

Abstract
Intracerebral haemorrhage (ICH) is a subtype of stroke that associated with neurological dysfunction and inflammation, which may be ameliorated by a neuroprotective strategy targeting the complement cascade. The protective effect of C5a-receptor antagonist (PMX53) solely and in combination with thrombin antagonist (argatroban) was investigated in the ICH mouse model, respectively. Adult male C57BL/6J wild-type (WT) mice and C3–/– mice were randomized to receive PMX53/argatroban 1, 3 and 5 days after ICH. A double injection technique was used to infuse 25 μl of autologous whole blood into the right striatum. Mice in the sham group received only needle insertion. Brain water content and mRNA of inflammatory factors were measured on the first, third and fifth days after ICH, respectively. Neurological dysfunction was assessed using a 28-point neurological scoring system in the three cohorts, namely, on days 1, 3 and 5 following ICH. Animals treated with PMX53/argatroban demonstrated significant improvements in neurological function and fewer neurological apoptosis detected by TUNEL [terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labelling] and βIII-tubulin dual-staining compared with vehicle-treated animals. Compared with sham-treated mice, the brain water content in argatroban/PMX53-treated mice was decreased significantly in both the ipsilateral cortex and ipsilateral striatum. Administration of PMX53/argatroban provided a synergistic neuroprotective effect via reducing inflammatory factors and brain oedema, leading to improvements in neurofunctional outcome. The results of this study indicated that simultaneous blockade of the thrombin and C5a receptors represent a promising neuroprotective strategy in haemorrhagic stroke.
Keywords: argatroban, C5aR, complement, intracerebral haemorrhage, microglia
Introduction
Intracerebral haemorrhage (ICH) is a subtype of stoke that can cause an instantaneous mass effect and early neurological death, which may result in significant morbidity and mortality. Brain injury due to ICH occurs initially within the first few hours as the result of a mass effect due to haematoma formation. However, there is increasing interest in the mechanisms of secondary brain injury, as many patients continue to deteriorate clinically despite no signs of re-haemorrhage or haematoma expansion. Until now, there have been no specific treatment modalities needed for human ICH. It is believed that thrombin (TM), a neurotoxic mediator, plays a major part in acute brain injury after ICH [1,2]. Thrombin is produced in the brain immediately after ICH (either primary or secondary to brain trauma), or after the blood brain barrier (BBB) breakdown that occurs in various kinds of brain injury [3]. Therapies including thrombin inhibitors, N-methyl-D-aspartate antagonists and anti-inflammatory drugs are currently under investigation for alleviating this secondary brain injury [4].
It was found that thrombin functions as a C5 convertase to initiate complement activation and C5a generation that occurs in the absence of C3, providing evidence for a direct linkage between the complement and the clotting pathways. C5a was known as an anaphylatoxin, which exerts its biological effect through interaction with its respective G-protein coupled receptors and the C5a receptor (C5aR) that belongs to the rhodopsin family of 7-transmembrane G-protein-coupled receptor.
Uncontrolled complement activation can lead to excessive tissue inflammation and damage, which contributes to many immune complex-mediated diseases [5]. This suggested that C5a receptor blockade decreased pain behaviours, due partially to prevention of cytokine production in inflammatory, neuropathic and postoperative pain, as well as incision and arthritis models [6,7]. Due to the inflammatory effects of complement activation under pathological conditions, blocking C5a/C5aR signalling pathway may be a favourable modality for the treatment of ICH disorders.
In ICH injury progression, the role of C5aR contributing to the detrimental outcome in brain tissue maintains unclear. In the current study, C5aR antagonist (PMX53) and thrombin antagonist (argatroban) were used to address the role of C5aR and thrombin in ICH pathological injury and a possible way was found to attenuate ICH injury.
Material and methods
Mice
The C57BL/6 wild-type (WT) and C3–/– mice, weighing between 22 and 24 g, purchased from Jackson Laboratories (Bar Harbor, ME, USA), were used in this study. All procedures on animal studies followed the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the National Institute of Health, and all implementations were approved by the Zunyi Hospital of Zunyi Animal Welfare Committee.
The mice were allowed free access to food and water. Animals were maintained at a constant ambient temperature (22 ± 1°C) under a 12-h light/dark cycle. Mice were anaesthetized with 4% chloralic hydras (0·2 ml/20 g) and their body temperature was maintained at 37 ± 0·5°C. They were allowed to recover fully after a procedure in an incubator maintained at 37°C, and were returned to their cages with free access to water and food afterwards.
ICH mouse model
The ICH induction procedure was implemented by the method reported previously [8]. Mice were anaesthetized and placed into a stereotaxic frame (Stoelting, Wood Sale, IL, USA). Approximately 25 μl of autologous blood was collected from the ventral tail artery or lateral tail veins. The withdrawal site was disinfected and a 25 G needle was inserted into the vein; a capillary tube was used to collect the blood from the hub. The cannula was positioned over the entry point (EP) and introduced into the left striatum (co-ordinates: 0·2 mm anterior, 2·3 lateral and 3·5 mm ventral to the bregma). First, 5 μl blood was injected into the target point at a rate of 2 μl/min. For control groups, the injection should be performed using the above-mentioned method, but the actual injection should be avoided. The remaining blood was injected into the target site at a rate of 2 μ/min, 7 min later, and the needle was retained for 10 min. After withdrawing the needle, the drilled hole was closed using bone-wax.
Argatroban and PMX53 treatment
Argatroban (Sigma-Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulphoxide (DMSO) and injected intraperitoneally into the mice at a dose of 9 mg/kg daily before induction of ICH from day –2. The mice then underwent argatroban intraperitoneal injection once a day for 5 days [9,10]. PMX53 (C5aR antagonist; Shanghai Jier Biochemistry Inc., Shanghai, China) has been found to be safe and well tolerated in Phase I clinical trials after either oral or local administration, making these compounds promising tools for further clinical research and development [11]. It was dissolved in normal sterile saline (0·9%) before use [6], and the saline vehicle was prepared in the same manner without adding the drug. PMX53 were administered daily at an oral dose of 10 mg/kg in PMX53-treated mice (n = 9) from day –2 [12], and then once daily for 5 days.
Neurological deficit and behaviour tests
Acute neurological deficits were assessed using a previously described 28-point scoring system [13,14] on days 1, 3 and 5 after ICH injuries. The tests included body symmetry, gait, climbing, circling behaviour, front limb symmetry, compulsory circling and whisker response. Each point was graded from 0 to 4, establishing a minimum deficit score of 0 (no apparent deficits) and maximum deficit score of 28 (apparent deficits). The tests were performed blind by two researchers, respectively.
Western blot
The brain tissue was washed in sterile phosphate-buffered saline (PBS), then lysed in 2% sodium dodecyl sulphate (SDS) (in deionized water) with a protease inhibitor cocktail (118836153001; Roche Diagnostics, Indianapolis, IN, USA). The lysate was then centrifuged at 12 000 g at 4°C for 15 min. The supernatant was collected and the protein concentration was measured using a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA); 35 μg samples were loaded onto 8% SDS-polyacrylamide gels. Thereafter proteins were transferred to polyvinylidene difluoride membranes (Millipore Corporation, Bedford, MA, USA) using a 150 V current for 1·5 h. The membranes were washed with Tris-buffered saline and Tween (TBS-T; 50 mmol/l Tris pH 7·4, 150 mmol/l NaCl and 0·1%Tween), followed by blocking in 5% non-fat milk-TBS-T overnight at 4°C. C5aR (Abcam, Cambridge, UK; ab117579, rat anti-mouse, diluted 1:500) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal antibodies were diluted in a solution of 3% milk-TBS-T, used at 4°C overnight, then washed three times with TBS-T and incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rat immunoglobulin (Ig)G secondary antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in TBS-T for 1·5 h at 25°C. The results were obtained using the chemiluminescent detection system (Super-Signal® West Pico chemiluminescent substrate detection system; Thermo Fisher Scientific Inc., Rockford, IL, USA).
Determination of brain water content (BWC)
Mice were selected randomly from each group and euthanized immediately 3 days post-haemorrhage for assay of BWC, as described previously [14]. Brain tissue was removed and the water on the surface of the two hemispheres was blotted with filter paper. The cortex of each hemisphere was then dissected carefully from the striatum. The cerebellum was separated and retained as a control. Each of the components was then weighed on an electronic analytical balance to determine the wet weights (HW). We then dried them for 48 h at a temperature of 95°C in an electro-Thermo oven and took their dry weights (DW). BWC (%) was calculated by using the equation 100 × (wet HW–DW)/HW (%).
Immunofluorescent staining of peri-haematoma tissue
Tissue sections (30 μm) were incubated overnight at 4°C with mouse monoclonal antibody against CD68 (Abcam; ab31630, mouse anti-mouse, diluted 1:800), which is widely regarded as a reactive marker for microglia [15]. Sections were then washed with PBS and incubated with Alexa Fluor® 488-coupled secondary antibodies (goat anti-mouse IgG, diluted 1:300) at 25°C for 1 h. Sections were incubated further with primary antibody C5aR (Abcam; ab117579, rat anti-mouse, diluted 1:200) followed by a PBS wash and then incubated with Alexa Fluor® 568-coupled secondary antibodies (goat anti-rat IgG, diluted 1:300) at 25°C for 1 h. All primary antibodies were purchased from Abcam, and all secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA). To serve as negative controls, sections were incubated with IgG instead of primary antibodies. 4′, 6-Diamidino-2-phenylindole (DAPI, 0·5 μg/ml) was used as the counterstaining label. Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labelling (TUNEL) and immunohistochemistry dual-label staining βIII-tubulin was used as the primary antibody for neurone detection (Abcam; ab14545, mouse anti-mouse, diluted 1:1000). The sections were then washed with PBS and incubated with Alexa Fluor® 568-coupled secondary antibodies (goat anti-rat IgG, diluted 1:300) at 25°C for 1 h, followed by TUNEL staining.
TUNEL
TUNEL assay was performed with a commercial kit that labels DNA strand breaks with fluorescein isothiocyanate (In Situ Cell Death Detection Kit; Roche Molecular Biochemicals, Mannheim, Germany). Sections were rinsed twice and pretreated with 20 μg/ml proteinase-K in 10 mmol/l Tris-HCl at 37°C for 15 min. After rinsing in distilled water and PBS, the sections were treated with a 0·3% hydrogen peroxidase solution. Each section was incubated with 50 μl of TUNEL reaction mixture, including terminal deoxynucleotidyl transferase for 60 min at 37°C in humidified conditions. All sections were observed and photographed under a fluorescence microscope (Olympus BX-51) with a blue (450–490 nm) excitation light. Negative controls were prepared under the same conditions, with the omission of the terminal deoxynucleotidyl transferase enzyme.
Determination of inflammatory factors by real-time PCR
To analyse the mRNA expression of cytokines, total RNA extraction and real-time PCR were performed as described previously [16]. Total RNA was extracted using RNAiso Plus (Takara Biotechnology, Dalian, China), according to the manufacturer's protocol. The RNA samples were treated with DNase I (Takara Biotechnology) to eliminate DNA contamination.
Total RNA was reverse-transcribed to cDNA using the PrimeScript RT reagent kit according to the manufacturer's instructions. Quantitative real-time PCR was performed with Taq polymerase (SYBR Premix Ex Taq II; Takara Biotechnology) in a final volume of 20 μl containing 0·5 μg of total RNA with SYBR Green PCR supermix (Takara Biotechnology), in accordance with the manufacturer's instructions.
Quantitative real-time PCR conditions were as follows: initial denaturation at 94°C for 5 min, 40 cycles with denaturation at 94°C for 5 s, annealing at 60°C for 20 s, followed by melting curve analysis. Primers used were as follows: β-actin (NM 007393·3): forward 5′-AGATTACTGCTCTGGCTCCTAGC, reverse 5′-ACTCATCGTACTCCTGCTTGCT; interleukin (IL)-1β NM_008361·3: forward 5′-TCCAGGATGAGGACAT, reverse 5′-GAACGTCACACACCAGCAGGTTA; IL-6 NM_031168·1: forward 5′-GAGGATACCACTCCCAACAGACC reverse, 5′-AAGTGCATCATCGTTGTTCATACA; inducible nitric oxide synthase (iNOS) NM_010927·3: forward 5′-AATTCGGCTGTGCTTTGATGG, reverse, 5′-GACTTGCGGGAGTCAGAATAGGAG; and tumour necrosis factor (TNF)-α NM_013693·2: forward 5′-TCCAGGCGGTGCCTATGT, reverse, 5′-CGATCACCCCGAAGTTCAGTA.
Statistical analysis
Analyses were performed using spss version 17·0 software (SPSS, Inc., Chicago, IL, USA), and all values are presented as the mean ± standard error of the mean (s.e.m.). Student's t-test and one-way analysis of variance (anova) followed by Scheffé's post-hoc test were used to compare differences between two and three or more groups, respectively. A P-value less than 0·05 was considered statistically significant.
Results
C5aR expression in peri-haematoma tissue in WT and C3–/– mice
It is becoming increasingly evident that neuroinflammation plays a crucial role in the development and progression of many neurodegenerative diseases. The complement system is involved in the procedure [14,17–19]. A mouse model of ICH was induced by stereotactic infusion of autologous blood collected from mouse tail artery. Sham-treated mice underwent a sterile saline injection. In an ICH model of WT mice, it was found that C5aR was up-regulated 3 days after ICH; however, it was decreased 5 days after ICH.
Complement component 3 is a key molecule in the complement system whose activation may affect haem metabolism and the inflammatory response after ICH, suggesting that C3 is an important factor causing ICH-induced brain injury. C3–/– mice, used to investigate the inflammation triggered by complement [20], failed to generate complement proteins C3a, C3b, C5a and MAC. It was demonstrated that ICH-induced forelimb-use asymmetry deficit and brain oedema are less severe in complement C3 mice. However, the neurological deficits and brain oedema were worse in complement C5-deficient mice [13]. In this study, it was confirmed that in the ICH model of C3–/– mice, C5aR reached maximum expression 3 days after ICH, whereas it decreased 1 and 5 days after ICH, depending on the time–course. There was no significant difference of C5aR expression 3 days after ICH between WT and C3–/– mice.
C5aR was up-regulated in microglia in WT and C3–/– mice
As shown by the results in WT and C3–/– mice, the highest level of C5aR expression was noted 3 days posterior to ICH. Microglia are resident innate immune cells in the central nervous system, which can be activated and produce diverse sets of diffusible neurotoxic factors that are potentially cytotoxic to neurones [21]. The previous study revealed that C5aR was thought to be expressed only on microglia [22]. Comparing the Western blot results of C5aR expression 1, 3 and 5 days after ICH (Fig. 1), it reached the highest level in brain tissue 3 days after ICH. Thus, C5aR distribution was investigated 3 days after ICH. The results suggest that CD68 was a specific marker activated by microglia, which was co-localized with C5aR at the peri-haematoma region 3 days after ICH both in the WT and C3–/– mouse groups. In the sham group, there was no C5aR and less CD68 expression (Fig. 2). It seems that C3 deficiency did not completely inhibit the C5aR expression. Results of the microglia quantification indicated that the number of activated microglia was at a baseline without difference, and in the 3-day ICH group the numbers of activated microglia (CD68-positive) were 146 ± 11 and 142 ± 21 per field in the WT and C3–/– groups, respectively, without difference. The number of C5aR-positive cells showed no significant difference between the WT and C3–/– groups.
Fig. 1.

The expression of C5aR was up-regulated 3 days post-intracerebral haemorrhage (ICH) either in the wild-type (WT) or C3–/– ICH groups by Western blot detection. C5aR expression was up-regulated from the sham to the 3 days post-ICH group, and then decreased 5 days post-ICH, as well as in the C3–/– group. There were significant differences of optical density (OD) value 3 days post-ICH versus sham, 1 and 5 days post-ICH groups (**P < 0·05 versus sham, 1 and 5 days post-ICH groups, n = 5). Similar results were found in the C3–/– group (**P < 0·05 versus sham, 1 and 5 days post-ICH groups, n = 5). Sham, saline injection group. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as inter-reference.
Fig. 2.

(a) C5aR is localized in CD68-positive microglia either in wild-type (WT) 3 days or C3–/– 3 days post-intracerebral haemorrhage (ICH) at the site of peri-haematoma tissue in a brain section with dual-label immunofluorescence staining. In the sham group, no positive cell was labelled with C5aR (red); however, positive cells labelled with activated microglial marker CD68 are indicated (green). The morphology of microglia was partly ramified and partly dot-like-shaped. There was no significant difference between the WT and C3–/– groups 3 days post-ICH. (b) Quantitization of CD68- and C5aR-positive cells in peri-haematoma tissue in ICH brain sections. Few CD68- and C5aR-positive cells were observed in the WT sham and C3–/– sham groups. Approximately 150 CD68- and C5aR-positive cells in the WT ICH 3 days and C3–/– ICH 3 days groups, but without significant differences (P < 0·05, n = 6).
Reduction of C5aR and brain oedema after ICH initiated by C5aR antagonist and thrombin antagonist
Complement activation is triggered by the binding of molecules via the classical, alternative and mannose-binding lectin pathways, with subsequent activation of complement proteins C3 and C5 in the terminal cascade. The small complement fragments generated during complement activation, C3a and C5a, are known as anaphylatoxins that induce several biological responses. They are able to exert a range of various biological effects. Uncontrolled complement activation can lead to excessive tissue inflammation and damage [5]. Therefore, in order to explore further the role of microglial C5aR in inflammation secondary to brain injury in ICH, the C5aR antagonist function (PMX53 in short, blocking the C5a complement signalling pathway) was investigated. The results showed that C5aR expression was down-regulated in the PMX53-treated mice 3 days after ICH induction (Fig. 3), implying that in the genetic absence of C3, thrombin is able to act as a substitute for the C3-dependent C5 convertase [23], suggesting that complement system activation might not be inhibited completely by deletion of C3. Argatroban, a clinically used thrombin inhibitor [9], was applied to suppress the progression of ICH-induced inflammation as well as the putative complement activation signalling pathway. Argatroban mildly decreased the C5aR synthesis and expression in brain tissue 3 days after ICH. It was found that PMX53 in combination with argatroban could reduce the C5aR expression significantly, and the difference was statistically significant in mouse brain 3 days after ICH induction (Fig. 3, n = 9, P < 0·05). These findings suggest that the combination of C5aR and thrombin antagonist ameliorated the ICH-induced neurological deficit.
Fig. 3.
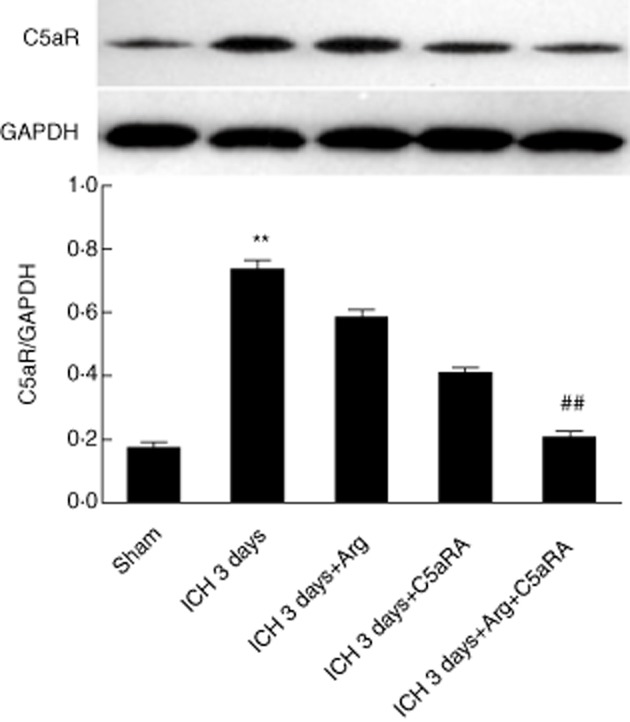
Argatroban (Arg) and C5aR antagonist (PMX53) inhibited C5aR expression when treated individually and together by Western blot. In the sham group, intracerebral haemorrhage (ICH) 3 days, argatroban treatment ICH 3 days, C5aRA (PMX53) treatment and combination treatment groups, the optical density (OD) values were 0·19 ± 0·03, 0·76 ± 0·02, 0·59 ± 0·027, 0·41 ± 0·025 and 0·22 ± 0·02, respectively. The expression of C5aR in the ICH 3 days group reached the maximum level, significantly differently from other groups (P < 0·05, n = 5, versus sham, ICH 3 days + Arg, ICH 3 days + C5aRA groups and combination treatment groups), and in the combination group reached a lower level compared with 3 days ICH, 3 days ICH + Arg and 3 days ICH + C5aRA groups (P < 0·05, n = 5, versus ICH 3 days, 3 days ICH + Arg and 3 days ICH + C5aRA groups).
Attenuation of brain oedema by combination treatment of PMX53 and argatroban
It was considered that red blood cell lysis plays an important role in oedema development after ICH [24]. Theoretically, less oedema may develop as a result of impairment in the thrombin generation, which typically increases vascular permeability [25]. Compared with the ICH group, the BWC was decreased significantly in the treatment group (ICH+Arg+PMX53) in the ipsilateral basal ganglia and ipsilateral cortex 1–5 days after injection of autologous blood (P < 0·01, n = 9) (Fig. 4). BWC of the argatroban (ICH+Arg) and PMX53 (ICH+PMX53) groups were mildly decreased compared with the ICH group 1–5 days after ICH modelling (Fig. 4). However, no significant differences were found in the ContCx, ContBG and Cereb groups between the ICH, ICH+Arg, ICH+PMX53 and ICH+Arg+PMX53 groups (P > 0·05, n = 9).
Fig. 4.

Brain water content (BWC) of intracerebral haemorrhage (ICH) and inhibitor treatment groups. (a) BWC in lpsiCX region of 1 day ICH mice brain were 76·1% ± 0·24, 79·1% ± 0·27, 77·1% ± 0·45, 76·8% ± 0·32 and 76·4% ± 0·28 in sham, ICH, ICH + argatroban (Arg), ICH + C5aRA and Arg + C5aR combination treatment groups. There were significant differences between ICH and other groups (P < 0·05, n = 8). BWC in the lpsi BG region of 1 day ICH mice brain were 76% ± 0·19, 78·8% ± 0·49, 77·8% ± 0·18, 77·6% ± 0·18 and 76·3% ± 0·19, in the sham, ICH, ICH + Arg, ICH + C5aRA and Arg + C5aR combination treatment groups (P < 0·05, n = 8). (b,c) Similar to (a), BWC in lpsi CX and lpsi BG regions changed significantly between the sham, ICH, ICH + Arg, ICH + C5aRA and Arg + C5aR combination treatment groups. BWC reached the maximum value in ICH groups compared to the other groups (P < 0·05, n = 8). Data are presented as mean relative expression ± standard deviation.
Inhibition of inflammatory factors released in the region of peri-haematoma by combination treatment of PMX53 and argatroban
Many byproducts generated during complement activation are mediators of inflammation, and uncontrolled complement activation can result in excessive tissue inflammation and damage [5]. By dissecting peri-haematoma tissue, we isolated specimens which contained inflammatory factor and found that the TNF-α, IL-6, iNOS and IL-1β mRNA were up-regulated on days 1 and 3 after ICH induction. There were significant differences in the expression of TNF-α, IL-6 and iNOS between the ICH group and other groups, either 1 or 3 days after ICH induction, except for IL-1β. No significant change of mRNA of inflammatory factors was recorded in the sham group (Fig. 5).
Fig. 5.

Relative fold change of inflammatory factor mRNA in peri-haematoma tissue. (a) Relative fold change of tumour necrosis factor (TNF)-α mRNA in peri-haematoma tissue in wild-type (WT) intracerebral haemorrhage (ICH), WT ICH + C5aRA, WT ICH + argatroban (Arg) and WT ICH + C5aRA + Arg (combination treatment) groups. Sham, saline injection group. One and 3 days ICH, 1 and 3 days post-autologous blood-induced ICH. (b) Relative fold change of inducible nitric oxide synthase (iNOS) mRNA in peri-haematoma tissue in the same groups and same time–course as (a). (c) Relative fold change of interleukin (IL)-6 mRNA expression in peri-haematoma in the same groups and same time–course as (a). Data are presented as mean relative expression ± standard deviation. **P < 0·05, WT ICH versus C5aRA, Arg and C5aRA + Arg group, n = 5.
Protection of the neurone against inflammation-induced neural death with PMX53 in combination with argatroban
It was revealed that functional inhibition of C5 and the C5a receptor has shown a neuroprotective effect against ischaemia–reperfusion injury in middle cerebral artery occlusion (MCAO) models [26,27]. Furthermore, the administration of C5a receptor antagonist, PMX53 and PMX205, could provide therapeutic benefits in neurodegenerative diseases [12]. In this study we used PMX53 and argatroban to detect whether or not they could protect neurone injury and death following ICH. The brain sections from different groups, including WT 3 days ICH, 3 days ICH+Arg, 3 days ICH+PMX53 and 3 days ICH+Arg+PMX53, were selected, respectively, and death neurones were identified by βIII-tubulin and TUNEL dual-label staining. The dual-label cells in several objective fields in the peri-haematoma region were counted blind by Image Pro Plus version 6·0 software. No TUNEL-labelled positive cells (data not shown) in the control group were observed. However, dual-labelled positive spots (apoptosis neurones) were seen in other groups (Fig. 6a, arrow). The quantification of positive cells was implemented using statistical methods. The results showed that there were fewer TUNEL-positive cells in Arg (43 ± 3), PMX53 (30 ± 4) and PMX53+Arg (16 ± 3) groups than those in the WT ICH groups in mice 3 days after ICH. The neuronal apoptosis numbers were fewer in the PMX53+Arg group than those in the Arg and PMX53 groups. In line with these observations, numerous cells were observed in the peri-haematoma region exhibiting nuclear fragmentation in response to ICH. In addition, combination treatment of PMX53 and argatroban prevented blood clot toxicity in the striatum, which suggested that the combined treatment of PMX53 and argatroban plays a beneficial role in protecting neurones from apoptosis after ICH induction.
Fig. 6.

Neuroprotective effect of argatroban (Arg) and/or PMX53 detected by TUNEL (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labelling) and βIII-tubulin dual-label staining in the peri-haematoma region of 3 days intracerebral haemorrhage (ICH) brain sections. (a) In the 3 days ICH lane, several neurones were labelled by βIII-tubulin (red) and co-labelled with TUNEL (green) in the peri-haematoma region which demonstrated that neurones were apoptosis. Nuclear were labelled by Hoechst (blue) and merged figures showed the three colours merged together. In the 3 days ICH + Arg lane, 3 days ICH + PMX53 and 3 days ICH + PMX53 + Arg lanes, neurones were labelled by βIII-tubulin (red) and co-labelled with TUNEL (green); nucleus was also labelled with Hoechst (blue) and merged figures showed the combination of βIII-tubulin (red), TUNEL (green) and Hoechst (blue). (b) Quantization of βIII-tubulin-positive (red), TUNEL-positive (green) and merged cells in observation fields. In different treatment groups, i.e. WT 3 days ICH, WT 3 days ICH + Arg, PMX53 and combination treatment groups, numbers of TUNEL positive and merged cells were decreased significantly (P < 0·05, n = 3); however, no significant difference in the βIII-tubulin-positive group (P > 0·05, n = 3). Arg, argatroban. PMX53, C5aR antagonist.
Combination treatment of PMX53 and argatroban promote neurological and behaviour recovery
Within 1 day of injection of autologous blood into the mouse brain, there were marked neurological deficits as assessed by 28-point scale. On days 1, 3 and 5 after ICH, all test groups consisting of the argatroban-treated group, the PMX53-treated group and the combined treatment group presented a gradual recovery of neurological function in C3–/– mice. On days 1 and 3 after ICH, significant differences in neurological deficits were found between the argatroban-treated group, the PMX53-treated group and the combined treatment group in the WT and C3–/– groups, respectively (Fig. 7; P < 0·05). Administration of agartroban and PMX53 were beneficial for animal behaviour recovery. However, 5 days after ICH no significant difference was found between the WT ICH and other groups which suggested that, to some extent, the self-recovery of mice occurred without argatroban and PMX53 treatment.
Fig. 7.
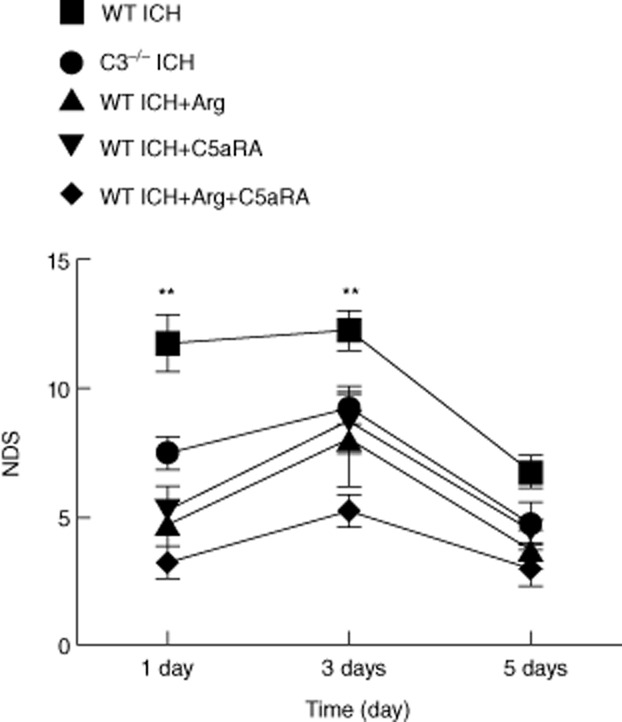
Neurological deficit score (NDS) of intracerebral haemorrhage (ICH), argatroban (Arg) and/or C5aRA treatment groups from 1, 3 and 5 days post-ICH. The NDS of the wild-type (WT) ICH group showed a significant difference compared with C3–/– ICH, WT ICH + Arg and/or C5aRA groups (P < 0·05, n = 9) on 1 and 3 days post-ICH; however, there was no significant difference between different groups on 5 days post-ICH (P < 0·05, n = 9).
Discussion
This study was designed to provide a novel insight into the treatment of ICH via complement and thrombin inhibition. Upon activation of the complement system, split fragments C5a and C3a augment inflammatory responses, e.g. contribute to the inflammatory factors release and facilitate the recruitment of neutrophils and monocytes to the inflamed tissues [28,29]. C5a is known as an anaphylotoxin, which exerts a detrimental function inducing tissue injury via C5aR [27,30]. Blockade of C5aR partially prevents cytokine production and inflammatory injury in models of incision, arthritis and acute lung injury [6,31]. In this study, C3–/– mice were utilized to explore the mechanism of the protective function and the role of the complement system in the ICH mouse model. Moreover, whether or not complement C3 deletion down-regulated the C5a receptor expression and further regulated the ICH inflammatory injury was also observed. From days 1–5 after ICH, C5aR expression was up-regulated 3 days after ICH both in the WT and the C3–/– groups. It seems that C3–/– deletion did not completely inhibit C5aR expression. The activation of complement pathway could be triggered by C5aR [5], and the coagulation and complement pathways simultaneously promote homeostasis in response to injury, whereas they cause tissue damage when up-regulated. This study found a new paradigm for complement activation in which thrombin and C5 convertase exerted a synergistic effect, enhancing the terminal pathway via the generation of newly uncovered C5 intermediates [23,32,33]. The results were verified further by results and findings from ICH pathological procedures.
The study revealed that C5aR reached maximum expression 3 days after ICH induction (Fig. 1), in which a time–course of C5aR expression was localized and up-regulated on microglia (Fig. 2a). C5aR was found to be up-regulated on microglia in the central nervous system in several pathological disorders, such as Alzheimer's disease [18,22] and pain hypersensitivity-induced inflammatory responses[34]. Activated microglia can be phagocytic, but they also can secrete several proinflammatory cytokines, as well as interleukins, reactive oxygen species and nitric oxide (NO) which, if not regulated, can generate a neurotoxic inflammatory environment that accelerates pathological and neuronal dysfunction [18]. The results of behavioural tests indicated that C3–/– mice gained better functional recovery on days 1, 3 and 5 after ICH. Treatment of PMX53 in combination with argatroban can also attenuate the brain injury secondary to ICH. The inhibition of microglial C5aR with antagonist would reduce C5a-induced inflammation in brain tissue [35]. It was confirmed that C3 deletion did not completely inhibit C5aR expression on microglia in the region of peri-haematoma tissue, revealing that C5a or other complement molecules can also activate the complement pathway even if C3 was deleted. This result was consistent with previous findings [23,33], which demonstrated that thrombin acting as C5 convertase promotes C5a and other complement molecule formation.
The breakdown of cerebral blood vessels induces leakage of intrinsic factors, including a serine protease thrombin into the brain parenchyma. Nevertheless, the coagulation cascade is activated, and an excessive amount of thrombin is transformed from prothrombin in the haematoma [36]. Thrombin activates immunocompetent microglia and increases the release of inflammatory cytokines under ICH induction. Furthermore, thrombin injection into the striatum evokes acute necrosis and delayed apoptosis of neurones [37]. With regard to the prevention of thrombin-induced brain injury, an ideal thrombin inhibitor is supposed to enter the haematoma and parenchyma easily, and also be a putative molecule that functions as a C5 convertase promoting intracerebral inflammation. In this study, inhibition of thrombin by argatroban suppresses C5aR expression (Fig. 3) and alleviates ICH-induced brain oedema (Fig. 4), suggesting that argatroban may inhibit either thrombin-induced inflammation or complement pathway activation. Argatroban is a direct thrombin inhibitor that safely augments recanalization achieved by tissue-type plasminogen activator (tPA) in animal stroke models [38]. Argatroban is a potent inhibitor of fibrin-bound and clot-bound thrombin because it has a hydrophobic binding site to thrombin, and can even bind to the bound thrombin [36]. Administration of argatroban carries a risk of bleeding tendency. In this study, autologous blood injection model was utilized to investigate whether argatroban can cause bleeding. The results showed no significant difference in haematoma size between the argatroban treatment group and the control group (data not shown), which was in accordance with the results found previously [9].
Although complement activation has been linked to oedema formation, the definitive mechanism has not been elucidated clearly. It was believed that C5b and subsequent formation of membrane attack complex (MAC) may contribute to the mechanism of oedema formation [39]. With dissecting different regions of mice brain, the BWC can be calculated precisely. As expected, the dosing regimens of argatroban and PMX53 that attenuated ICH-induced brain oedema were also effective in improving the performance of animals in neurological deficit score. The 28-point NDS system has been used extensively in ICH models and has proved to be useful for evaluating the extent of brain injury [40]. Neurological deficit score is an important component of ICH investigation in proving clinical relevance.
Argatroban is superior to other thrombin inhibitors due to its short half-life, which allows rapid offset of action in case of bleeding and ease of monitoring its anti-thrombotic effect [41]. However, argatroban only partially inhibited thrombin-induced cortical cell death [42]. No evidence has demonstrated that a combination of argatroban and PMX53 is detrimental to neural recovery and ICH secondary inflammation. In this study, administration of PMX53 in combination with argatroban was used for the first time to treat ICH. The dual blockage of C5aR and thrombin may be more clinically effective than the single suppression of either inhibitor. Dual-labelling of TUNEL staining and βIII-tubulin immunohistochemistry indicated a neuroprotective effect of both argatroban and PMX53. It was found that a transient increase in PI fluorescence indicative of the occurrence of necrosis is observed after thrombin infusion in the striatal region of slice cultures [42]. It was also indicated that thrombin-induced neuronal apoptosis could be examined using MAP-2 and TUNEL double-labelling in vitro [43], which provided verification for our findings. Thrombin may cause apoptosis by stimulating post-mitotic cortical neurones to re-enter the cell cycle, which indicated that mitogenic signalling triggered by thrombin and aberrant cell cycle re-entry may be involved in the same mechanism [44]. Thus, it is concluded that combination of the two inhibitors attenuate neurone death, but the mechanism needs to be explored thoroughly in future research.
Animal experiments indicate that the cerebral thrombin is associated with secondary brain damage after intracerebral haemorrhage (ICH) [42]. Delayed argatroban treatment reduced oedema in a rat model of ICH, and did not increase collagenase-induced haematoma volume when given into the clot after 3 h or given systemically at 6 h [36,45], which indicated that the time-window for administration of argatroban is extremely important. In patients, thrombin–anti-thrombin complex (TAT) levels of plasma and haematoma fluid correlate positively with the National Institutes of Health stroke scale and the severity of ICH [46]. As opposed to conventional therapeutic approaches, the administration of argatroban could effectively limit oedema formation and neuronal damage, improving survival and functional outcomes [3]. Furthermore, diabigatran, another thrombin inhibitor, did not enlarge haematoma volume in experimental ICH [47], which demonstrated that it is possible to treat ICH with anti-thrombin treatment. However, anti-thrombin drugs used in mice and humans are different ICH situations, and there are still controversial results and conclusions regarding the function and usage of anti-thrombin drugs. It remains to be studied whether this anti-coagulant could be useful in a clinical situation. However, the safety and ethical issues concerning clinical usage need to be discussed further. If possible, therefore, more experiments should be conducted in other types of animals, such as rabbits, dogs and monkeys.
Based on our findings, we have proposed a possible pathphysiological mechanism for ICH-induced brain injury. In this attempt, C3 deletion did not completely inhibit C5aR expression and function. Thrombin is a putative molecule which is able to take effect as C5a convertase that promotes complement system activation, even if C3 is deleted in ICH-induced brain injury. However, the complement system activation can be inhibited by argatroban. The combination of C5aR antagonist and argatroban blocks the two different but intercrossed complement pathways, delivering a theoretical basis for novel approaches to effective therapeutic intervention in ICH-induced brain injuries.
Acknowledgments
This work was supported by the Major Program of Science and Technology Foundation of Zunyi (Guizhou) Technology Bureau. We would like to thank Dr Sen Lin for the language rewrite.
Disclosure
The authors declared that there are no conflicts of interest.
References
- 1.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 2.Xue M, Balasubramaniam J, Parsons KA, McIntyre IW, Peeling J, Del Bigio MR. Does thrombin play a role in the pathogenesis of brain damage after periventricular hemorrhage? Brain Pathol. 2005;15:241–249. doi: 10.1111/j.1750-3639.2005.tb00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuoka H, Hamada R. Role of thrombin in CNS damage associated with intracerebral haemorrhage: opportunity for pharmacological intervention? CNS Drugs. 2002;16:509–516. doi: 10.2165/00023210-200216080-00001. [DOI] [PubMed] [Google Scholar]
- 4.Babu R, Bagley JH, Di C, Friedman AH, Adamson C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg Focus. 2012;32:E8. doi: 10.3171/2012.1.FOCUS11366. [DOI] [PubMed] [Google Scholar]
- 5.Liang DY, Li X, Shi X, et al. The complement component C5a receptor mediates pain and inflammation in a postsurgical pain model. Pain. 2012;153:366–372. doi: 10.1016/j.pain.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark JD, Qiao Y, Li X, Shi X, Angst MS, Yeomans DC. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology. 2006;104:1274–1282. doi: 10.1097/00000542-200606000-00024. [DOI] [PubMed] [Google Scholar]
- 7.Woodruff TM, Strachan AJ, Dryburgh N, et al. Antiarthritic activity of an orally active C5a receptor antagonist against antigen-induced monarticular arthritis in the rat. Arthritis Rheum. 2002;46:2476–2485. doi: 10.1002/art.10449. [DOI] [PubMed] [Google Scholar]
- 8.Rynkowski MA, Kim GH, Komotar RJ, et al. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–128. doi: 10.1038/nprot.2007.513. [DOI] [PubMed] [Google Scholar]
- 9.Ohnishi M, Katsuki H, Fujimoto S, Takagi M, Kume T, Akaike A. Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp Neurol. 2007;206:43–52. doi: 10.1016/j.expneurol.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Asanuma K, Wakabayashi H, Hayashi T, et al. Thrombin inhibitor, argatroban, prevents tumor cell migration and bone metastasis. Oncology. 2004;67:166–173. doi: 10.1159/000081004. [DOI] [PubMed] [Google Scholar]
- 11.Woodruff TM, Pollitt S, Proctor LM, et al. Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J Pharmacol Exp Ther. 2005;314:811–817. doi: 10.1124/jpet.105.086835. [DOI] [PubMed] [Google Scholar]
- 12.Woodruff TM, Crane JW, Proctor LM, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20:1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura T, Xi G, Hua Y, Schallert T, Hoff JT, Keep RF. Intracerebral hemorrhage in mice: model characterization and application for genetically modified mice. J Cereb Blood Flow Metab. 2004;24:487–494. doi: 10.1097/00004647-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Garrett MC, Otten ML, Starke RM, et al. Synergistic neuroprotective effects of C3a and C5a receptor blockade following intracerebral hemorrhage. Brain Res. 2009;1298:171–177. doi: 10.1016/j.brainres.2009.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavisse S, Guillermier M, Herard AS, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32:10809–10818. doi: 10.1523/JNEUROSCI.1487-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirakawa H, Sakimoto S, Nakao K, et al. Transient receptor potential canonical 3 (TRPC3) mediates thrombin-induced astrocyte activation and upregulates its own expression in cortical astrocytes. J Neurosci. 2010;30:13116–13129. doi: 10.1523/JNEUROSCI.1890-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J Neuroimmunol. 2010;221:46–52. doi: 10.1016/j.jneuroim.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fonseca MI, Ager RR, Chu SH, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. 2009;183:1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ducruet AF, Zacharia BE, Hickman ZL, et al. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp Neurol. 2009;219:398–403. doi: 10.1016/j.expneurol.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang S, Nakamura T, Hua Y, et al. The role of complement C3 in intracerebral hemorrhage-induced brain injury. J Cereb Blood Flow Metab. 2006;26:1490–1495. doi: 10.1038/sj.jcbfm.9600305. [DOI] [PubMed] [Google Scholar]
- 21.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 22.Ager RR, Fonseca MI, Chu SH, et al. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer's disease. J Neurochem. 2010;113:389–401. doi: 10.1111/j.1471-4159.2010.06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 24.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 25.Sansing LH, Kaznatcheeva EA, Perkins CJ, Komaroff E, Gutman FB, Newman GC. Edema after intracerebral hemorrhage: correlations with coagulation parameters and treatment. J Neurosurg. 2003;98:985–992. doi: 10.3171/jns.2003.98.5.0985. [DOI] [PubMed] [Google Scholar]
- 26.Costa C, Zhao L, Shen Y, et al. Role of complement component C5 in cerebral ischemia/reperfusion injury. Brain Res. 2006;1100:142–151. doi: 10.1016/j.brainres.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 27.Kim GH, Mocco J, Hahn DK, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery. 2008;63:122–125. doi: 10.1227/01.NEU.0000335079.70222.8D. discussion 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank MM, Fries LF. The role of complement in inflammation and phagocytosis. Immunol Today. 1991;12:322–326. doi: 10.1016/0167-5699(91)90009-I. [DOI] [PubMed] [Google Scholar]
- 29.Wetsel RA. Structure, function and cellular expression of complement anaphylatoxin receptors. Curr Opin Immunol. 1995;7:48–53. doi: 10.1016/0952-7915(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 30.Tokodai K, Goto M, Inagaki A, et al. C5a-inhibitory peptide combined with gabexate mesilate prevents the instant blood-mediated inflammatory reaction in a rat model of islet transplantation. Transplant Proc. 2010;42:2102–2103. doi: 10.1016/j.transproceed.2010.05.100. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Guo RF, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J. 2009;23:3808–3818. doi: 10.1096/fj.09-133694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120:1717–1725. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ, Leung LL. Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood. 2007;109:1992–1997. doi: 10.1182/blood-2006-03-012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffin RS, Costigan M, Brenner GJ, et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–8708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleming SD, Phillips LM, Lambris JD, Tsokos GC. Complement component C5a mediates hemorrhage-induced intestinal damage. J Surg Res. 2008;150:196–203. doi: 10.1016/j.jss.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagatsuna T, Nomura S, Suehiro E, Fujisawa H, Koizumi H, Suzuki M. Systemic administration of argatroban reduces secondary brain damage in a rat model of intracerebral hemorrhage: histopathological assessment. Cerebrovasc Dis. 2005;19:192–200. doi: 10.1159/000083466. [DOI] [PubMed] [Google Scholar]
- 37.Fujimoto S, Katsuki H, Ohnishi M, Takagi M, Kume T, Akaike A. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience. 2007;144:694–701. doi: 10.1016/j.neuroscience.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 38.Barreto AD, Alexandrov AV, Lyden P, et al. The argatroban and tissue-type plasminogen activator stroke study: final results of a pilot safety study. Stroke. 2012;43:770–775. doi: 10.1161/STROKEAHA.111.625574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xi G, Wagner KR, Keep RF, et al. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke. 1998;29:2580–2586. doi: 10.1161/01.str.29.12.2580. [DOI] [PubMed] [Google Scholar]
- 40.Belayev L, Saul I, Curbelo K, et al. Experimental intracerebral hemorrhage in the mouse: histological, behavioral, and hemodynamic characterization of a double-injection model. Stroke. 2003;34:2221–2227. doi: 10.1161/01.STR.0000088061.06656.1E. [DOI] [PubMed] [Google Scholar]
- 41.Sugg RM, Pary JK, Uchino K, et al. tPA stroke study: study design and results in the first treated cohort. Arch Neurol. 2006;63:1057–1062. doi: 10.1001/archneur.63.8.1057. [DOI] [PubMed] [Google Scholar]
- 42.Fujimoto S, Katsuki H, Kume T, Akaike A. Thrombin-induced delayed injury involves multiple and distinct signaling pathways in the cerebral cortex and the striatum in organotypic slice cultures. Neurobiol Dis. 2006;22:130–142. doi: 10.1016/j.nbd.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Liu DZ, Cheng XY, Ander BP, et al. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol Dis. 2008;30:201–211. doi: 10.1016/j.nbd.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao HV, Thirumangalakudi L, Desmond P, Grammas P. Cyclin D1, cdk4, and Bim are involved in thrombin-induced apoptosis in cultured cortical neurons. J Neurochem. 2007;101:498–505. doi: 10.1111/j.1471-4159.2006.04389.x. [DOI] [PubMed] [Google Scholar]
- 45.Kitaoka T, Hua Y, Xi G, Hoff JT, Keep RF. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke. 2002;33:3012–3018. doi: 10.1161/01.str.0000037673.17260.1b. [DOI] [PubMed] [Google Scholar]
- 46.Wu CH, Yang RL, Huang SY, et al. Analysis of thrombin–antithrombin complex contents in plasma and hematoma fluid of hypertensive intracerebral hemorrhage patients after clot removal. Eur J Neurol. 2011;18:1060–1066. doi: 10.1111/j.1468-1331.2010.03336.x. [DOI] [PubMed] [Google Scholar]
- 47.Lauer A, Cianchetti FA, Van Cott EM, et al. Anticoagulation with the oral direct thrombin inhibitor dabigatran does not enlarge hematoma volume in experimental intracerebral hemorrhage. Circulation. 2011;124:1654–1662. doi: 10.1161/CIRCULATIONAHA.111.035972. [DOI] [PMC free article] [PubMed] [Google Scholar]