

Abstract
Appendicitis followed by appendectomy (AA) at a young age protects against inflammatory bowel disease (IBD). We wanted to characterize the role of the T helper type 17 (Th17) system involved in this protective effect. AA was performed on 5-week-old male BALB/c mice and distal-colon samples were harvested. Mice with two laparotomies each served as sham–sham (SS) controls. RNA was extracted from four individual colonic samples per group (AA and SS groups) and each sample microarray-analysed and reverse transcription–polymerase chain reaction (RT–PCR)-validated. Gene-set enrichment analysis (GSEA) showed that the Th17 recruitment factor gene CCL20 was significantly suppressed at both 3 days post-AA and 28 days post-AA. Although Th17 cell development differentiation factor genes TGF-β2 and TGF-β3 were significantly up-regulated 3 days post-AA, GSEA 28 days post-AA showed that AA down-regulated 29 gene-sets associated with TGF-β1, TGF-β2 and TGF-β3 in contrast to none up-regulated with any of these genes. GSEA showed substantial down-regulation of gene-sets associated with Th17 lymphocyte recruitment, differentiation, activation and cytokine expression in the AA group 28 days post-AA. We conclude that Th17-system cytokines are kept under control by AA via down-regulation of proinflammatory CCL20, a rapid down-regulation of pro-Th17 cell differentiation genes TGF-β2 and TGF-β3, suppression of RORC-associated gene-sets, increased protective STAT1 expression and suppression of 81 ‘pro-Th17’ system gene-sets. AA suppresses the Th17 pathway leading to colitis amelioration. Further characterization of Th17-associated genes and biological pathways will assist in the development of better therapeutic approaches in IBD management.
Keywords: appendectomy, appendicitis, colitis, Th17 cells
Introduction
The appendix, replete with abundant lymphoid tissue, is constantly exposed to manifold intestinal flora. The incidence of appendicitis is approximately 7%, making it the most common abdominal emergency requiring surgery [1]. The peak incidence of appendicitis without perforation is in the second and third decades of life [2].
The complex interplay between genetic proclivity, gut flora and intestinal immunity in inflammatory bowel disease (IBD) (ulcerative colitis and Crohn's disease) has not been delineated satisfactorily. The critical role of appendicitis followed by appendectomy (AA) in ameliorating or preventing development of human ulcerative colitis [3–5] and Crohn's disease [4,6] is limited to patients having surgery before turning 20 [5]. In mice, the major caecal lymphoid patch is the equivalent of the human appendix. The removal of the caecum prevented the development of experimental colitis in three murine colitis models, namely T cell receptor-α mutants [7], the dextran sulphate sodium model [8] and the adoptive T cell transfer colitis model [9].
The newly described interleukin (IL)-17-secreting subset of CD4+ T cells, called T helper type 17 (Th17) cells, has been increasingly implicated in the pathogenesis of inflammatory changes in inflammatory/autoimmune diseases, including IBD. Th17 cells are not homogeneous. Contingent upon the cytokine milieu, and depending upon co-expression of other cytokines such as IL-10 or interferon (IFN)-γ, Th17 cells may turn out to be protective or damaging [10].
In an adoptive transfer model of colitis, it was shown that retinoic acid-related orphan receptor (ROR)γt controls IL-17A and IL-17F production in the induction of colitis pathology [11]. In the dextran sulphate sodium murine colitis model, IL-17F deficiency results in decreased intensity of colitis [12]. IBD gut mucosa contain an excess of IL-17-producing lymphocytes [13]. IL-17F gene expression expression is up-regulated in IBD mucosa [14].
In contrast, a few studies have shown that Th17 cytokines may have some protective effects in the gastrointestinal tract. Adoptive transfer of CD45RBhiCD25–CD4+ T cells from IL-17A knock-out mice induces severe colitis in recipient immunodeficient mice [15], suggesting that anti-inflammatory effects of IL-17A in the gut may be linked to the negative regulation of Th1 cell responses. One of the Th17 cytokines, IL-22, enhances epithelial defensin and mucus production, posting a protective gut counter-regulation [16].
IL-23 is necessary for conferring pathogenicity to Th17 cells, as it has been shown that in the absence of IL-23, Th17 cells may develop IL-10-related regulatory functions [17].
The first murine model of AA was developed by us [18]. In this model, the appendiceal pathology closely resembled that of human appendicitis, and AA offers an age-dependent protection against trinitrobenzene sulphonic acid (TNBS) colitis [18].
Gene expression analysis strategies have been used successfully to elucidate differences in individual gene expression between two experimental groups. However, this approach does not take into account the biological reality of cellular processes cohesively or contiguously effecting changes in gene-sets. These changes, subtle at the level of individual genes, are explicit at the level of gene-sets.
This study uses microarray analysis and gene-set enrichment analysis (GSEA) [19] to identify, characterize and link possible roles of the Th17 system and its biological pathways involved in the protective effect of AA in experimental colitis.
This study is a topically circumscribed but intricate follow-up to our previous study published in Clinical and Experimental Immunology [20]. A robust set of reverse transcription–polymerase chain reaction (RT–PCR) results have already been published [20], validating our microarray results. This has been indicated with the reference or summarized succinctly in the Materials and methods section. Repeating the methodologies and results in detail here would therefore be redundant and repetitive. The raw microarray data in toto have already been submitted to the Gene Expression Omnibus. The Accession number for microarray data reported here is GSE23914. We do not intend to overshoot our speculative boundaries in Fig. 1. The figure merely depicts a pictorial summary of the conceptual linkage between the salient findings in this study.
Fig. 1.
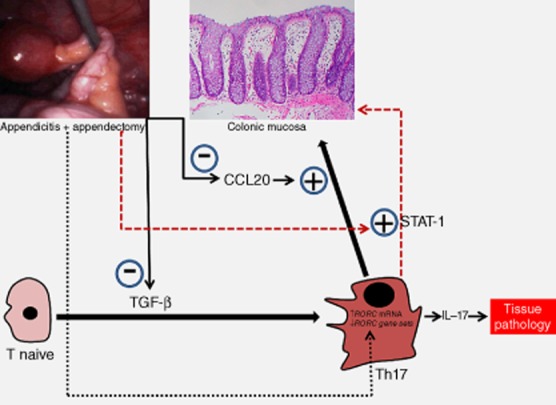
Pathways utilized by appendicitis–appendectomy (AA) to suppress T helper type 17 (Th17) responses. Th17 effector cytokine outflow with its pathogenic potential is suppressed by AA via (1) down-regulation of proinflammatory Th17 cell recruitment-factor CCL20; (2) a rapid down-regulation of pro-Th17 cell differentiation genes TGF-β1, TGF-β2 and TGF-β3; (3) down-regulated RORC-associated gene-sets, despite increased RORC expression per se; and (4) increased protective STAT1 expression.
Materials and methods
Animal experiments
Specific pathogen-free Balb/c mice (male, 5 weeks) were purchased from the Animal Resource Centre, Perth, Western Australia and kept in the University of New South Wales holding and care facility in physical containment level 2 rooms. All experiments were approved and monitored by the University of New South Wales Animal Care and Ethics Committee. Mice were anaesthetized with xylazine (5 mg/kg) and ketamine (100 mg/kg) intraperitoneally (i.p.) followed by allocation into two treatment groups, the appendicitis group or the sham surgery group [18]. Mice were randomized to have either appendicitis or sham operation. Appendicitis was induced by constructing an appendiceal pouch from the caecal lymphoid patch. This murine appendix was obstructed by rubber band ligation using standardized negative aspiration. Sham surgery entailed a similar procedure, but without continuous obstruction by band ligation of the caecal patch and the placement of a sterile rubber band in the abdominal cavity as a control for foreign body reaction. Seven days following initial surgery, appendicitis mice underwent appendectomy [appendicitis and appendectomy (AA) group], while sham mice underwent a second sham surgery [sham and sham (SS) group]. All mice were monitored daily.
Processing of colonic specimens for RNA extraction
Transmural colonic samples were cleaned of faecal contents with normal saline and transferred immediately to TRIzol® reagent (50–75 mg of tissue in 600 μl of TRIzol® reagent; Invitrogen Australia Pty Limited, Mulgrave, Australia), snap-frozen in liquid nitrogen and stored at −80°C until the microarray analysis. Further extraction entailed chloroform and isopropanol treatment and centrifugation followed by washing the resultant pellet with 75% ethanol, air-drying and final reconstitution in nuclease-free H2O. Concentration and purity of RNA were determined by automated optical density evaluation [optical density (OD) 260/OD 280 ≥ 1·8 and OD 260/OD 230 ≥ 1·8] using Nanodrop ND-1000 (Nanodrop Technologies, Wilmington, DE, USA). The degree of RNA degradation was analysed by the Agilent electrophoresis bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA, USA) with the RNA integrity number (RIN) values consistently above 7.
Experimental design of microarray study
All experiments were designed to be compliant with minimum information about a microarray experiment (MIAME) standards [21,22]. For Affymetrix array experiments, four individual test samples (for 3-day post-SS/AA time-point) or three individual test samples (for 28-day post-SS/AA time-point) were used per group (AA group versus SS group; one colonic sample per mouse) with each sample hybridized to an individual slide.
Affymetrix array process – labelling, hybridization, scanning and normalization
RNA from distal colonic tissue obtained from mice was not pooled. RNA from each mouse specimen was taken individually through the microarray process. For Affymetrix arrays, 100 ng of RNA from each sample was labelled using the Whole Transcript Sense Target Labelling Assay, as described (Affymetrix). Labelled cRNA samples were then hybridized to Affymetrix Mouse Gene version 1.0 ST arrays (28 853 well-annotated genes) (Ramaciotti Centre for Gene Function Analysis, University of New South Wales, Australia) before being scanned using a Affymetrix GCS3000 7G four-colour gene array scanner with autoloader (Affymetrix). The Gene Expression Omnibus Accession number for microarray data reported here, inclusive of MIAME-compliant experimental details [21,22], is GSE23914, and the relevant link is http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23914.
Microarray preprocessing and filtering
All non-control probesets from the eight arrays were imported into Partek (version 6.4; Partek Inc., St Louis, MO, USA), and then normalized using Robust Multichip Average (RMA) [23]. By principle components analysis, a batch effect was evident in principle component 1, which was removed using the batch removal tool in Partek, using default parameters. The probability of each probeset being expressed was determined using the detected above background procedure, using Affymetrix Power Tools (version 1.10.2), excluding 13 probes from probeset 10338063 which had very low guanine–cytosine (GC), and thus did not have matched controls. Probesets were excluded if none of the samples were detected above background (P-value = 10−5). To assess the degree of differential expression between AA and SS groups, a two-way analysis of variance (anova) on treatment and batch was fitted to each probeset using Partek. To correct for multiple hypothesis testing, we used the qvalue/positive false discovery rate (FDR) [24].
Gene-set enrichment analysis (GSEA)
We compared gene expression profiles to the c2? all collection of curated gene-sets from the molecular signatures database (version 2.5) [19]. This collection contains gene-sets that are experimentally derived, as well as from expert curated pathway databases. A preranked file was created, containing the average difference between AA and SS for each probeset, sorted from most up-regulated in SS to most down-regulated. We used the na28 annotation csv file from http://www.affymetrix.com to determine the gene symbol for each probeset and collapsed probesets to unique genes using the default, max_probe option, resulting in 18 600 unique genes. GSEA (version 2.0) [19] was run in preranked mode, using default parameters (gene-set sizes between 15 and 500 leaving 1387 gene-sets, 1000 permutations, images on the top 50 gene-sets).
Enrichment of Th17 system associated gene-sets
We utilized GSEA developed by Mootha et al. [25], which merges data from groups of gene-sets described previously in the literature to detect significant expression differences. These gene-set groups were Kegg Pathways (150 gene-sets), microRNAs (200 gene-sets), transcription factors (579 gene-sets), biological processes (536 gene-sets) and others (1387 gene-sets). We utilized stringent statistical cut-offs (FDR values < 1 % and P-value < 0·001) to delineate Th17 system-associated gene-sets, which were also consistently altered in the distal colons of all AA mice when compared to the all the control SS mice.
Quantitative RT–PCR validation of microarray study
We selected 14 genes for confirmation of our gene expression studies. These genes broadly belonged to four major groups: innate immunity (slpi, s100A8, lbp, CD68); immune mediators (IL18R1, IL33), cell migration chemokines (ccl8, cxcl10, ccl12 or mcp5, pf4, cxcl5, ccl7 or mcp3) and cell migration receptors (fpr1, ccr5). Specific methodologies and the RT–PCR study validation results have already been published [20].
Statistics
Group comparisons were analysed using the Mann–Whitney U-test with GraphPad Prism (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± standard error of the mean and the differences were considered to be significant if the P-value was < 0·05.
Results
Individual distal colonic gene expression of important Th1- and Th2-associated genes
We examined 33 Th1 system genes at two time-points – 3 days post-AA and 28 days post-AA. The 33 genes were: CD4, CD27, CD28, CD80, CD86, IL2, IL10, IL12A, IL12B, IL18, IL2RA, IL18R1, TNF, INHA, INHBA, IRF4, SFTPD, SPP1, TLR4, TLR6, CXCR3, CCR5, CSF2, GLMN, HAVCR2, IFNG, IGSF6, IRF1, SOCS1, SOCS5, STAT1, STAT4 and TBX21. Only one gene (IL2RA) among the 33 Th1 system genes evaluated showed changes (slightly down-regulated) 3 days post-AA in distal colonic samples (P-value = 0·031). Only one gene (STAT1) among the 33 Th1 system genes evaluated showed changes (up-regulated) 28 days post-AA in distal colonic samples (P-value = 0·004)
We examined 33 Th2 system genes at two time-points – 3 days post-AA and 28 days post-AA. The 33 genes were: CCL5, CCL7, CCL11, CCR2, CCR3, CCR4, CEBPB, NFATC2IP, GATA3, GFI1, GPR44, ICOS, IL4, IL5, IL9, IL10, IL13, IL18, IL13RA1, IL1R1, IL1R2, IL4RA, IL4RB, IRF4, JAK1, MAF, NFATC1, NFATC2, NFATC2IP, PCGF2, STAT6, TMED1 and CD86. Only one gene (MAF) among the 33 Th2 system genes evaluated showed changes (up-regulated) 3 days post-AA in distal colonic samples (P-value = 0·012). Only one gene (CCL5) among the 33 Th2 system genes evaluated showed changes (up-regulated) 28 days post-AA in distal colonic samples (P-value = 0·049).
Individual distal colonic gene expression of key Th17 system-associated genes
The genes chosen (Table 1) for expression-level evaluation are those with well-documented direct links with the Th17 system with or without links to IBD. The Th17 recruitment factor gene CCL20, which has been shown to be up-regulated in IBD [26], was suppressed significantly at both 3 days post-AA and 28 days post-AA. The Th17 cell development transcription factor genes RORC and STAT1 were up-regulated significantly only at 28 days post-AA, but not at 3 days post-AA. The Th17 cell development differentiation factor genes TGF-β1 and TGF-β2 were up-regulated significantly only 3 days post-AA, but not later at 28 days post-AA. However, none of the Th17 system growth/stabilization factors or the effector Th17 system interleukins was modulated at either time-point.
Table 1.
Individual distal colonic gene expression of key T helper type 17 (Th17) system-associated genes 3 days post-appendicitis–appendectomy (AA) and 28 days post-AA
| 3 days post-AA | 28 days post-AA | |||||
|---|---|---|---|---|---|---|
| Th17 system gene | Function of gene-product (Th17 system-related) | Selected references | Fold-change | P-value | Fold-change | P-value |
| Recruitment factors for Th17 cells | ||||||
| CCR6 | ↑ on Th17 cells, IBD association | [27,29] | – | – | 0·85 | 0·145 |
| CCL20 | ↑ on inflamed gut mucosa, CCR6-ligand, IBD association | [26,30] | 0·60‡ | 0·028* | 0·634‡ | 0·023* |
| Transcription factors involved in Th17 cell development | ||||||
| RORC | ↑ Th17 responses via IL-17A and IL-17F, IBD association | [11] | 0·93 | 0·443 | 1·24‡ | 0·037* |
| STAT1 | ↓ Th17 responses | [28] | 1·41† | 0·279 | 1·47† | 0·004* |
| STAT2 | ? | – | 1·58† | 0·053 | 1·34† | 0·065 |
| STAT3 | ↑ Th17 responses via IL-17 and RORC, IBD association | [27,31] | 1·07 | 0·251 | 0·98 | 0·804 |
| Differentiation factors involved in Th17 cell development | ||||||
| TGF-β1 | ↑ Th17 differentiation | [32] | 1·11 | 0·383 | 0·89 | 0·328 |
| TGF-β2 | ↑ Th17 differentiation | [32] | 1·23† | 0·002* | 0·98 | 0·768 |
| TGF-β3 | ↑ Th17 differentiation | [32] | 1·40† | 0·026* | 0·86 | 0·212 |
| IL-6 | ↑ Th17 differentiation | [32] | 1·21† | 0·494 | 1·00 | 0·942 |
| IL-21 | ↑ Th17 differentiation | [33] | – | – | 0·95 | 0·462 |
| Growth and stabilization for Th17 cell development | ||||||
| IL-23A | ↑ Pathogenic potential of Th17 cells | [17] | 0·97 | 0·686 | 1·00 | 0·876 |
| IL-23R | ↑ Pathology via IL-23, IBD association | [27,34] | – | – | 1·12 | 0·385 |
| Effector cytokines expressed by Th17 cells | ||||||
| IL-17A | ↑ Pathology | [11] | 1·11 | 0·280 | 1·03 | 0·617 |
| IL-17B | ? | – | – | – | 1·06 | 0·356 |
| IL-17C | ? | – | – | – | 1·12 | 0·282 |
| IL-17D | ? | – | 1·14 | 0·419 | 1·09 | 0·298 |
| IL-17F | ↑ Pathology | [11] | 0·89 | 0·114 | 1·02 | 0·778 |
| IL-22 | ↓ Pathology via ↑ gut barrier integrity | [16] | – | – | 1·08 | 0·265 |
The Th17 recruitment factor gene CCL20 was suppressed significantly at both 3 days post-AA and 28 days post-AA. The Th17 cell development transcription factor genes RORC and STAT1 were up-regulated significantly only at 28 days post-AA, but not at 3 days post-AA. The Th17 cell development differentiation factor genes TGF-β1 and TGF-β2 were up-regulated significantly only 3 days post-AA but not later at 28 days post-AA. However, none of the Th17 system growth/stabilization factors or the effector Th17 system interleukins was modulated at either time-point. SS group: sham and sham group; AA group: appendicitis and appendectomy group. The 3 days post-AA study used four AA mice versus four SS mice. The 3 days post-AA study involved three AA mice versus three SS mice.
P-value < 0·05;
fold-change > 1·2;
fold-change < 0·7.
Enrichment of Th17 system-associated gene-sets
Utilizing stringent statistical cut-offs for GSEA (FDR values < 5% and P-value < 0·001), we delineated 28-day post-AA Th17 system-associated gene-sets which were also consistently altered in the distal colons of each of the AA mice when compared to that from each of the control SS mice (Table 2). Only 17 gene-sets were up-regulated in the AA group, compared to 81 down-regulated gene-sets. Of the 17 up-regulated gene-sets, 13 were associated with STAT1, a down-regulator of Th17 responses. The down-regulation of AA-associated gene-sets spans genes associated with Th17 recruitment, differentiation, activation and effector interleukin expression. Pertaining to RORC-associated gene-sets (Table 2), no gene-sets were up-regulated in AA, in contrast to 12 gene-sets down-regulated by AA.
Table 2.
Differentially regulated gene-sets associated with the T helper type 17 (Th17) system in the distal colon at 28 days appendicitis–appendectomy (AA)
| Gene-sets in AA | ||
|---|---|---|
| Gene | Up-regulated | Down-regulated |
| CCR6 | 0 | 1 |
| CCL20 | 1 | 0 |
| RORC | 0 | 12 |
| STAT1 | 13 | 3 |
| STAT2 | 0 | 2 |
| STAT3 | 1 | 18 |
| TGF-β1 | 0 | 12 |
| TGF-β2 | 0 | 8 |
| TGF-β3 | 0 | 9 |
| IL-6 | 2 | 5 |
| IL-21 | 0 | 0 |
| IL-23A | 0 | 5 |
| IL-23R | 0 | 0 |
| IL-17A | 0 | 0 |
| IL-17B | 0 | 3 |
| IL-17C | 0 | 1 |
| IL-17D | 0 | 0 |
| IL-17F | 0 | 1 |
| IL-22 | 0 | 1 |
| Total | 17 | 81 |
The gene-set groups chosen for further evaluation had stringent cut-off values (FDR < 5% and P < 0·001). Each entry had a P-value of 0·000. Using these criteria, only 17 Th17 system-associated gene-sets were up-regulated in the AA group in contrast to 81 down-regulated gene-sets. Please see Supporting information for a detailed version of this table.
Discussion
Utilizing a new murine appendicitis model developed by us [18], we showed that AA provided significant protection against subsequent experimental colitis. We used GSEA to elucidate the pathways involved in this protective effect, and RT–PCR for validation of the study. Microarray data derived from distal colonic expression from the 3-day and 28-day post-surgery time-points indicate that different genes and gene-sets are responsible for the durable protective effect of AA against colitis [20]. Perturbations in Th1 and Th2 system genes were minimal.
We chose well-defined genes associated with the Th17 system (Table 1) for microarray evaluation of gene expression levels. The Th17 recruitment factor CCL20 [26] found on inflamed gut mucosa and its ligand CCR6 [27], found on Th17 cells, are both up-regulated in IBD.
In our study, the CCL20 gene was down-regulated significantly at both 3 days post-AA and 28 days post-AA, pointing to its direct, immediate and sustained inhibition by AA. The Th17 cell development differentiation factor genes TGF-β2 and TGF-β3 were up-regulated significantly only 3 days post-AA, but not later at 28 days post-AA, suggesting that the initial proinflammatory push towards Th17 cell differentiation was expeditiously curbed. Two Th17 cell development transcription factor genes, namely RORC and STAT1, were up-regulated not at 3 days post-AA but only at 28 days post-AA. There was an up-regulation of RORC at the latter 28 days post-AA time-point, encoding the transcription factor and putative Th17-marker RORγt. It is indeed noteworthy that STAT1, which was up-regulated only at 28 days post-AA, is known to inhibit Th17 responses [28], and hence could reflect immunological efforts to curtail extant Th17 responses. Despite all these Th17 system gene modulations, none of the Th17 system growth/stabilization factors or the effector Th17 system interleukins was up-regulated at both 3 days post-AA and 28 days post-AA.
GSEA 28 days post-AA (Table 2) flowed in the direction of individual Th17 system gene expression data, revealing that only 17 gene-sets were up-regulated by AA, in stark contrast to 81 gene-sets down-regulated by AA. Of the 17 up-regulated gene-sets, 13 were associated with signal transducer and activator of transcription-1 (STAT-1), a down-regulator of Th17 responses [28]. The substantial down-regulation of AA-associated gene-sets spreads across genes associated with Th17 lymphocyte recruitment, differentiation, activation and cytokine expression.
GSEA 28 days post-AA showed that AA down-regulated 12 gene-sets associated with TGF-β1, eight gene-sets associated with TGF-β2 and nine gene-sets associated with TGF-β3 in contrast to none up-regulated with any of these genes. These point to a marked and rapid down-regulation of TGF-β1/β2/β3-associated gene-sets at the 28-day post-AA time-point, in contrast to the 3-day post-AA time-point, where TGF-β2 and TGF-β3 were up-regulated.
There was an up-regulation of the RORC gene at the latter 28 days post-AA time-point. However, pertaining to RORC-associated gene-sets elaborated by GSEA analysis (Table 2), no gene-sets were up-regulated in AA, in contrast to 12 gene-sets down-regulated by AA. All or a few of these 12 gene-sets enumerated, most of which are transcription-factor-associated gene cohorts, may contribute towards suppressing Th17 system activity.
Of special interest to us, 18 STAT3-associated gene-sets were down-regulated 28 days post-AA in contrast to just one up-regulated gene-set. STAT3 is associated strongly with IBD [27].
Cumulatively, the data imply that the Th17 effector cytokine outflow with its pathogenic potential was kept under strict check by AA (Fig. 1) via down-regulation Th17 cell-recruitment factor CCL20, and/or down-regulation (after initial up-regulation) of TGF-β2 and TGF-β3, and/or down-regulated RORC-associated gene-sets (in spite of enhanced RORC gene expression), and/or STAT1 up-regulation, and/or down-regulation of other Th17-associated gene-sets.
Figure 1 depicts a summary of the key findings in this study, which need to be ‘functionally confirmed’ with various Th17-system gene knock-out animal models and in-vitro studies. However, these would be strictly dependent upon future funding possibilities.
Previous studies have portrayed the Th17 system as either a boon or a curse in IBD. In contrast, our novel study presented in this paper identifies the complex role of Th17 system ‘components’ and associated biological pathways involved in the protective effect of AA in experimental colitis. Our study indicates categorically that AA substantially curbs Th17 recruitment, differentiation, activation and effector interleukin expression, thereby contributing significantly to suppressing Th17 pathway-mediated immunopathologial damage as in IBD or experimental colitis. Elucidating the pathways involved in these changes will lead to the development of techniques to manipulate different components of these pathways, resulting in improved IBD management.
Acknowledgments
We acknowledge National Health and Medical Research Council (NHMRC) for funding this study. We acknowledge Warren Kaplan and Mark J. Cowley from Peter Wills Bioinformatics Centre, Garvan Institute of Medical Research, Sydney, Australia, who assisted us with Gene Set Enrichment Analysis. This study was funded by the National Health and Medical Research Council (NHMRC).
Disclosures
None to report.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Table S1. Differentially regulated gene-sets associated with the T helper type 17 (Th17) system in the distal colon at 28 days post-appendicitis-appendectomy (AA).
References
- 1.Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132:910–925. doi: 10.1093/oxfordjournals.aje.a115734. [DOI] [PubMed] [Google Scholar]
- 2.Marudanayagam R, Williams GT, Rees BI. Review of the pathological results of 2660 appendicectomy specimens. J Gastroenterol. 2006;41:745–749. doi: 10.1007/s00535-006-1855-5. [DOI] [PubMed] [Google Scholar]
- 3.Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis: a critical review. Inflamm Bowel Dis. 2002;8:277–286. doi: 10.1097/00054725-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Lopez Ramos D, Gabriel R, Cantero Perona J, Moreno Otero R, Fernandez Bermejo M, Mate Jimenez J. Association of MALTectomy (appendectomy and tonsillectomy) and inflammatory bowel disease: a familial case–control study. Rev Esp Enferm Dig. 2001;93:303–314. [PubMed] [Google Scholar]
- 5.Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy and protection against ulcerative colitis. N Engl J Med. 2001;344:808–814. doi: 10.1056/NEJM200103153441104. [DOI] [PubMed] [Google Scholar]
- 6.Radford-Smith GL, Edwards JE, Purdie DM, et al. Protective role of appendicectomy on onset and severity of ulcerative colitis and Crohn's disease. Gut. 2002;51:808–813. doi: 10.1136/gut.51.6.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizoguchi A, Mizoguchi E, Chiba C, et al. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J Exp Med. 1996;183:847–856. doi: 10.1084/jem.183.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krieglstein CF, Cerwinka WH, Laroux FS, et al. Role of appendix and spleen in experimental colitis. J Surg Res. 2001;101:166–175. doi: 10.1006/jsre.2001.6223. [DOI] [PubMed] [Google Scholar]
- 9.Farkas SA, Hornung M, Sattler C, et al. Preferential migration of CD62L cells into the appendix in mice with experimental chronic colitis. Eur Surg Res. 2005;37:115–122. doi: 10.1159/000084543. [DOI] [PubMed] [Google Scholar]
- 10.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leppkes M, Becker C, Ivanov II, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Yang XO, Chang SH, Park H, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn's disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2008;14:437–445. doi: 10.1002/ibd.20339. [DOI] [PubMed] [Google Scholar]
- 15.O'Connor W, Jr, Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 18.Watson Ng WS, Hampartzoumian T, Lloyd AR, Grimm MC. A murine model of appendicitis and the impact of inflammation on appendiceal lymphocyte constituents. Clin Exp Immunol. 2007;150:169–178. doi: 10.1111/j.1365-2249.2007.03463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheluvappa R, Luo AS, Palmer C, Grimm MC. Protective pathways against colitis mediated by appendicitis and appendectomy. Clin Exp Immunol. 2011;165:393–400. doi: 10.1111/j.1365-2249.2011.04434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brazma A. Minimum information about a microarray experiment (MIAME) – successes, failures, challenges. Scient World J. 2009;9:420–423. doi: 10.1100/tsw.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brazma A, Hingamp P, Quackenbush J, et al. Minimum information about a microarray experiment (MIAME) – toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 23.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Kaser A, Ludwiczek O, Holzmann S, et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24:74–85. doi: 10.1023/B:JOCI.0000018066.46279.6b. [DOI] [PubMed] [Google Scholar]
- 27.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villarino AV, Gallo E, Abbas AK. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J Immunol. 2010;185:6461–6471. doi: 10.4049/jimmunol.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanida S, Yoshitomi H, Nishitani K, et al. CCL20 produced in the cytokine network of rheumatoid arthritis recruits CCR6+ mononuclear cells and enhances the production of IL-6. Cytokine. 2009;47:112–118. doi: 10.1016/j.cyto.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Yang XP, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das J, Ren G, Zhang L, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fina D, Sarra M, Fantini MC, et al. Regulation of gut inflammation and Th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 34.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.