recurrence and decreased patient survival independent of other factors (8). Methylation of the DCC gene is significantly associated with bone invasion by gingival tumors, aggressive invasiveness of tumors of the tongue, and reduced survival (9). Invasion and metastasis of oral SCC cells have been shown to be dependent on methylation of the E-cadherin promoter (10). There is also evidence of a statistically significant association between DNA hypermethylation of the ADAM23 gene and progression in laryngeal cancer (11). And finally, a highly significant difference in DNA hypermethylation of the DAPK gene promoter is observed between laryngeal cancer patients with and without lymph node metastasis (12). Both MLH1 and CDKN2A are also known to play an important role in laryngeal cancer development and progression (13).
Here, we present a genome-wide view of differential DNA methylation in primary HNSCC tumors from 118 patients at Montefiore Medical Center in Bronx, NY. The goals of this study were several-fold. First, we hoped to identify novel genes affected by aberrant DNA methylation that may play a role in head and neck tumorigenesis but have not been previously identified in this disease. Second, we wanted to determine whether or not HNSCC primary tumors originating from different anatomic sites (oral cavity, larynx, and oropharynx) showed similar DNA methylation changes, or whether some DNA methylation events were specific to a single anatomic site. In each of these cases, the identification of genes specifically affected by DNA methylation represents a way forward to the identification of novel tumor suppressor genes with relevance as potential clinical biomarkers in HNSCC.
Materials and Methods
Study population
Patients recruited for this study were undergoing treatment for histologically confirmed head and neck squamous cell carcinoma at Montefiore Medical Center, Bronx, NY (Table 1). All patients consented to participation in this study under protocols approved by the Institutional Review Boards of the participating institutions. All primary tumor and histologically normal adjacent tissues were snap-frozen in liquid nitrogen within 30 minutes of surgical resection or biopsy and kept at −80°C until further processing. Adjacent mucosa was taken at a grossly unremarkable site away from the tumor identified by the surgeon (incisional biopsy) intraoperatively or the pathologist (resection specimen) when processing the specimen for frozen section or final diagnosis. In the case of tumor resections, this represented histologically normal tissue approximately 1 cm away from the primary tumor. In the case of tumor biopsies, this represented tissue taken from the contralateral side. The distributions of normal tissues for each anatomic site were as follows: oral cavity (32 adjacent, 3 contralateral), oropharynx (19 adjacent, 27 contralateral), and larynx (16 adjacent, 21 contralateral).
Table 1.
Clinical characteristics of recruited head and neck cancer patients.
| Oral Cavity (N=35) |
Oropharynx (N=46) |
Larynx (N=37) |
||||
|---|---|---|---|---|---|---|
| N | (%) | N | (%) | N | (%) | |
| Male | 24 | 70% | 37 | 80% | 21 | 57% |
| Female | 11 | 30% | 9 | 20% | 16 | 43% |
| Median Age (Range) | 61 | (22 - 85) | 62 | (40 - 84) | 62 | (33 - 91) |
| Race | ||||||
| White | 19 | 54% | 26 | 57% | 22 | 59% |
| Black or African | ||||||
| American | 10 | 28% | 17 | 37% | 12 | 32% |
| Asian | 1 | 3% | 0 | 0% | 0 | 0% |
| Unknown | 5 | 14% | 3 | 7% | 3 | 8% |
| Ethnicity | ||||||
| Hispanic/Latino | 11 | 31% | 9 | 20% | 10 | 27% |
| Non-Hispanic/Latino | 23 | 66% | 35 | 76% | 25 | 68% |
| Unknown | 1 | 3% | 2 | 4% | 2 | 5% |
| Tumor Stage | ||||||
| I | 9 | 26% | 6 | 13% | 0 | 0% |
| II | 6 | 17% | 5 | 11% | 7 | 19% |
| III | 3 | 8% | 4 | 9% | 10 | 27% |
| IV | 17 | 49% | 31 | 67% | 19 | 51% |
| Node Status | ||||||
| N0 | 16 | 45% | 11 | 24% | 16 | 43% |
| N1 | 2 | 9% | 5 | 10% | 4 | 11% |
| N2 | 14 | 40% | 23 | 50% | 14 | 38% |
| N3 | 0 | 0% | 3 | 7% | 0 | 0% |
| HPV Status | ||||||
| HPV (+) | 6 | 17% | 17 | 37% | 8 | 22% |
| HPV (−) | 27 | 77% | 16 | 35% | 22 | 59% |
| Indeterminate | 2 | 6% | 13 | 28% | 7 | 19% |
| %Tumor cell content | ||||||
| Samples less than 40% | 4 | 11% | 8 | 17% | 2 | 5% |
| Mean % tumor | 56% | 58% | 70% | |||
Histological confirmation of tumors was performed by a single pathologist assessing tumor grade, TNM staging based on the AJCC classification, and degree of lymphocytic infiltration. Control samples for all tumors were acquired; microscopy slides stained with hematoxylin and eosin were assessed for percentage of tumor, extent of necrosis and degree of lymphocytic infiltration as described previously (14). The mean percentage tumor content for samples from each anatomic site, as well as the number of samples with less than 40% tumor content, are both shown in Table 1.
All patient samples were tested for HPV infection using multiple testing methods. The presence of HPV DNA and HPV 16 DNA were assayed by MY09/11-PCR (degenerate and type-specific HPV16-L1 fragments) utilizing AmpliTaq Gold and dot-blot hybridization on matched-fresh frozen specimens as described previously (15). HPV 16 was also assayed using in situ hybridization of samples on formalin fixed, paraffin embedded tissue blocks with multiple probes (15). And finally, the presence of HPV E6 and E7 RNA transcripts was detected and quantitated using TaqMan RT-PCR. Only patients testing positive by all methods were classified as positive (Table 1). Only patients testing negative for all methods were classified as HPV negative. All others were classified as indeterminate.
Genome-wide methylation profiling of tissue genomic DNA
Genome-wide profiling of differential DNA methylation in tumor samples was carried out as described previously (14). Briefly, genomic DNA was isolated from snap-frozen tissue using the DNeasy tissue kit (Qiagen). Quantity and purity of isolated DNA was measured spectrophotometrically using a NanoDrop ND-1000 spectrophotometer. Bisulfite conversion of tissue genomic DNA was carried out using the EZ DNA Gold methylation kit (Zymo Research). Profiling of genome-wide DNA methylation in genomic DNA (500 ng) was carried out using the Illumina Infinium assay with the HumanMethylation27 DNA Analysis BeadChip. The methylation level at each CpG loci on the beadchip was determined by measuring the methylation fraction (beta), defined as the fraction of methylated signal over the total signal (unmethylated + methylated fractions) in each genomic DNA sample. This value ranged continuously from 0 (unmethylated) to 1 (fully methylated) for each CpG locus. A complete description of individual beadchip controls on the Illumina beadchip platform is available from the manufacturer (Illumina, San Diego, CA). Validation of the beadchip measurements was also carried out on a subset of identified CpG loci using matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) mass spectrometry using EpiTyper by massARRAY (Sequenom) on bisulfite-converted DNA to verify the accuracy of beadchip measurements (Supplementary Figure S1).
Data preparation and supervised analysis
Normalized M values were generated from primary HumanMethylation27 beadchip beta values using the R package HumMeth27KQCReport function, including the X chromosome data and using an average probe p-value of 0.03 as the cutoff for sample inclusion (16). The resulting normalized M values represent a continuous variable from −6 (unmethylated) to 6 (methylated). Differences in methylation (ΔM, Mtumor-Mnormal) in which both the tumor and adjacent mucosal samples passed quality control were calculated and combined with patient clinical information on nodal metastasis at diagnosis. For identifying differentially methylated CpG loci for each tumor/normal dataset, we selected CpG loci with a Storrey corrected p-value of less than 0.05 and an average ΔM of greater than 1.4 or less than —1.4 across a given dataset, as described previously for this type of data (17). A cutoff of 1.4 was established as the minimum threshold for distinguishing methylated from unmethylated DNA using this type of transformed data (17).
Whole genome expression analysis of tissue samples
For each primary HNSCC and corresponding normal tissue sample pair, linear amplification and biotin-labeling of total RNA (500 ng) were carried out using the Illumina TotalPrep RNA Amplification Kit (Ambion). Whole-genome expression analysis was carried out by hybridization of amplified RNA to an Illumina HumanHT-12 v3 Expression BeadChip. With this beadchip, we interrogated greater than 48,000 probes per sample, targeting genes and known alternative splice variants from the RefSeq database release 17 and UniGene build 188. Controls for each RNA sample (greater than 1000 bead types) confirmed sample RNA quality, labeling reaction success, hybridization stringency, and signal generation. All expression data were quantile normalized and background-subtracted prior to analysis using BeadStudio software (Illumina).
Results
Patient demographics and clinical characteristics
We recruited 118 patients undergoing treatment for HNSCC at Montefiore Medical Center in the Bronx, a high-risk area of New York City with a high incidence of the disease (Table 1). All pathologic grades of tumor were represented although the majority of cases (58%) had stage IV disease at presentation. Seventy-three patients (63%) had a positive nodal status at presentation. For all patients, we also collected histologically normal adjacent mucosa in parallel with primary tumor tissue collection. In the case of tumor resections, this represented histologically normal tissue approximately 1 cm away from the primary tumor. In the case of tumor biopsies, this represented tissue taken from the contralateral side.
Differential DNA methylation in HNSCC tumors
The goal of our first analysis was a genome-wide view of differential DNA methylation to identify novel hyper- and hypo-methylated CpG loci and their associated genes from each patient dataset (oral cavity SCC, oropharyngeal SCC, laryngeal SCC), and to examine the degree to which these tumors share common epigenetic alterations. We focused our lists for each patient set to significantly altered CpG loci (p<0.05) where the absolute difference in the average ΔM values (M̄ tumor − M̄ adjacent) was set at a threshold of 1.4 as described previously (17). The results were 293 differentially methylated CpG loci (119 hypermethylated; 174 hypomethylated) in oral cavity SCC, 219 differentially methylated CpG loci (75 hypermethylated; 144 hypomethylated) in laryngeal SCC, and 460 differentially methylated CpG loci (385 hypermethylated; 75 hypomethylated) in oropharyngeal SCC (Table 2). Overall, these numbers represented less than 2% of the total CpG loci surveyed using the HumanMethylation27 beadchip.
Table 2.
Summary of Differentially Methylated CpG Loci in Head and Neck Cancer
| Significant (p<0.05) and ΔM̄ >1.4 | ||
|---|---|---|
| CGI | Non-CGI | |
|
|
||
| Oral Cavity | ||
| Hypermethylated | 116 | 3 |
| Hypomethylated | 37 | 137 |
| Total: 293 CpG loci | ||
|
|
||
| Larynx | ||
| Hypermethylated | 68 | 7 |
| Hypomethylated | 49 | 95 |
| Total: 219 CpG loci | ||
|
|
||
| Oropharynx | ||
| Hypermethylated | 380 | 5 |
| Hypomethylated | 25 | 50 |
| Total: 460 CpG loci | ||
|
|
||
Within each of the oral cavity, larynx, and oropharynx SCC datasets, the distribution of these CpG loci (hyper- versus hypo-methylated) coincided primarily with their chromosomal position within, or outside of, classically defined CpG islands (18). For example, of the 119 CpG loci identified as hypermethylated in our oral cavity SCC patient population, 116 loci (97%) were located within classically defined CpG island sequences in the promoters of genes. In contrast, the vast majority of the 174 hypomethylated CpG loci were located predominantly outside of CpG island sequences (137 loci, 79%). However, it should be noted that a significant number of hypomethylated CpG loci (37 loci; 21%) did reside within CpG island sequences and may represent genes regulated by DNA hypomethylation in head and neck cancer.
Overall, our analysis also showed that head and neck tumors originating from different anatomic sites do share some common epigenetic alterations. An initial overlap of the three datasets identified 44 CpG loci (28 hypermethylated; 16 hypomethylated) that showed significantly altered DNA methylation across all three anatomic sites of HNSCC (Table 3). Interestingly, only a subset of these genes had been previously studied in head and neck cancer. As with our previous study, the most overrepresented group of genes in this list were the Kruppel family ZNF genes on chromosome 19q13, most notably ZNF132, ZNF154, ZNF542, ZNF545/ZFP82, ZNF671 and ZNF781(14). Validation of the beadchip measurements for ZNF132 and ZNF154 using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry using EpiTyper by massARRAY (Sequenom) on bisulfite-converted DNA demonstrated an increase in DNA methylation across multiple CpG loci associated with these genes in primary HNSCC tumors (Supplementary Figure S6). Genes such as ATP10A, RLN3R1, RYR2, SORCS3, SPARCL1, TBX5, and the Hox genes HOXD9, HOXD10, and HOXD12 also showed hypermethylation across anatomic sites. Of the 16 hypomethylated CpG loci identified across all three anatomic sites of HNSCC cancer cases, only 4 loci resided within defined CpG island sequences. These genes also had not previously studied in HNSCC tumors, and several, including claudin-18 (CLDN18) and the orphan G protein-coupled receptor GPR55, have been seen as contributing to, or even promoting, tumor progression in other cancers (19, 20).
Table 3.
Summary of differentially methylated CpG loci common across all HNSCC sites. Information for each loci include Illumina TargetID, chromosomal position, associated gene symbol and gene name, relative DNA methylation (tumor versus adjacent mucosa), and relative gene expression (tumor versus adjacent mucosa).
| TargetID | Chromosome | Position | Symbol | Gene Product | Relative DNA Methylation (Average ΔM) |
Relative Gene Expression in HNSCC |
||
|---|---|---|---|---|---|---|---|---|
| Oral Cavity | Oropharynx | Larynx | ||||||
| Hypermethylated CpG loci common across all HNSCC sites: | ||||||||
| cg03238797 | 16q23 | 76026394 | ADAMTS18 | ADAM metallopeptidase with thrombospondin type 1 motif; 18 isoform 2 preproprotein |
1.89 | 1.76 | 2.11 | ns |
| cg13912117 | 8q24 | 132123737 | ADCY8 | Adenylate cyclase 8 | 2.10 | 2.34 | 1.52 | ns |
| cg19326876 | 15q11.2 | 23658751 | ATP10A | ATPase; Class V; type 10A | 2.16 | 2.37 | 2.26 | Increased* |
| cg17793621 | 15q11.2 | 23659177 | ATP10A | ATPase; Class V; type 10A | 2.04 | 2.47 | 2.17 | Increased* |
| cg21475402 | 1q31 | 154878764 | BCAN | Brevican isoform 2 | 1.86 | 1.51 | 2.45 | ns |
| cg08047907 | 1q24 | 167663482 | C1orf114 | Hypothetical protein LOC57821 | 1.71 | 2.57 | 2.05 | ns |
| cg10088985 | 4q13.3 | 75083177 | CXCL5 | Chemokine (C-X-C motif) ligand 5 precursor | 1.63 | 1.95 | 2.20 | Decreased*** |
| cg15540820 | 3p24.1 | 27740287 | EOMES | Eomesodermin | 1.55 | 2.05 | 1.92 | ns |
| cg21591742 | 2q31.1 | 176689244 | HOXD10 | Homeobox D10 | 1.41 | 1.85 | 1.67 | Increased*** |
| cg03874199 | 2q31.1 | 176672702 | HOXD12 | Homeobox D12 | 1.82 | 2.50 | 1.82 | Decreased* |
| cg14991487 | 2q31.1 | 176695650 | HOXD9 | Homeobox D9 | 1.90 | 2.55 | 1.68 | Increased*** |
| cg07748540 | 11q22.3 | 103539829 | PDGFD | Platelet derived growth factor D isoform 1 precursor | 1.64 | 1.98 | 2.18 | Decreased*** |
| cg03909500 | 5p15.1-p14 | 33972354 | RLN3R1 | Relaxin 3 receptor 1 | 1.67 | 2.02 | 1.74 | Decreased* |
| cg12238343 | 5p15.1-p14 | 33972159 | RLN3R1 | Relaxin 3 receptor 1 | 1.74 | 2.06 | 1.70 | Decreased* |
| cg11657808 | 1q43 | 235272573 | RYR2 | Ryanodine receptor 2 | 2.25 | 2.69 | 1.91 | ns |
| cg04072323 | 2q36 | 228754569 | SKIP | Sphingosine kinase type 1-interacting protein | 1.69 | 1.98 | 1.63 | ns |
| cg04490714 | 16q12.2 | 54248065 | SLC6A2 | Solute carrier family 6 member 2 | 1.69 | 2.07 | 2.46 | Increased*** |
| cg16787600 | 10q23-q25 | 106390870 | SORCS3 | VPS10 domain receptor protein SORCS 3 | 1.94 | 2.29 | 1.52 | Decreased** |
| cg19466563 | 4q22.1 | 88669530 | SPARCL1 | SPARC-like 1 | 1.48 | 1.41 | 1.89 | Decreased*** |
| cg21907579 | 12q24.1 | 113330251 | TBX5 | T-box 5 isoform 2 | 1.45 | 1.61 | 1.83 | ns |
| cg01009664 | 3q13.3-q21 | 131176303 | TRH | Thyrotropin-releasing hormone | 1.58 | 2.38 | 2.27 | ns |
| cg13877915 | 19q13.4 | 63643484 | ZNF132 | Zinc finger protein 132 (clone pHZ−12) | 2.66 | 3.12 | 3.10 | Decreased*** |
| cg21790626 | 19q13.4 | 62912306 | ZNF154 | Zinc finger protein 154 (pHZ−92) | 1.66 | 2.31 | 2.44 | Decreased*** |
| cg08668790 | 19q13.4 | 62912474 | ZNF154 | Zinc finger protein 154 (pHZ−92) | 1.81 | 2.72 | 2.41 | Decreased*** |
| cg26309134 | 19q13.43 | 61571383 | ZNF542 | Zinc finger protein 542 | 2.34 | 2.94 | 2.49 | Decreased*** |
| cg25886284 | 19q13.12 | 41601258 | ZNF545/ZFP82 | Zinc finger protein 545 | 2.09 | 2.56 | 2.75 | Decreased* |
| cg19246110 | 19q13.43 | 62930740 | ZNF671 | Zinc finger protein 671 | 1.88 | 3.11 | 2.46 | Decreased* |
| cg25875213 | 19q13.12 | 42874895 | ZNF781 | Zinc finger protein 781 | 2.13 | 3.39 | 3.57 | Decreased* |
| Hypomethylated CpG loci common across all HNSCC sites: | ||||||||
| cg10636246 | 1q22 | 157313597 | AIM2 | Absent in melanoma 2 | −1.50 | −1.64 | −1.73 | Increased*** |
| cg25659818 | 17q12 | 31455860 | CCL4 | Chemokine C-C motif ligand 4 precursor | −1.45 | −1.46 | −1.41 | ns |
| cg13765621 | 1q22-q23 | 156415852 | CD1D | CD1D antigen; d polypeptide | −1.48 | −1.65 | −1.45 | ns |
| cg23181133 | 19q13.2 | 46992652 | CEACAM3 | Carcinoembryonic antigen-related cell adhesion molecule 3 | −1.96 | −1.45 | −2.40 | ns |
| cg17298704 | 3q22.3 | 139200297 | CLDN18 | Claudin 18 isoform 2 | −1.69 | −1.58 | −1.47 | Decreased** |
| cg05037688 | 9q34.3 | 138676375 | EGFL7 | EGF-like-domain; multiple 7 | −1.80 | −2.17 | −1.85 | Decreased* |
| cg14696870 | 1q23 | 157525501 | FCER1A | Fc fragment of IgE; high affinity I; receptor for; alpha polypeptide precursor |
−1.52 | −1.54 | −1.44 | Decreased*** |
| cg24169915 | 14q32.2 | 98247740 | FLJ25773 | Hypothetical protein LOC283598 | −2.09 | −1.44 | −1.43 | ns |
| cg20287234 | 2q37 | 231497709 | GPR55 | G protein-coupled receptor 55 | −1.66 | −1.69 | −2.11 | ns |
| cg24423088 | 21q22.1 | 31107236 | KRTAP8−1 | Keratin associated protein 8−1 | −1.59 | −1.58 | −1.53 | ns |
| cg07545232 | Xq28 | 151688898 | MAGEA3 | Melanoma antigen family A; 3 | −1.85 | −1.72 | −1.64 | Increased*** |
| cg15408454 | Xq28 | 151617883 | MAGEA6 | Melanoma antigen family A; 6 | −1.76 | −1.56 | −1.72 | Increased*** |
| cg18750756 | 17p13.2 | 4283980 | MGC29671 | Hypothetical protein LOC201305 | −1.59 | −1.41 | −1.50 | na |
| cg04491443 | 16p12.3 | 20323467 | PDILT | Protein disulfide isomerase-like protein of the testis | −1.84 | −1.51 | −1.62 | ns |
| cg27342801 | 2p12 | 79240781 | REG3A | Pancreatitis-associated protein precursor | −2.28 | −1.73 | −1.65 | Increased** |
| cg07405796 | 6p22.1 | 30212531 | TRIM40 | Tripartite motif-containing 40 | −1.71 | −1.92 | −1.81 | ns |
Abbreviations:
p<0.05,
p<0.01,
p<0.001,
na not available, ns not significant.
The significance of these DNA methylation events was further explored by examining their impact on the expression of proximal genes. Using microarray gene expression data from these same patients, we cross-referenced these CpG loci with global gene expression profiles, and extracted gene expression data for all of the genes associated with the differentially methylated CpG loci. We found that of the 28 hypermethylated CpG loci identified in our analysis, 14 of the associated genes also showed a significant reduction in gene expression in the primary tumor compared to adjacent mucosa when combining all HNSCC cases (Table 3). These included all of the Kruppel family ZNF genes on chromosome 19q13. Surprisingly, several genes actually showed an increase in gene expression in the primary tumor, such as ATP10A, HOXD9, HOXD10 and SLC6A2, although the reasons for this remain unclear. We utilized genome-wide DNA methylation data from the Cancer Genome Atlas (TCGA) in order to externally validate the hyper- and hypo-methylation properties of these loci (http://cancergenome.nih.gov/cancersselected/headandneck). In independent analysis, all 28 commonly hypermethylated CpG loci also showed a statistically significant increase in DNA methylation in the primary tumors compared to available normal mucosal tissue in these cohort of patients (N=290 tumors, 45 normal, unpaired t-test, p<0.001)(Supplementary Table S4). Of the 16 hypomethylated CpG loci, 11 CpGs had usable data available in the TCGA repository, and all showed a significant decrease in DNA methylation in the primary tumors compared to the available normal mucosa (N=290 tumors, 45 normal, unpaired t-test, p<0.001)(Supplementary Table S5).
Two examples of epigenetic silencing in primary HNSCC tumors were the Kruppel family zinc finger proteins ZNF132 and ZNF154 which showed both elevated DNA hypermethylation as well as reduced gene expression in head and neck primary tumors compared to adjacent mucosa from the patient (paired t-test, p<0.05) (Figure 1). In fact, our results showed that DNA hypermethylation of the Kruppel family ZNF genes on chromosome 19q13, which was demonstrated in our previous study on oropharyngeal SCC, now extends to all anatomic sites of HNSCC, and is accompanied by a corresponding decrease in gene expression in the primary tumor. The role of these genes in head and neck carcinogenesis is still unknown, and represents a novel avenue of exploration as new tumor suppressor candidates. Complete lists hyper- and hypo-methylated CpG loci from each anatomic site of HNSCC are available as Supplementary Tables S1, S2 and S3.
Figure 1.

(A) Dot plots of relative DNA methylation levels (tumor to adjacent normal tissue), measured as a change in M value (ΔM), for CpG loci cg13877915 (ZNF132), cg08668790 (ZNF154) and cg21790626 (ZNF154) for specimen pairs originating from oral cavity (OC), oropharyngeal (OP) and laryngeal (LX) SCC. The median level of differential methylation is indicated by a bar for each patient population. (B) Relative gene expression (tumor to adjacent normal tissue) of corresponding target genes ZNF132 and ZNF154, as obtained by beadchip probe fluorescence measurements in corresponding gene expression microarray datasets, for specimen pairs originating from oral cavity (OC), oropharyngeal (OP) and laryngeal (LX) SCC. The median level of differential expression is indicated by a bar for each patient population.
Ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) (UCHL1) shows oral cavity-specific DNA hypermethylation as well as DNA hypomethylation in HNSCC
While some epigenetic changes were found in all anatomic sites, other CpG loci had significantly altered hyper- (or hypo-) methylation in primary tumors from only one anatomic site. This did not imply a lack of differential methylation in the other sites, only that it was not of a sufficient frequency to attain statistical significance for that specific patient dataset. The most striking example of this phenomenon was ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) (UCHL1), also known as PGP9.5, a controversial tumor suppressor gene previously identified as hypermethylated in multiple cancers, including head and neck cancer (21). While UCHL1 was identified as hypermethylated in over 54% of oral cavity cases, we observed hypermethylation frequencies of only 16% and 4% for laryngeal and oropharyngeal SCC cases, respectively (Figure 2A). Furthermore, hypomethylation of UCHL1 was also seen at different frequencies in all three anatomic sites of HNSCC, with the highest frequencies of hypomethylation seen in laryngeal (27%) and oropharyngeal (13%) SCC compared to oral cavity SCC primary tumors (6%).
Figure 2.
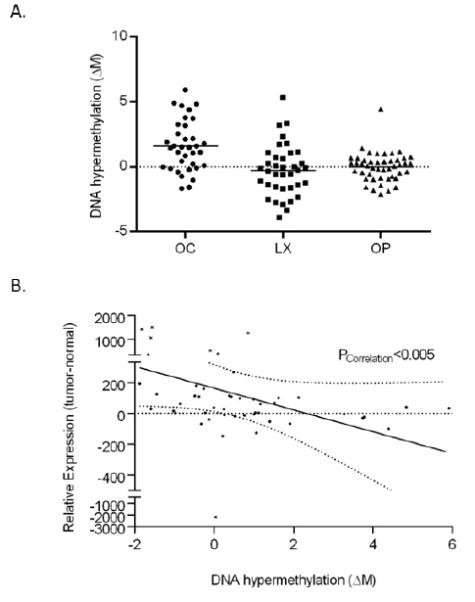
(A) Dot plot of relative DNA methylation levels (tumor to adjacent mucosa), measured as a change in M value (ΔM) for CpG loci cg08319991 (UCHL1) for specimen pairs originating from oral cavity (OC), oropharyngeal (OP) and laryngeal (LX) SCC. The median level of differential methylation is indicated by a bar for each patient population. Statistical significance (p<0.05) is indicated by an asterisk. (B) Plot of relative gene expression (tumor to adjacent mucosa) of UCHL1 (beadchip probe fluorescence measurements) versus relative DNA methylation (ΔM) for all HNSCC patients. Outliers are indicated by an ‘x’. Correlation between cg08319991 methylation and UCHL1 gene expression is calculated by linear regression analysis.
Differential methylation associated with the UCHL1 gene also had a corresponding effect on UCHL1 gene expression and tumor behavior. As with the previous genes, we cross-referenced relative methylation of the CpG loci associated with the UCHL1 gene (cg08319991) with corresponding UCHL1 gene expression data extracted from microarray datasets for these same patients. From these data, we observed a significant correlation between DNA methylation status of the UCHL1 CpG loci (cg08319991) and expression of the UCHL1 gene (Figure 2B). The results suggest that expression of UCHL1 is affected by both hyper- and hypo-methylation, and that the mechanism of choice is specific to the anatomic site of HNSCC. Furthermore, these epigenetic changes manifest as a change in expression of the UCHL1 gene in HNSCC tumor cells.
Oropharyngeal-specific DNA hypermethylation is strongly associated with the HPV status of the patient
Different anatomic sites of HNSCC also differed in terms of the overall number of differentially methylated CpG loci we identified using our genome-wide approach. Overall, we observed the highest number of differentially methylated CpG loci (hyper- and hypo-methylated) in primary tumors originating from the oropharynx (Table 2). Given the high propensity of HPV infection in this patient population, we investigated whether or not the number of differentially methylated CpG loci might be influenced by HPV status of the patient. In order to eliminate anatomic site of the primary tumor as a potential confounding variable, we focused only on oropharyngeal cases, stratified these patients into two separate populations according to HPV status, and then repeated the earlier differential methylation analysis. From this new analysis, we identified a total of 204 differentially methylated CpG loci in the HPV− oropharyngeal cases (Table 4). In general, these differentially methylated loci shared the most similarities with tumors from the oral cavity and larynx, both in terms of the number of differentially methylated loci we identified (204 total CpG loci) and the degree of overlap with other sites (32% of loci were also seen in oral cavity cases; 25% of loci were also seen in larynx cases). However, this same analysis in HPV+ oropharyngeal cases identified more than twice as many differentially methylated loci (564 total CpG loci). These included the majority of the differentially methylated loci seen in HPV− oropharyngeal cancer cases (eg. 74% of the hypermethylated loci); however, we also observed an additional 360 CpG loci in HPV+ oropharyngeal cancer cases not seen previously in HPV− cases. Among the 28 CpG loci whose methylation status best differentiated HPV− from HPV+ oropharyngeal cases were three loci within the CpG island of CDKN2A, and two loci located within the CpG island of the GALR1 gene (22, 23) (Figure 3). Also at the top of our list were two loci associated with the homeodomain transcription factor known as pancreatic and duodenal homeobox 1 (Pdx1/Ipf1), which had previously been observed to be methylated in breast and gastric cancer (24, 25). Overall, these results suggest that a higher level of aberrant DNA hypermethylation in HPV+ oropharyngeal cancer cases, and that this increased hypermethylation resulted in the targeting of genes not affected in tumors of HPV− cases.
Table 4.
Differentially Methylated CpG Loci in Oropharyngeal Cancer by HPV status.
| Significant (p<0.05) and ΔM̄ >1.4 | ||
|---|---|---|
| CGI | Non-CGI | |
|
|
||
| Oropharynx (HPV−) | ||
| N=16 patients | ||
| Hypermethylated | 161 | 3 |
| Hypomethylated | 12 | 28 |
| Total: 204 CpG loci | ||
|
|
||
| Oropharynx (HPV+) | ||
| N=17 patients | ||
| Hypermethylated | 428 | 9 |
| Hypomethylated | 49 | 78 |
|
|
||
| Total: 564 CpG loci | ||
|
|
||
Figure 3.

Dot plot of relative DNA methylation levels (tumor to adjacent mucosa) (ΔM) in oropharyngeal SCC patients for each of the 28 CpG loci whose methylation status best differentiated HPV− from HPV+ oropharyngeal cases. HPV+ oropharyngeal cases are indicated by red dots; HPV− oropharyngeal cases are indicated by green dots. Details for each CpG loci include Illumina Target ID, associated gene symbol, as well as the average ΔM for HPV− and HPV+ patient sets. Abbreviations: *p<0.05, **p<0.01, ***p<0.001, ns not significant.
Discussion
Identification of epigenetically affected genes has become an important tool for understanding aberrant gene expression and mechanisms of tumorigenesis. Here we report a whole-genome analysis of DNA methylation profiles in fresh, frozen HNSCC tissues and normal adjacent mucosa samples using the Illumina HumanMethylation27 Beadchip with patient genomic DNA. Overall, our findings highlighted many new targets identified across multiple anatomic sites of HNSCC, as well as loci related more specifically to the anatomic site of the tumor, and HPV status of the patient.
With a highly stringent criterion for assessing differential DNA methylation between primary HNSCC tumors and adjacent mucosal tissue, we identified hundreds of significantly altered CpG loci in three anatomic sites of this disease. Of the 28 genes we identified as hypermethylated across all three anatomic sites, many have not been extensively studied in HNSCC and thus represent possible new avenues of research. Among these examples, it has been previously reported that methylation of RLN3R1 has been associated with increased microsatellite instability in endometrial cancer (26). SPARC-like protein 1 (SPARCL1) is an extracellular matrix glycoprotein that has been recently been shown to be an independent prognostic factor in gastric adenocarcinoma patients, with loss of SPARCL1 expression being a negative event for gastric cancer progression and prognosis (27). The novel tumor suppressor gene T-box transcription factor 5 (TBX5) has been shown to be methylated and epigenetically silenced in colon cancer, and patients with TBX5 methylation have a significantly poorer overall survival than other colon cancer patients (28). All of these genes may play equally significant roles in the mechanism of head and neck carcinogenesis.
Our results also showed that epigenetic silencing of the Kruppel family ZNF genes on chromosome 19q13 which was previously identified in oropharyngeal tumors now appears to extend to all anatomic sites of HNSCC, and are among the most frequently hypermethylated genes observed in our study. The role of these proteins in HNSCC carcinogenesis remains unclear. However, we know that ZNF132 has been identified as epigenetically silenced by DNA hypermethylation in prostate cancer, and that reduced ZNF132 immunoreactivity was significantly associated with high Gleason score and advanced T stage in prostate cancer patient cohorts (29). Furthermore, hypermethylation of ZNF154 has recently been identified as part of a panel of early detection biomarkers in DNA from voided urine of bladder cancer patients, along with two other hypermethylated genes (HOXA9 and EOMES) identified in our HNSCC patients (30). And most recently, ZNF545/ZFP82 has been identified as a tumor suppressor gene frequently methylated in multiple primary tumors of nasopharyngeal, esophageal, lung, gastric, colon, and breast, inducing tumor cell apoptosis, repressing ribosome biogenesis and inhibiting both AP-1 and NF-κB signaling (31). The exact role of these transcriptional repressors in HNSCC carcinogenesis will be an area of future study by our group.
Of the hypo-methylated genes we observed across anatomic sites, claudin 18 and the G protein-coupled receptor GPR55 were of most interest. Claudin 18 expression had previously been shown to be affected by DNA methylation in pancreatic cancer cells (20). GPR55 had previously been shown to promote cancer cell proliferation and be expressed in an aggressiveness-related manner in several cancers, suggesting it also might be a target for therapeutic intervention (32, 33).
Overall, the differential methylation events observed in our study also contained significant overlap with previous studies employing the Illumina HumanMethylation27 beadchip. For example, of the 28 hypermethylated CpG loci observed in our study, 27 of these were also observed in at least one of three DNA methylation “clusters” reported previously by Poage and colleagues (34). Only loci cg122238343 (one of two CpG loci corresponding to the RLN3R1gene) was not previously reported. All six ZNF genes identified by our study were also observed in the study by Poage and colleagues, with five of the six genes (ZNF132, ZNF154, ZNF542, ZNF545/ZFP82 and ZNF781) appearing in all three methylation clusters. Of the 16 hypomethylated CpG loci we identified across all three anatomic sites, 13 of these were observed in at least one of three DNA methylation “clusters” reported previously (34). CLDN18 was included in the low methylation cluster, while GPR55 hypomethylation was observed across all three methylation clusters. When looking specifically at a previously published oral cavity SCC dataset, we observed less overlap between our identified gene lists and those described by Guerrero-Preston and colleagues (35). In this case, 48 of the 119 oral cavity hypermethylated genes (40%) and only 11 of the 174 hypomethylated genes (6%) were common to both datasets. However, this was likely due to the fact that this study incorporated gene expression changes and cross-referenced gene lists with known methylation events as part of the selection process (35). In spite of these differences in analytic methods, both HOXA9 and NID2 were also observed in our oral cavity datasets, and both ZNF132 and ZNF154 were observed to be hypermethylated in the Guerrero-Preston oral cavity SCC dataset.
The methylation of the CpG locus associated with the ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) (UCHL1/PGP9.5) gene appeared to be dependent on the anatomic site of the primary tumor. We observed both DNA hyper- and hypo-methylation of UCHL1, with most hypermethylation identified specifically in oral cavity SCC cases. Furthermore, we observed that this differential DNA methylation was directly correlated with expression of the gene. UCHL1 has been observed to be epigenetically regulated in other cancers, and is believed to induce G0/G1 cell cycle arrest and apoptosis by stabilization of p53 (21, 36). Furthermore, the methylation status of UCHL1 has been shown to have prognostic significance in several cancers (37-39). The role of UCHL1 in HNSCC and the mechanisms responsible for its oral-cavity SCC-specific hypermethylation pattern will be explored in future studies.
Profiles of DNA hypermethylation were greatly affected by HPV status in oropharyngeal cancer patients. We observed a significant increase in the level of differential DNA methylation (tumor versus normal) in HPV+ oropharyngeal cases compared to HPV- cases. Much of this was due to the increased DNA hypermethylation of CpG loci in HPV+ cases, corresponding to genes such as CDKN2A and GALR1 (22, 23). Among the 28 most differentially methylated CpGs corresponding to HPV status were two CpG loci corresponding to the insulin promoter factor 1/homeodomain transcription factor (IPF1), a transcriptional activator that is involved in the early development of the pancreas and plays a major role in glucose-dependent regulation of insulin gene expression (40). Overexpression of PDX1 has been previously observed in gastric and esophageal carcinoma and shows a significant correlation with CDX2 expression among phenotypic classifications of gastric carcinomas, thereby suggesting a similar function to that gene (41, 42). Also included in the group of differentially methylated genes correlated to HPV status were three additional homeodomain-containing genes, including ALX4, CUTL2, and HOXA7, as well as previously identified cancer-related genes such as FBX039, IGSF4, PLOD2 and SLITRK3 (43-49). However, the differential methylation of PDX1 and these other genes, and their relationship to the mechanism of carcinogenesis in HPV+ cancers is not yet understood. It is somewhat surprising that of these 28 genes identified in our study, only two (CDKN2A and HOXA7) were also identified in a similar study that examined DNA methylation differences between HPV+ and HPV− HNSCC cell lines (50). This might be attributable to differences when comparing cell lines to whole tissues, or the relatively high levels of DNA methylation observed in many HNSCC cell lines when compared to corresponding patient samples (51). Our analysis also filtered CpG loci based on the magnitude of the observed methylation changes; removal of that magnitude filter would have expanded the overlap to include loci corresponding to DIO2, CCNA1, EREG, IL6ST, COL5A2 and C3orf14.
A study of this type involving human specimens with high-throughput profiling platforms can be susceptible to measurement bias from a variety of sources. First, we are aware of the fact that a portion of the DNA methylation and gene expression signals we collected in this study contained measurement error due to non-tumor content in the processed tissue sample. While samples containing less than 40% tumor were included in the initial identification of hyper- and hypo-methylated CpG loci, these samples were excluded from subsequent analysis for the nodal metastasis signature and HPV status. Furthermore, we confirmed that: i) there were no statistically significant associations between any of the CpG loci identified in this manuscript and the percentage tumor content, and ii) there were no statistically significant associations between any clinical parameters (tumor stage, HPV status, patient outcome) and percentage of tumor content for these primary HNSCC samples. Second, we are aware of the potential for measurement bias due to the presence of methylation and genetic alterations in histologically normal mucosa adjacent to primary HNSCC tumors. The normal tissue samples in this study included both samples taken from sites adjacent to the primary tumor, as well as those taken from the contralateral side. We therefore confirmed that for all of the CpG loci included in this manuscript, there were no significant associations between methylation (M value) measurements and the source of normal tissue (adjacent normal tissue versus contralateral side).
In summary, our genome-wide survey of differential DNA methylation has revealed many new avenues of exploration as potential mechanisms promoting tumorigenesis and influencing tumor behavior in this disease, including many novel genes not previously studied in this disease. Furthermore, we have solidified the significance of HPV as a factor correlating with increased changes in DNA methylation in primary HNSCC tumors. It is hoped that further elucidation of these pathways can be useful both as potential diagnostic and prognostic biomarkers and as potential therapeutic targets for this disease in the future.
Supplementary Material
Acknowledgements
The authors would like to thank the Albert Einstein College of Medicine Genomics Core for help with the Sequenom EpiTyper validation assays for cytosine methylation. This work was supported by grant R21CA131648 (TJB) from the National Cancer Institute. DNA methylation and gene expression data for all of the tissue samples used in this study have been deposited into the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/).
Footnotes
Translation Relevance
Squamous cell cancers of the head and neck constitute an anatomically heterogeneous group of solid tumors arising from the oral cavity, oropharynx, hypopharynx, larynx and nasopharynx. Certain anatomic sites have a predilection for early metastasis and treatment planning depends almost entirely upon anatomical staging of the disease at presentation. Relatively few molecular markers have been found that can be reliably used either in early detection of head and neck cancer, or as indicators of prognosis. Therefore, better molecular classification of head and neck tumors is required to provide prognostic as well as mechanistic information to improve patient care.
Disclosure of Potential Conflicts of Interest
There are no potential conflicts of interest to disclose.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Belbin TJ, Singh B, Barber I, Socci N, Wenig B, Smith R, et al. Molecular classification of head and neck squamous cell carcinoma using cDNA microarrays. Cancer research. 2002;62:1184–90. [PubMed] [Google Scholar]
- 3.Takes RP, Baatenburg de Jong RJ, Schuuring E, Hermans J, Vis AA, Litvinov SV, et al. Markers for assessment of nodal metastasis in laryngeal carcinoma. Archives of otolaryngology--head & neck surgery. 1997;123:412–9. doi: 10.1001/archotol.1997.01900040048008. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA: a cancer journal for clinicians. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. The lancet oncology. 2006;7:77–82. doi: 10.1016/S1470-2045(05)70540-4. [DOI] [PubMed] [Google Scholar]
- 6.Shaw R. The epigenetics of oral cancer. International journal of oral and maxillofacial surgery. 2006;35:101–8. doi: 10.1016/j.ijom.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Reboiras-Lopez MD, Gandara Rey JM, Garcia-Garcia A. Genetic and molecular alterations associated with oral squamous cell cancer (Review) Oncology reports. 2009;22:1277–82. doi: 10.3892/or_00000565. [DOI] [PubMed] [Google Scholar]
- 8.Zuo C, Ai L, Ratliff P, Suen JY, Hanna E, Brent TP, et al. O6-methylguanine-DNA methyltransferase gene: epigenetic silencing and prognostic value in head and neck squamous cell carcinoma. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2004;13:967–75. [PubMed] [Google Scholar]
- 9.Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, et al. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3164–71. [PubMed] [Google Scholar]
- 10.Kudo Y, Kitajima S, Ogawa I, Hiraoka M, Sargolzaei S, Keikhaee MR, et al. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous beta-catenin. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:5455–63. doi: 10.1158/1078-0432.CCR-04-0372. [DOI] [PubMed] [Google Scholar]
- 11.Calmon MF, Colombo J, Carvalho F, Souza FP, Filho JF, Fukuyama EE, et al. Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer genetics and cytogenetics. 2007;173:31–7. doi: 10.1016/j.cancergencyto.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Kong WJ, Liu Z. Promoter hypermethylation of DNA repair gene O6-methylguanine DNA methyltransferase in laryngeal squamous cell carcinoma. Zhonghua er bi yan hou ke za zhi. 2004;39:152–6. [PubMed] [Google Scholar]
- 13.Sasiadek MM, Stembalska-Kozlowska A, Smigiel R, Ramsey D, Kayademir T, Blin N. Impairment of MLH1 and CDKN2A in oncogenesis of laryngeal cancer. British journal of cancer. 2004;90:1594–9. doi: 10.1038/sj.bjc.6601679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lleras RA, Adrien LR, Smith RV, Brown B, Jivraj N, Keller C, et al. Hypermethylation of a cluster of Kruppel-type zinc finger protein genes on chromosome 19q13 in oropharyngeal squamous cell carcinoma. The American journal of pathology. 2011;178:1965–74. doi: 10.1016/j.ajpath.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlecht NF, Brandwein-Gensler M, Nuovo GJ, Li M, Dunne A, Kawachi N, et al. A comparison of clinically utilized human papillomavirus detection methods in head and neck cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:1295–305. doi: 10.1038/modpathol.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mancuso FM, Montfort M, Carreras A, Alibes A, Roma G. HumMeth27QCReport: an R package for quality control and primary analysis of Illumina Infinium methylation data. BMC research notes. 2011;4:546. doi: 10.1186/1756-0500-4-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, et al. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30:245–52. doi: 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Kojima T, Yamaguchi H, Kyuno D, Kimura Y, Imamura M, et al. Transcriptional regulation of claudin-18 via specific protein kinase C signaling pathways and modification of DNA methylation in human pancreatic cancer cells. Journal of cellular biochemistry. 2011;112:1761–72. doi: 10.1002/jcb.23095. [DOI] [PubMed] [Google Scholar]
- 21.Tokumaru Y, Yamashita K, Kim MS, Park HL, Osada M, Mori M, et al. The role of PGP9.5 as a tumor suppressor gene in human cancer. International journal of cancer Journal international du cancer. 2008;123:753–9. doi: 10.1002/ijc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao G, Chernock RD, Gay HA, Thorstad WL, Zhang TR, Wang H, et al. A novel RTPCR method for quantification of human papillomavirus transcripts in archived tissues and its application in oropharyngeal cancer prognosis. International journal of cancer Journal international du cancer. 2012 doi: 10.1002/ijc.27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanazawa T, Misawa K, Carey TE. Galanin receptor subtypes 1 and 2 as therapeutic targets in head and neck squamous cell carcinoma. Expert opinion on therapeutic targets. 2010;14:289–302. doi: 10.1517/14728221003598922. [DOI] [PubMed] [Google Scholar]
- 24.Ma J, Wang JD, Zhang WJ, Zou B, Chen WJ, Lam CS, et al. Promoter hypermethylation and histone hypoacetylation contribute to pancreatic-duodenal homeobox 1 silencing in gastric cancer. Carcinogenesis. 2010;31:1552–60. doi: 10.1093/carcin/bgq140. [DOI] [PubMed] [Google Scholar]
- 25.Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, et al. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Molecular oncology. 2011;5:61–76. doi: 10.1016/j.molonc.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang YW, Luo J, Weng YI, Mutch DG, Goodfellow PJ, Miller DS, et al. Promoter hypermethylation of CIDEA, HAAO and RXFP3 associated with microsatellite instability in endometrial carcinomas. Gynecologic oncology. 2010;117:239–47. doi: 10.1016/j.ygyno.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li P, Qian J, Yu G, Chen Y, Liu K, Li J, et al. Down-regulated SPARCL1 is associated with clinical significance in human gastric cancer. Journal of surgical oncology. 2012;105:31–7. doi: 10.1002/jso.22025. [DOI] [PubMed] [Google Scholar]
- 28.Yu J, Ma X, Cheung KF, Li X, Tian L, Wang S, et al. Epigenetic inactivation of T-box transcription factor 5, a novel tumor suppressor gene, is associated with colon cancer. Oncogene. 2010;29:6464–74. doi: 10.1038/onc.2010.370. [DOI] [PubMed] [Google Scholar]
- 29.Abildgaard MO, Borre M, Mortensen MM, Ulhoi BP, Torring N, Wild P, et al. Downregulation of zinc finger protein 132 in prostate cancer is associated with aberrant promoter hypermethylation and poor prognosis. International journal of cancer Journal international du cancer. 2012;130:885–95. doi: 10.1002/ijc.26097. [DOI] [PubMed] [Google Scholar]
- 30.Reinert T, Modin C, Castano FM, Lamy P, Wojdacz TK, Hansen LL, et al. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:5582–92. doi: 10.1158/1078-0432.CCR-10-2659. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Y, Liang P, Geng H, Wang Z, Li L, Cheng SH, et al. A novel 19q13 nucleolar zinc finger protein suppresses tumor cell growth through inhibiting ribosome biogenesis and inducing apoptosis but is frequently silenced in multiple carcinomas. Molecular cancer research : MCR. 2012;10:925–36. doi: 10.1158/1541-7786.MCR-11-0594. [DOI] [PubMed] [Google Scholar]
- 32.Hu G, Ren G, Shi Y. The putative cannabinoid receptor GPR55 promotes cancer cell proliferation. Oncogene. 2011;30:139–41. doi: 10.1038/onc.2010.502. [DOI] [PubMed] [Google Scholar]
- 33.Pineiro R, Maffucci T, Falasca M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene. 2011;30:142–52. doi: 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- 34.Poage GM, Butler RA, Houseman EA, McClean MD, Nelson HH, Christensen BC, et al. Identification of an epigenetic profile classifier that is associated with survival in head and neck cancer. Cancer research. 2012;72:2728–37. doi: 10.1158/0008-5472.CAN-11-4121-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerrero-Preston R, Soudry E, Acero J, Orera M, Moreno-Lopez L, Macia-Colon G, et al. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev Res (Phila) 2011;4:1061–72. doi: 10.1158/1940-6207.CAPR-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang T, Li L, Yin X, Yuan C, Tan C, Su X, et al. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PloS one. 2012;7:e29783. doi: 10.1371/journal.pone.0029783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akishima-Fukasawa Y, Ino Y, Nakanishi Y, Miura A, Moriya Y, Kondo T, et al. Significance of PGP9.5 expression in cancer-associated fibroblasts for prognosis of colorectal carcinoma. American journal of clinical pathology. 2010;134:71–9. doi: 10.1309/AJCPRJP39MIDSGBH. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, Zhao M, Zhong J, Shi L, Luo Q, Liu J, et al. Proteomic profiling of proteins associated with lymph node metastasis in colorectal cancer. Journal of cellular biochemistry. 2010;110:1512–9. doi: 10.1002/jcb.22726. [DOI] [PubMed] [Google Scholar]
- 39.Mitsui Y, Shiina H, Hiraki M, Arichi N, Hiraoka T, Sumura M, et al. Tumor suppressor function of PGP9.5 is associated with epigenetic regulation in prostate cancer--novel predictor of biochemical recurrence after radical surgery. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2012;21:487–96. doi: 10.1158/1055-9965.EPI-11-0970. [DOI] [PubMed] [Google Scholar]
- 40.Babu DA, Deering TG, Mirmira RG. A feat of metabolic proportions: Pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Molecular genetics and metabolism. 2007;92:43–55. doi: 10.1016/j.ymgme.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oz Puyan F, Can N, Ozyilmaz F, Usta U, Sut N, Tastekin E, et al. The relationship among PDX1, CDX2, and mucin profiles in gastric carcinomas; correlations with clinicopathologic parameters. Journal of cancer research and clinical oncology. 2011;137:1749–62. doi: 10.1007/s00432-011-1044-7. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi O, Hamada J, Abe M, Hata S, Asano T, Takahashi Y, et al. Dysregulated expression of HOX and ParaHOX genes in human esophageal squamous cell carcinoma. Oncology reports. 2007;17:753–60. [PubMed] [Google Scholar]
- 43.Chang H, Mohabir N, Done S, Hamel PA. Loss of ALX4 expression in epithelial cells and adjacent stromal cells in breast cancer. Journal of clinical pathology. 2009;62:908–14. doi: 10.1136/jcp.2009.067298. [DOI] [PubMed] [Google Scholar]
- 44.Tanzer M, Balluff B, Distler J, Hale K, Leodolter A, Rocken C, et al. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PloS one. 2010;5:e9061. doi: 10.1371/journal.pone.0009061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:7296–304. doi: 10.1158/1078-0432.CCR-07-0861. [DOI] [PubMed] [Google Scholar]
- 46.Aruga J, Yokota N, Mikoshiba K. Human SLITRK family genes: genomic organization and expression profiling in normal brain and brain tumor tissue. Gene. 2003;315:87–94. doi: 10.1016/s0378-1119(03)00715-7. [DOI] [PubMed] [Google Scholar]
- 47.Kikuchi S, Iwai M, Sakurai-Yageta M, Tsuboi Y, Ito T, Maruyama T, et al. Expression of a splicing variant of the CADM1 specific to small cell lung cancer. Cancer science. 2012;103:1051–7. doi: 10.1111/j.1349-7006.2012.02277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noda T, Yamamoto H, Takemasa I, Yamada D, Uemura M, Wada H, et al. PLOD2 induced under hypoxia is a novel prognostic factor for hepatocellular carcinoma after curative resection. Liver international : official journal of the International Association for the Study of the Liver. 2012;32:110–8. doi: 10.1111/j.1478-3231.2011.02619.x. [DOI] [PubMed] [Google Scholar]
- 49.Song MH, Ha JC, Lee SM, Park YM, Lee SY. Identification of BCP-20 (FBXO39) as a cancer/testis antigen from colon cancer patients by SEREX. Biochemical and biophysical research communications. 2011;408:195–201. doi: 10.1016/j.bbrc.2011.02.077. [DOI] [PubMed] [Google Scholar]
- 50.Sartor MA, Dolinoy DC, Jones TR, Colacino JA, Prince ME, Carey TE, et al. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics : official journal of the DNA Methylation Society. 2011;6:777–87. doi: 10.4161/epi.6.6.16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, et al. A systematic profile of DNA methylation in human cancer cell lines. Cancer research. 2003;63:1114–21. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.