

Abstract
The Janus kinase inhibitor tofacitinib is currently being investigated as a disease-modifying agent in rheumatoid arthritis (RA). We investigated the in-vivo effects of tofacitinib treatment for 4 weeks on elevated circulating acute-phase serum amyloid (SAA) levels in 14 Japanese patients with RA. SAA levels fell from 110·5 ± 118·5 μg/ml (mean ± standard deviation) at treatment initiation to 15·3 ± 13·3 μg/ml after 4 weeks treatment with tofacitinib. The reduction in SAA levels was greater in patients receiving tofacitinib plus methotrexate compared with those receiving tofacitinib monotherapy. Tofacitinib was also associated with reduced serum interleukin (IL)-6, but had no effect on serum levels of soluble IL-6 receptor. Patients were divided into groups with adequate (normalization) and inadequate SAA responses (without normalization). Serum IL-6 levels were reduced more in the group with adequate SAA response compared with those with inadequate SAA response. These results suggest that tofacitinib down-regulates the proinflammatory cytokine, IL-6, accompanied by reduced serum SAA levels in patients with active RA. The ability to regulate elevated serum IL-6 and SAA levels may explain the anti-inflammatory activity of tofacitinib.
Keywords: interleukin-6, Janus kinase, rheumatoid arthritis, serum amyloid A, tofacitinib
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by persistent synovitis, systemic inflammation and ultimately by joint destruction [1]. Several different cellular responses are involved in the pathogenesis of RA, including activation of immune-inflammatory cells and the production of proinflammatory cytokines from these cells [2]. Biological disease-modifying anti-rheumatic drugs (DMARDs) may interact with the biological targets such as circulating cytokines, including tumour necrosis factor (TNF)-α and interleukin (IL)-6 or their receptors, or with co-stimulatory cell surface molecules such as CD80/CD86 [3]. However, further treatments are needed for patients with an inadequate response to methotrexate (MTX) and these biological DMARDs [4].
Small-molecule inhibitors of signalling mediators have been introduced recently, with intracellular targets such as the Janus kinase (JAK) family of tyrosine kinases [5]. Tofacitinib (CP-690, 550) is a novel oral JAK inhibitor developed as a targeted immunomodulator and disease-modifying therapy for RA [6]. Tofacitinib plus MTX has produced rapid and clinically meaningful improvements in patients refractory to treatment with MTX or TNF-blockers, suggesting that it might represent an effective treatment option for refractory RA [7–11].
C-reactive protein (CRP) and acute-phase serum amyloid (SAA) are the major acute-phase reactants in RA, but have different functions and are regulated differentially [12]. Previous studies have demonstrated that SAA has a wider dynamic range and may be the better marker for assessing inflammatory joint disease [13]. IL-6 is one of the major factors affecting the induction of SAA in RA, and monoclonal antibodies to IL-6R-mediated signalling have proved clinically beneficial in patients with AA amyloidosis, by normalizing elevated SAA levels [14]. Binding of IL-6 to the IL-6 receptor (IL-6R) results in the activation of JAK-1/JAK-2, which in turn results in the recruitment of signal transducer and activator of transcription-3 (STAT-3) [15]. JAK-mediated phosphorylation of STAT-3) is crucial for its nuclear translocation and for activating transcription of SAA [16]. We previously demonstrated that CP690,550 abrogated IL-6-mediated A-SAA mRNA expression by preventing JAK-2/STAT-3 activation in rheumatoid synoviocytes [17]. In the current study, we investigated the in-vivo effects of tofacitinib on circulating SAA levels in Japanese patients with active RA.
Material and methods
Patients
Eligible patients were age ≥18 years with a diagnosis of active RA, based on the American College of Rheumatology 1987 revised criteria. Active disease was defined as ≥ 6 tender/painful joints (68-joint count) and ≥ 6 swollen joints (66-joint count), and an erythrocyte sedimentation rate (ESR) (Westergren method) > 28 mm/h or a CRP level of > 7 mg/l (reference range 0–10 mg/l). Patients were also required to have evidence of ≥ 3 distinct joint erosions on posteroanterior hand and wrist radiographs or anteroposterior foot radiographs, as determined by the investigator. If radiographic evidence of joint erosions was unavailable, immunoglobulin (Ig)M rheumatoid factor (RF) positivity or the presence of antibodies to cyclic citrullinated peptide were considered as acceptable evidence.
Study design and treatment
This study was conducted at multiple centres in Japan, in compliance with the ethical principles of the Declaration of Helsinki and with all International Conference on Harmonization Good Clinical Practice guidelines. The study is registered with ClinicalTrials.gov. The study protocol was approved by the institutional review board and/or independent ethics committee of each study centre, and the patients provided written informed consent.
One included study was a Phase III, randomized, double-blind, placebo-controlled study of tofacitinib in combination with MTX (Pfizer protocol A3921044, NCT00687193) conducted in North America, South America, Europe, Asia and Australia between 31 March 2009 and 1 April 2011. This analysis included 12-month data for all patients. Patients were randomized in a ratio of 4:4:1:1 to one of four treatments: tofacitinib 5 mg twice daily; tofacitinib 10 mg twice daily; placebo to tofacitinib 5 mg twice daily; and placebo to tofacitinib 10 mg twice daily, all combined with stable doses of MTX. MTX doses were 15–25 mg weekly for ≥6 weeks, although stable doses < 15 mg were allowed if there were safety issues at higher doses. Stable doses of low-dose corticosteroids (≤ 10 mg/day prednisone or equivalent) and non-steroidal anti-inflammatory drugs were also allowed. Prior use of biological or non-biological DMARDs was permitted.
A second included study was a Phase III, randomized, double-blind, placebo-controlled trial of tofacitinib monotherapy (Pfizer protocol A3921040, NCT00847613). Patients were assigned randomly in a 1:1:1:1:1:1 ratio to 1, 3, 5, 10 or 15 mg of tofacitinib twice daily or placebo for 3 months.
Procedures
Consecutive pairs of sera (before and after 4 weeks) were taken for each patient and assessed without knowledge of the patient's identity, treatment or laboratory results. No patient recruited into the study was lost before the baseline assessment. SAA concentrations were determined by the latex agglutination nephelometric immunoassay test (LZ test ‘Eiken’ SAA; Eiken Kagaku Co. Ltd, Tokyo, Japan). Serum IL-6 and soluble IL-6 receptor (sIL-6R) levels were measured using a sensitive sandwich enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems Inc., Minneapolis, MN, USA). The detection limit of the IL-6 ELISA assay is 0·04 pg/ml in sera. All measurements were made in duplicate, and the average values were used in the statistical analyses.
Statistical analysis
Laboratory data were expressed as mean ± standard deviation (s.d.) of tested samples. Statistical analysis used the paired t-test. The overall effect of tofacitinib was evaluated with the paired t-test on the basis of changes from baseline level observed after tofacitinib administration. Data were analysed using spss software (SPSS Inc., Chicago, IL, USA). P-values less than 0·05 were considered significant.
Results
Subjects
Fifteen subjects with active RA were enrolled. All enrolled subjects completed all the planned evaluations for efficacy and safety analyses. The 15 patients included 10 from study 3920144 and five from study A3921040. One patient enrolled in the A3921040 study received placebo, and the remaining 14 patients received various doses of tofacitinib. The patient who received placebo was excluded following the analysis of pretreatment and post-treatment sera. The baseline characteristics of the RA patients who received tofacitinib are shown in Table 1. The mean age of the patients ranged from 26–62 years and the mean duration of RA ranged from 4 to 27 years. A total of 92·9% (13 of 14) of the patients were women. The mean baseline disease-activity score (DAS)28 (CRP) was 5·66 ± 0·77, indicating that the patients enrolled in the trial had moderate or high RA disease activities. Baseline C-reactive protein (CRP) levels were 1·34 ± 1·05 mg/dl in the enrolled patients.
Table 1.
Baseline characteristics of the patients
| Tofacitinib (n = l4) | |
|---|---|
| Female sex, n (%) | 13 (92·9) |
| Age (years) | 47·8 ± 9·5 |
| Stature (cm) | 158·0 ± 6·5 |
| Body weight (kg) | 57·2 ± 12·0 |
| Stage | |
| II | 3 |
| III | 5 |
| IV | 6 |
| Duration of rheumatoid arthritis (years) | |
| Mean (range) | 10·6 ± 5·9 (4–27) |
| Tender and swollen joints, mean n | |
| Tender | 30·4 ± 16·4 |
| Swollen | 24·3 ± 12·5 |
| DAS28 (CRP) | 5·66 ± 0·77 (4·05–6·46) |
| Erythrocyte sedimentation rate (mm/h, range) | 32·1 ± 14·3 (15–56) |
| C-reactive protein (mg/dl, range | 1·34 ± 1·05 (0·23–3·84) |
| Prior treatment | |
| TNF inhibitor – n (%) | 2 (14·3) |
| IL-6 inhibitor – n (%) | 1 (7·1) |
| Prednisolone – n (%) | 14 (100) |
| Dosage (mg/day) | 6·2 ± 2·5 |
| Methotrexate – n (%) | 10 (71·4) |
| Dosage (mg/week) | 7·8 ± 1·8 |
CRP: C-reactive protein; IL-6: interleukin-6; TNF: tumour necrosis factor.
SAA values at treatment initiation and after 4 weeks
We have demonstrated previously that tofacitinib abrogated the A-SAA mRNA expression by inhibiting IL-6-mediated JAK-2/STAT-3 activation in rheumatoid synovial fibroblasts. We therefore measured serum SAA levels after 4 weeks of tofacitinib treatment and compared them with pretreatment levels using SAA-specific latex agglutination nephelometric immunoassay. As shown in Fig. 1, baseline levels of SAA were elevated (mean ± s.d.; 110·5 ± 118·5 μg/l). Significant reductions in SAA occurred after 4 weeks' treatment with different doses of tofacitinib (15·3 ± 13·3 μg/ml). In patients receiving background MTX, SAA levels fell from 136·4 ± 132·0 μg/ml at treatment initiation to 12·8 ± 12·0 μg/ml after 4 weeks. In patients receiving tofacitinib alone (without MTX), SAA levels fell from 45·9 ± 28·3 μg/ml at treatment initiation to 21·4 ± 16·4 μg/ml after 4 weeks' treatment. The reduction in SAA levels was greater in the MTX plus tofacitinib group compared with the tofacitinib alone group (Fig. 2).
Fig. 1.
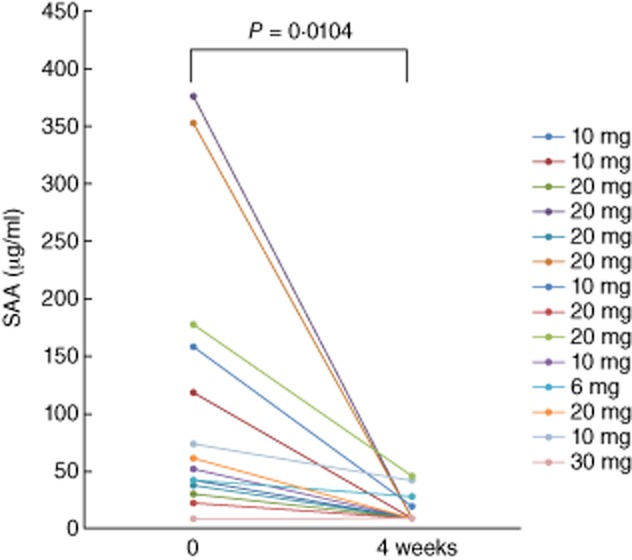
The serum levels of acute-phase serum amyloid (SAA) from rheumatoid arthritis (RA) patients receiving tofacitinib treatment. SAA was measured before and at 4 weeks after tofacitinib treatment. There was a significant difference between the SAA levels before and at 4 weeks.
Fig. 2.
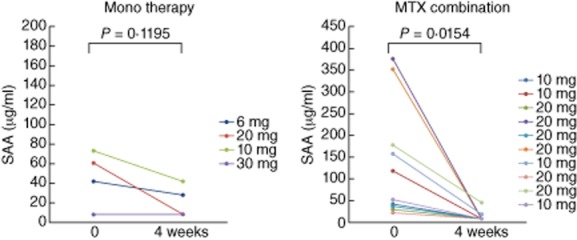
The serum levels of acute-phase serum amyloid (SAA) from rheumatoid arthritis (RA) patients receiving methotrexate (MTX) plus tofacitinib or tofacitinib alone. SAA was measured before and at 4 weeks after tofacitinib treatment.
Serum IL-6 values at treatment initiation and after 4 weeks
IL-6 is a proinflammatory cytokine, which induces SAA. We measured serum IL-6 levels after 4 weeks of tofacitinib treatment and compared them with pretreatment levels using IL-6-specific ELISA kits. As shown in Fig. 3, baseline levels of IL-6 were elevated (mean ± s.d.; 39·2 ± 38·6 pg/ml), but significant reductions in IL-6 occurred after 4 weeks treatment with different doses of tofacitinib (7·9 ± 8·9 pg/ml).
Fig. 3.
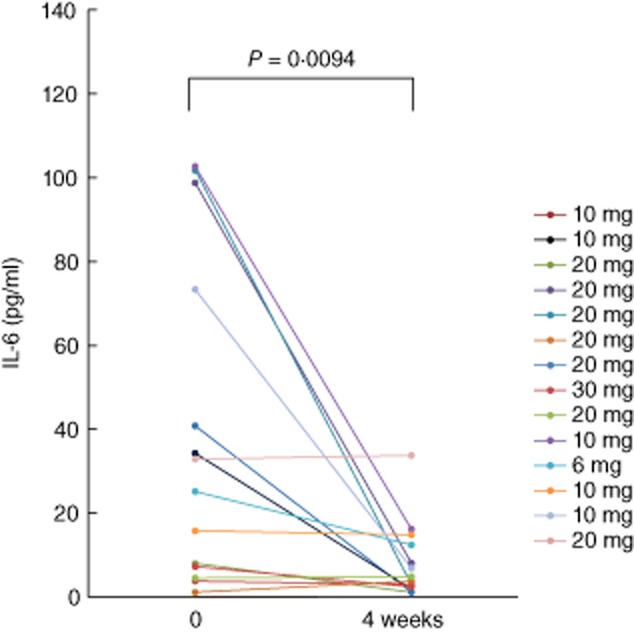
The serum levels of interleukin (IL)-6 from rheumatoid arthritis (RA) patients receiving tofacitinib treatment. IL-6 was measured before and at 4 weeks after tofacitinib treatment. There was a significant difference between the IL-6 levels before and at 4 weeks.
We further characterized the relationship between SAA and serum IL-6 by grouping patients according to SAA normalization (<8 μg/ml) during treatment with tofacitinib. Among the RA patients who received tofacitinib, 71·4% (10 of 14) had an adequate decline in SAA (<8 μg/ml, normal range), whereas 28·6% (four of 14) had an inadequate SAA decline. Changes in serum IL-6 levels were compared between both groups. Serum IL-6 levels fell from 51·6 ± 11·6 pg/ml at treatment initiation to 11·5 ± 7·1 pg/ml after 4 weeks in the SAA normalization group, and from 51·7 ± 11·4 pg/ml to 21·5 ± 11·7 pg/ml in the group without SAA normalization (Fig. 4). The decrease of serum IL-6 levels was greater in the group with SAA normalization. Similarly, the reduction of DAS28 (CRP) was greater in the group without SAA normalization compared to the group without SAA normalization (Fig. 5).
Fig. 4.
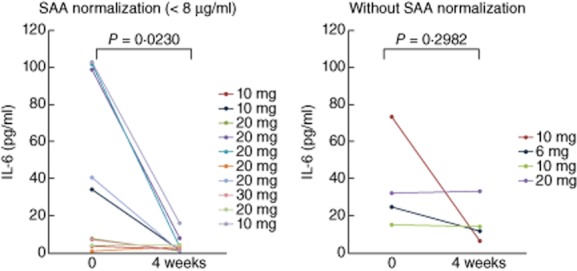
The serum levels of interleukin (IL)-6 from rheumatoid arthritis (RA) patients with or without acute-phase serum amyloid (SAA) normalization. RA patients receiving tofacitinib were divided into patients with an adequate decline in SAA (normalization; <8 μg/ml) and patients with an inadequate SAA decline (without normalization, ≥8 μg/ml) at 4 weeks. SAA was measured before and at 4 weeks after tofacitinib treatment in both patient groups. There was a significant difference between the IL-6 levels before and at 4 weeks in the patients with SAA normalization.
Fig. 5.
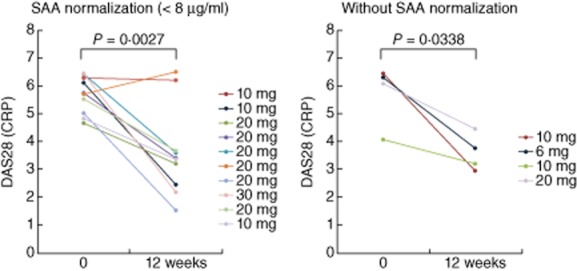
DAS28 [C-reactive protein (CRP)] in from rheumatoid arthritis (RA) patients with or without acute-phase serum amyloid (SAA) normalization. RA patients receiving tofacitinib were divided into patients with an adequate decline in SAA (normalization; <8 ug/ml) and patients with an inadequate SAA decline (without normalization, ≥8 μg/ml) at 4 weeks. DAS28 (CRP) was measured before and at 12 weeks after tofacitinib treatment in both patient groups. There was a significant difference between DAS28 (CRP) before and at 12 weeks in the patients with SAA normalization.
Circulating and soluble IL-6R during tofacitinib treatment
Changes in circulating and soluble IL-6R (sIL-6R) levels are considered to be clinically meaningful, because IL-6/sIL-6R signalling plays an important role in SAA induction in inflammatory diseases. However, tofacitinib did not reduce the serum sIL-6R levels after 4 weeks' treatment with tofacitinib (Fig. 6).
Fig. 6.
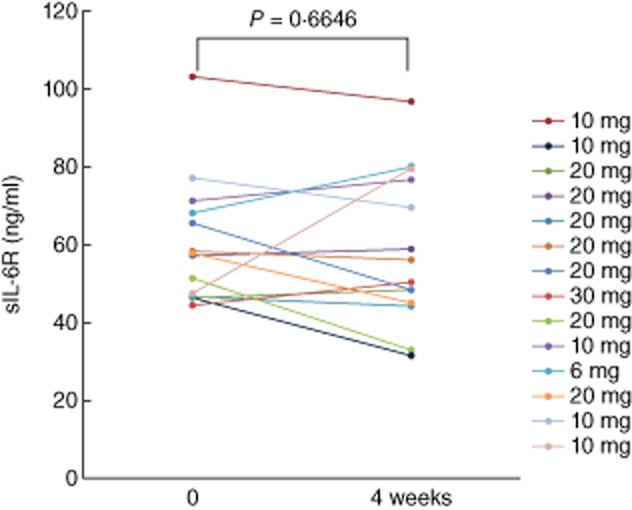
The serum levels of soluble interleukin (sIL)-6R from rheumatoid arthritis (RA) patients receiving tofacitinib treatment. sIL-6 receptor (sIL-6R) was measured before and at 4 weeks after tofacitinib treatment. There was no significant difference between the sIL-6R levels before and at 4 weeks.
Discussion
Tofacitinib is a potent, orally active small-molecule inhibitor of JAK with specificities for JAK-1, JAK-2 and JAK-3 at clinically relevant concentrations [18]. Although tofacitinib has demonstrated an ability to control rheumatoid synovitis, its immunological mechanism of action in active arthritis remains unclear. In the present study, we investigated the effects of tofacitinib on serum levels of acute-phase reactants. Serial laboratory measurements revealed a reduction in the acute-phase protein SAA following tofacitinib administration. Several cytokines contribute to the induction of the acute-phase response, of which IL-6 is of particular importance [19]. Serum IL-6 is elevated in RA patients and correlates with rheumatoid disease activity [20]. In the current study, tofacitinib reduced high baseline circulating IL-6 levels in RA patients in parallel with the fall in circulating SAA levels. This is the first evidence to demonstrate the effective regulation of IL-6 and the acute-phase reactant SAA by tofacitinib in patients with active RA.
Although the number of the cases is limited, in patients with SAA normalization after tofacitinib treatment the reduction of serum IL-6 levels and DAS28 was greater compared to those without SAA normalization. The correlation between serum levels of SAA and IL-6 was demonstrated in RA patients [21]. Recent studies also demonstrated that A-SAA levels were correlated significantly with clinical disease activity in inflammatory arthritis [20]. These findings may contribute to our data, in which normalization of SAA was associated with the decrease of circulating IL-6 levels or RA disease activity after tofacitinib treatment. Additionally, there was heterogeneity in the changes of serum IL-6 levels after tofacitinib. In a few patients, the reduction of serum IL-6 was marginal. Elevated serum levels of IL-6 in RA patients were shown to be decreased in responders for biological treatments [22]. In patients with an inadequate response to tofacitinib or patients without elevated IL-6 levels at the baseline, the changes of serum IL-6 could be minimal after tofacitinib treatments, compared to those with adequate response.
In general, the interaction between a cytokine and its membrane receptor results in intracellular signalling that triggers a cascade involving activation of JAKs followed by phosphorylation of STAT proteins [15]. Tofacitinib was first identified as an inhibitor of JAK-3, which is involved in signal transduction by the cytokine receptor family employing the common γ chain (IL-2R, IL-4R, IL-7R, IL-15R, IL-9R, IL-21R). These receptors are essential for activation of T cells [5,23]. In addition to the T cell-associated cytokines, JAK are also associated with other cytokine receptors such as IL-6, IL-12 and IL-23 in non-lymphoid cells [24], including macrophages and synoviocytes. We previously demonstrated that treatment of synoviocytes with tofacitinib reduced expression of the IL-6-mediated acute-phase reactant, SAA, by blocking JAK-2/STAT-3 signalling [17]. The combined inhibition of JAK-1/-2/-3 with tofacitinib may account for its anti-inflammatory activity against IL-6-mediated proinflammatory pathways.
Compounds that inhibit tyrosine kinase pathways involved in cellular signalling have been shown to be effective for the treatment of RA [5]. Tofacitinib, formerly designated CP690,550, is one of the JAK inhibitors that has entered clinical practice for RA and has been approved by the Food and Drug Administration (FDA) and Pharmaceuticals and Medical Devices Agency, Japan (PMDA), to treat patients with moderately active RA who have had an inadequate response to or who are intolerant of MTX. Tofacitinib potentially inhibits cγ cytokines, but also blocks interferon (IFN)-γ and IL-6 [25]. Indeed, we demonstrated that tofacitinib treatment reduced elevated SAA levels in RA patients, probably by interrupting an IL-6-mediated STAT-3 signalling pathway required for SAA induction. However, tofacitinib unexpectedly also suppressed elevated IL-6 levels in patients with active RA.
The precise anti-inflammatory mechanisms of tofacitinib remain unclear. IL-6 plays a pivotal role in the pathogenesis of RA, and anti-IL-6 receptor antibodies have been shown to be therapeutically useful in RA [26]. The mechanism of tofacitinib-induced down-regulation of circulating IL-6 in RA patients remains unknown. A previous study showed that tofacitinib inhibited human IL-6 derived from RA synovium implanted in severe combined immunodeficient (SCID) mice [27]. However, it did not directly affect IL-6 production by RA synoviocytes, CD14+ monocytes and CD4+ T cells. Meanwhile, IL-6 production from synovial fibroblasts was inhibited by conditioned medium from CD4+ T cells cultured with tofacitinib, indicating an indirect effect of tofacitinib on rheumatoid synovial fibroblasts [27]. More recently, Yarilina et al. investigated the effects of tofacitinib on IFN signalling using rheumatoid synovial macrophages and found that tofacitinib suppressed the TNF responses by interfering with nuclear factor-κB signalling, resulting in the inhibition of IL-6 production [28]. These findings suggest that tofacitinib may suppress IL-6 induction by affecting the TNF-mediated innate immune response. Similarly, tofacitinib treatment resulted in significant and rapid decreases in both plasma and tissue levels of IL-6 in a rat adjuvant-induced arthritis model [29]. The ability of tofacitinib to modulate innate responses to lipopolysaccharide (LPS) or IFN-γ by affecting STAT-1 signalling has been demonstrated [26]. It is possible that direct or indirect inhibition of cytokine-dependent IL-6 production may play a role in the anti-inflammatory activity of tofacitinib. Many cytokines are elevated in RA patients, which then act on macrophages and innate immune cells that are implicated in the pathogenesis of RA [1,2]. Given that IL-6 blockade is an effective therapy for RA, IL-6 represents a candidate for explaining the efficacy of tofacitinib. These results raise the possibility that inhibition of IL-6 production, as well as IL-6-mediated signalling, may help to explain the therapeutic efficacy of tofacitinib.
In conclusion, tofacitinib can reduce serum levels of SAA in patients with active RA. In addition, tofacitinib may also modulate serum levels of the proinflammatory cytokine IL-6. The immunomodulatory activity of tofacitinib against these proinflammatory molecules provides a mechanistic basis to support the anti-inflammatory properties of this small-molecule compound.
Acknowledgments
This work was supported by a grant from the Japan Society for the Promotion of Science (Grant-in-Aid for Scientist Research C23591452).
Disclosures
The authors declare that they have no competing interests.
References
- 1.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 2.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 3.Buch MH, Emery P. New therapies in the management of rheumatoid arthritis. Curr Opin Rheumatol. 2011;23:245–251. doi: 10.1097/BOR.0b013e3283454124. [DOI] [PubMed] [Google Scholar]
- 4.Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. 2013;9:154–163. doi: 10.1038/nrrheum.2013.8. [DOI] [PubMed] [Google Scholar]
- 5.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–142. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka Y, Maeshima K, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis. 2012;71:i70–74. doi: 10.1136/annrheumdis-2011-200595. [DOI] [PubMed] [Google Scholar]
- 7.Kremer JM, Bloom BJ, Breedveld FC, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 8.Coombs JH, Bloom BJ, Breedveld FC, et al. Improved pain, physical functioning and health status in patients with rheumatoid arthritis treated with CP-690,550, an orally active Janus kinase (JAK) inhibitor: results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2010;69:413–416. doi: 10.1136/ard.2009.108159. [DOI] [PubMed] [Google Scholar]
- 9.Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- 10.Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- 11.Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64:617–629. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- 12.Malle E, De Beer FC. Human serum amyloid A (SAA) protein: a prominent acute-phase reactant for clinical practice. Eur J Clin Invest. 1996;26:427–435. doi: 10.1046/j.1365-2362.1996.159291.x. [DOI] [PubMed] [Google Scholar]
- 13.Cunnane G, Grehan S, Geoghegan S, et al. Serum amyloid A in the assessment of early inflammatory arthritis. J Rheumatol. 2000;27:58–63. [PubMed] [Google Scholar]
- 14.Okuda Y, Takasugi K. Successful use of a humanized anti-interleukin-6 receptor antibody, tocilizumab, to treat amyloid A amyloidosis complicating juvenile idiopathic arthritis. Arthritis Rheum. 2006;54:2997–3000. doi: 10.1002/art.22118. [DOI] [PubMed] [Google Scholar]
- 15.Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 16.Hagihara K, Nishikawa T, Sugamata Y, et al. Essential role of STAT3 in cytokine-driven NF-kappaB-mediated serum amyloid A gene expression. Genes Cells. 2005;10:1051–1063. doi: 10.1111/j.1365-2443.2005.00900.x. [DOI] [PubMed] [Google Scholar]
- 17.Migita K, Koga T, Komori A, et al. Influence of Janus kinase inhibition on interleukin 6-mediated induction of acute-phase serum amyloid A in rheumatoid synovium. J Rheumatol. 2011;38:2309–2317. doi: 10.3899/jrheum.101362. [DOI] [PubMed] [Google Scholar]
- 18.Migita K, Komori A, Torigoshi T, et al. CP690,550 inhibits oncostatin M-induced JAK/STAT signaling pathway in rheumatoid synoviocytes. Arthritis Res Ther. 2011;13:R72. doi: 10.1186/ar3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, Yoshizaki K. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem Biophys Res Commun. 2004;314:363–369. doi: 10.1016/j.bbrc.2003.12.096. [DOI] [PubMed] [Google Scholar]
- 20.Connolly M, Mullan RH, McCormick J, et al. Acute-phase serum amyloid A regulates tumor necrosis factor α and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012;64:1035–1045. doi: 10.1002/art.33455. [DOI] [PubMed] [Google Scholar]
- 21.van Leeuwen MA, Westra J, Limburg PC, van Riel PL, van Rijswijk MH. Clinical significance of interleukin-6 measurement in early rheumatoid arthritis: relation with laboratory and clinical variables and radiological progression in a three year prospective study. Ann Rheum Dis. 1995;54:674–677. doi: 10.1136/ard.54.8.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimamoto K, Ito T, Ozaki Y, et al. Serum interleukin 6 before and after therapy with tocilizumab is a principal biomarker in patients with rheumatoid arthritis. J Rheumatol. 2013;40:1074–1081. doi: 10.3899/jrheum.121389. [DOI] [PubMed] [Google Scholar]
- 23.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choe JY, Park KY, Park SH, Lee SI, Kim SK. Regulatory effect of calcineurin inhibitor, tacrolimus, on IL-6/sIL-6R-mediated RANKL expression through JAK2-STAT3-SOCS3 signaling pathway in fibroblast-like synoviocytes. Arthritis Res Ther. 2013;15:R26. doi: 10.1186/ar4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. 2012;71:440–447. doi: 10.1136/ard.2011.150284. [DOI] [PubMed] [Google Scholar]
- 26.Schoels MM, van der Heijde D, Breedveld FC, et al. Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta-analysis informing a consensus statement. Ann Rheum Dis. 2013;72:583–589. doi: 10.1136/annrheumdis-2012-202470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeshima K, Yamaoka K, Kubo S, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012;64:1790–1798. doi: 10.1002/art.34329. [DOI] [PubMed] [Google Scholar]
- 28.Yarilina A, Xu K, Chan C, Ivashkiv LB. Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum. 2012;64:3856–3866. doi: 10.1002/art.37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin TH, Hegen M, Quadros E, et al. Selective functional inhibition of JAK-3 is sufficient for efficacy in collagen-induced arthritis in mice. Arthritis Rheum. 2010;62:2283–2293. doi: 10.1002/art.27536. [DOI] [PubMed] [Google Scholar]