

Abstract
We report the enantiospecific total synthesis of N-methylwelwitindolinone D isonitrile. Our route features a double C-H functionalization event involving a keto oxindole substrate to introduce the tetrahydrofuran ring of the natural product.
Keywords: C-H functionalization, natural products, nitrene insertion, total synthesis, welwitindolinone
The welwitindolinone family of natural products (e.g., 1–2, Scheme 1) has attracted tremendous attention from the synthetic community over the past two decades.[1,2,3,4,5] Interest in these compounds stems from their promising biological profiles, in addition to their compact, yet daunting structures. Synthetic efforts toward the welwitindolinones have led to at least ten methods for building the bicyclo[4.3.1] core that is common to most of these natural products.[1,4] However, the sheer difficulty associated with late-stage manipulations has plagued most synthetic routes and only a few completed syntheses have been reported in recent years.[5]
Scheme 1.
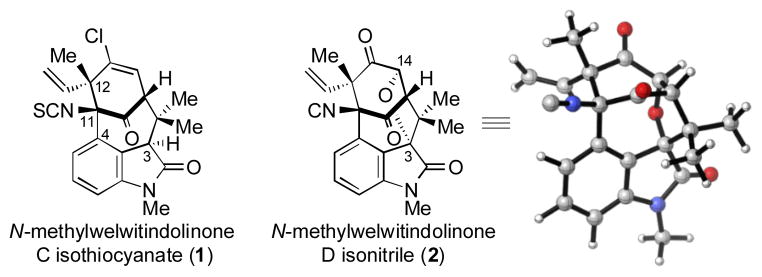
Welwitindolinones 1 and 2.
One exceptionally challenging synthetic target is N-methylwelwitindolinone D isonitrile (2).[6, 7] The compound possesses five stereocenters, two quaternary carbons, and a heavily substituted cyclohexyl ring. Compared to other related family members, 2 also possesses an ether linkage between C3 and C14. Thus, a successful synthesis of 2 would not only have to assemble the congested oxindole-fused bicyclo[4.3.1] framework, but would also have to allow for introduction of the ethereal linkage on the sterically congested face of the bicycle. Highlights of synthetic efforts toward 2 include the Wood group’s assembly of the spirocyclic oxindole[8] and Rawal’s elegant total synthesis of (±)-2 in 2011.[5a] Herein, we report our synthetic forays toward 2, which culminate in an enantiospecific synthesis.
Our retrosynthetic plan for the synthesis of 2 is presented in Scheme 2. The natural product would be accessed from 3 via late-stage manipulations. In a key disconnection, the tetrahydrofuran ring would be installed from keto-oxindole derivative 4. Of note, the ability to elaborate 4 to 3 would hinge on our ability to perform chemoselective and diastereoselective manipulations adjacent to the two carbonyls. The cyclic carbamate was thought to be accessible using an intramolecular nitrene insertion reaction[9] involving oxindole substrate 5. Substrate 5 would be derived from ketone 6, which in turn can be readily prepared from known carvone derivative 7[10] in just four steps using our previously established procedure involving an indolyne cyclization.[5b,11]
Scheme 2.

Retrosynthetic analysis of 2.
Our approach toward implementing the retrosynthetic plan is highlighted in Scheme 3. Indole 6 was converted to oxindole 8 using a one-pot oxidation/hydrolysis sequence. As the acidic conditions led to desilylation, reprotection of the alcohol was necessary to provide 9. Deuteride reduction and carbamoylation proceeded without event to furnish 5 in quantitative yield. To our delight, exposure of 5 to Ag-promoted nitrene insertion conditions[12,5e] furnished 10 in 70% yield. It should be noted that attempts to use the proteo analog of 5 gave only 44% yield of the corresponding insertion product, along with 19% of recovered ketone 9. Thus, consistent with our previous findings on an alternate substrate,[5e] the strategic use of deuterium minimizes an undesirable competitive reaction, thus giving synthetically useful yields of the desired insertion product 10. From 10, a standard deprotection/oxidation sequence delivered key intermediate 4.
Scheme 3.

Elaboration of 6 to keto oxindole 4; TBS=tert-butyldimethylsilyl, NBS=N-bromosuccinimide, DMAP=4-dimethylaminopyridine, DMF=dimethylformamide, THF=tetrahydrofuran, Tf=trifluoromethanesulfonyl, OAc=acetate, bathophenanthroline=4,7-diphenyl-1,10-phenanthroline, Dess–Martin=1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one.
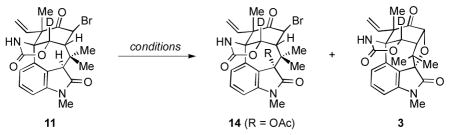
Many attempts to introduce the tetrahydrofuran ring from 4 were put forth. Unfortunately, efforts toward site-selective functionalization of one carbonyl over the other via enol ethers were unsuccessful. After considerable experimentation, it was found that the keto carbonyl could be α-functionalized first upon treatment of 4 with CuBr2 in THF at ambient temperature to yield 11 as a single diastereomer (Scheme 4). It was hoped that C3-oxidation would provide an alcohol intermediate that would cyclize to give the necessary tetrahydrofuran ring. However, upon treatment of 11 with C3 oxidation conditions,[5b] the desired oxidation and cyclization did not occur. Instead, we unexpectedly obtained cyclobutane 13 in high yield, presumably via direct cyclization of the oxindole enolate (see transition structure 12).[13] X-ray analysis of a single crystal of 13 validated our structural assignment.[14,7]
Scheme 4.

Unexpected formation of cyclobutane 13; THF = tetrahydrofuran.
As a workaround, we opted to introduce a protected hydroxyl group directly onto C3 of substrate 11. Mn(OAc)3 was deemed a potential reagent for selective C3-acetoxylation, based on its use in benzylic acetoxylation reactions.[15] As shown in Table 1, treatment of oxindole 11 with Mn(OAc)3 in AcOH at 80 °C provided acetoxylated product 14 (entry 1). Interestingly, when the corresponding reaction was conducted at 150 °C, we obtained a 53% yield of 3, which possesses the desired tetrahydrofuran ring. Alternatively, 3 could also be prepared in one-pot by performing the acetoxylation at 80 °C, removing the volatiles, and exposing the crude intermediate to K2CO3 in MeOH and H2O at 70 °C.
Table 1.
Conversion of 11 to acetate 14 and cyclized product 3.
| |||
|---|---|---|---|
| entry | conditions | conversion to products 14 and 3 | |
| 1 | Mn(OAc)3 (4.0 equiv), AcOH, 80 °C | 74 | 0 |
| 2 | Mn(OAc)3 (4.0 equiv), AcOH, 150 °C | 2 | 53 |
| 3 | Mn(OAc)3 (4.0 equiv), AcOH, 80 °C; K2CO3, MeOH, H2O, 70 °C | 0 | 56 |
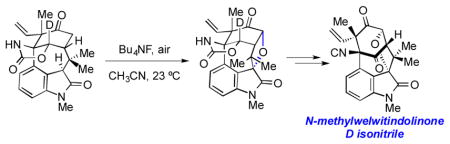
We also explored the feasibility of directly converting keto oxindole 4 to 3 (Scheme 5). Of note, the Wood group was able to elegantly install a tetrahydrofuran ring from a keto oxindole substrate using basic conditions and O2.[8] Despite the modest yield, this key precedent laid the groundwork for additional experimentation. To our delight, we found that simple exposure of 4 to tetrabutylammonium fluoride in acetonitrile in the presence of air efficiently delivered 3.[16] In previous studies, we[17] and others[18] have found that TBAF/air can facilitate C3 oxidation of oxindoles containing the welwitindolinone scaffold, but the use of TBAF/air to build an ethereal linkage via double C-H functionalization was unknown. It should be noted that the use of other bases in place of TBAF, such as K2CO3 and Cs2CO3, also promoted the formation of 3, albeit in lower yields. It is likely that this efficient method for introducing the tetrahydrofuran ring proceeds by initial diastereoselective C3 oxidation, followed by cyclization.[19] Related C3-peroxy compounds have been observed in our studies[20] and in Wood’s.[8]
Scheme 5.
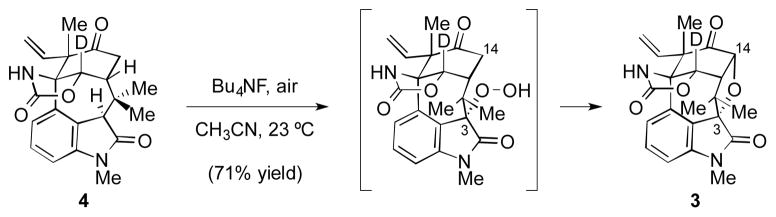
Double C-H functionalization of substrate 4 to install the tetrahydrofuran ring.
To complete the total synthesis, it remained to elaborate the cyclic carbamate to the ketone and isonitrile functional groups present in 2 (Scheme 6). Unexpectedly, attempted hydrolysis of 3 led to cyclohexyl ring fragmentation, a process that was attributed to the reactivity of the ketone. To circumvent this, ketone 3 was reduced to alcohol 15 with LiAlH4. Fortunately, upon exposure of 15 to hydrolysis conditions, cyclohexyl ring fragmentation was not observed. Hydrolysis gave the desired diol intermediate, which was oxidized with IBX to provide diketone 16. Finally, formylation provided 17, which was directly exposed to standard dehydration conditions to deliver (+)-2.
Scheme 6.

Completion of (+)-2; THF=tetrahydrofuran, dioxane=1,4-dioxane, IBX=2-iodoxybenzoic acid, TFA=trifluoroacetic acid, DMSO=dimethylsulfoxide, Burgess reagent=methyl N-(triethylammoniumsulfonyl)carbamate.
In summary, we have completed the enantiospecific total synthesis of N-methylwelwitindolinone D isonitrile. Several unexpected hurdles, including the formation of the unusual cyclobutane-containing compound 13 were overcome en route to the natural product. Our total synthesis features a double C-H functionalization event of keto oxindole 4 to introduce the tetrahydrofuran ring of 2 and is achieved in 17 steps from readily available carvone derivative 7.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH-NIGMS (R01 GM090007), Boehringer Ingelheim, DuPont, Eli Lilly, Amgen, AstraZeneca, Roche, the A. P. Sloan Foundation, the S.T. Li Foundation, the Dreyfus Foundation, the University of California, Los Angeles, Bristol–Myers Squibb (A.D.H.), the NSF (N.A.W., DGE-1144087), and the Foote Family (A.D.H. and E.D.S.) for financial support. We thank the Garcia–Garibay laboratory (UCLA) for access to instrumentation and Dr. John Greaves (UC Irvine) for mass spectra. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For pertinent reviews, see: Wood JL. Nat Chem. 2012;4:341. doi: 10.1038/nchem.1335.Huters AD, Styduhar ED, Garg NK. Angew Chem. 2012;124:3820. doi: 10.1002/anie.201107567.Angew Chem Int Ed. 2012;51:3758.Brown LE, Konopelski JP. Org Prep Proced Int. 2008;40:411.Avendaño C, Menéndez JC. Curr Org Synth. 2004;1:65.
- 2.a) Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GML, Shaffer S, Smith CD, Smitka TA. J Am Chem Soc. 1994;116:9935. [Google Scholar]; b) Jimenez JI, Huber U, Moore RE, Patterson GML. J Nat Prod. 1999;62:569. doi: 10.1021/np980485t. [DOI] [PubMed] [Google Scholar]
- 3.For total syntheses of welwitindolinone A isonitrile, see: Baran PS, Richter JM. J Am Chem Soc. 2005;127:15394. doi: 10.1021/ja056171r.Reisman SE, Ready JM, Hasuoka A, Smith CJ, Wood JL. J Am Chem Soc. 2006;128:1448. doi: 10.1021/ja057640s.
- 4.For progress toward the synthesis of bicyclo[4.3.1]-welwitindolinones, see: Konopelski JP, Deng H, Schiemann K, Keane JM, Olmstead MM. Synlett. 1998:1105.Wood JL, Holubec AA, Stoltz BM, Weiss MM, Dixon JA, Doan BD, Shamji MF, Chen JM, Heffron TP. J Am Chem Soc. 1999;121:6326.Kaoudi T, Quiclet-Sire B, Seguin S, Zard SZ. Angew Chem. 2000;112:747.Angew Chem Int Ed. 2000;39:731.Deng H, Konopelski JP. Org Lett. 2001;3:3001. doi: 10.1021/ol016379r.Jung ME, Slowinski F. Tetrahedron Lett. 2001;42:6835.López-Alvarado P, García-Granda S, Álvarez-Rúa C, Avendaño C. Eur J Org Chem. 2002:1702.MacKay JA, Bishop RL, Rawal VH. Org Lett. 2005;7:3421. doi: 10.1021/ol051043t.Baudoux J, Blake AJ, Simpkins NS. Org Lett. 2005;7:4087. doi: 10.1021/ol051239t.Greshock TJ, Funk RL. Org Lett. 2006;8:2643. doi: 10.1021/ol0608799.Lauchli R, Shea KJ. Org Lett. 2006;8:5287. doi: 10.1021/ol0620747.Guthikonda K, Caliando BJ, Du Bois J. Abstr. Pap., 232nd ACS National Meeting; September, 2006; p. abstr ORGN-002.Xia J, Brown LE, Konopelski JP. J Org Chem. 2007;72:6885. doi: 10.1021/jo071156l.Boissel V, Simpkins NS, Bhalay G, Blake AJ, Lewis W. Chem Commun. 2009:1398. doi: 10.1039/b820674k.Boissel V, Simpkins NS, Bhalay G. Tetrahedron Lett. 2009;50:3283.Tian X, Huters AD, Douglas CJ, Garg NK. Org Lett. 2009;11:2349. doi: 10.1021/ol9007684.Trost BM, McDougall PJ. Org Lett. 2009;11:3782. doi: 10.1021/ol901499b.Brailsford JA, Lauchli R, Shea KJ. Org Lett. 2009;11:5330. doi: 10.1021/ol902173g.Freeman DB, et al. Tetrahedron. 2010;66:6647. doi: 10.1016/j.tet.2010.04.131.Heidebrecht RW, Jr, Gulledge B, Martin SF. Org Lett. 2010;12:2492. doi: 10.1021/ol1006373.Ruiz M, López-Alvarado P, Menéndez JC. Org Biomol Chem. 2010;8:4521. doi: 10.1039/c0ob00382d.Bhat V, Rawal VH. Chem Commun. 2011;47:9705. doi: 10.1039/c1cc13498a.Bhat V, MacKay JA, Rawal VH. Org Lett. 2011;13:3214. doi: 10.1021/ol201122f.Bhat V, MacKay JA, Rawal VH. Tetrahedron. 2011;67:10097. doi: 10.1016/j.tet.2011.09.088.Zhang M, Tang W. Org Lett. 2012;14:3756. doi: 10.1021/ol301614v.Cleary L, Brailsford JA, Launchli R, Shea KJ. Abstr. Pap. 245th ACS National Meeting; April, 2013; p. abstr ORGN-391.
- 5.For complete and formal total syntheses of bicyclo[4.3.1]-welwitindolinones, see: Bhat V, Allan KM, Rawal VH. J Am Chem Soc. 2011;133:5798. doi: 10.1021/ja201834u.Huters AD, Quasdorf KW, Styduhar ED, Garg NK. J Am Chem Soc. 2011;133:15797. doi: 10.1021/ja206538k.Fu T-h, McElroy WT, Shamszad M, Martin SF. Org Lett. 2012;14:3834. doi: 10.1021/ol301424h.Allan KW, Kobayashi K, Rawal VH. J Am Chem Soc. 2012;134:1392. doi: 10.1021/ja210793x.Quasdorf KW, Huters AD, Lodewyk MW, Tantillo DJ, Garg NK. J Am Chem Soc. 2012;134:1396. doi: 10.1021/ja210837b.Fu Th, McElroy WT, Shamszad M, Heidebrecht RW, Jr, Gulledge B, Martin SF. Tetrahedron. 2013;69:5588. doi: 10.1016/j.tet.2013.03.010.
- 6.3D representation of 2 was obtained using B3LYP/6-31G* calculations (geometry optimization), using MacSpartan software.
- 7.Image prepared using CYLview: Legault CY. CYLview, 1.0b. Université de Sherbrooke; Québec, Montreal, Canada: 2009. http://www.cylview.org.
- 8.Holubec AA. PhD Dissertation. Yale University; New Haven, CT: 2000. Progress Toward the Total Synthesis of the Welwitindolinone Alkaloids: Efficient Construction of the Carbocyclic Skeleton. [Google Scholar]
- 9.For a recent review on C–N bond forming reactions involving C(sp3)-H bonds, see: Jeffrey JL, Sarpong R. Chem Sci. 2013 doi: 10.1039/c3sc51420j.
- 10.Sakagami M, Muratake H, Natsume M. Chem Pharm Bull. 1994;42:1393. [Google Scholar]
- 11.For our laboratory’s recent studies involving indolynes and other heterocyclic arynes, see: Bronner SM, Bahnck KB, Garg NK. Org Lett. 2009;11:1007. doi: 10.1021/ol802958a.Cheong PHY, Paton RS, Bronner SM, Im GYJ, Garg NK, Houk KN. J Am Chem Soc. 2010;132:1267. doi: 10.1021/ja9098643.Im GYJ, Bronner SM, Goetz AE, Paton RS, Cheong PHY, Houk KN, Garg NK. J Am Chem Soc. 2010;132:17933. doi: 10.1021/ja1086485.Bronner SM, Goetz AE, Garg NK. J Am Chem Soc. 2011;133:3832. doi: 10.1021/ja200437g.Goetz AE, Bronner SM, Cisneros JD, Melamed JM, Paton RS, Houk KN, Garg NK. Angew Chem. 2012;124:2812. doi: 10.1002/anie.201108863.Angew Chem Int Ed. 2012;51:2758.Goetz AE, Garg NK. Nat Chem. 2013;5:54. doi: 10.1038/nchem.1504.Bronner SM, Goetz AE, Garg NK. Synlett. 2011:2599.
- 12.a) Li Z, Capretto DA, Rahaman R, He C. Angew Chem. 2007;119:5276. doi: 10.1002/anie.200700760. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:5184. [Google Scholar]; b) Cui Y, He C. Angew Chem. 2004;116:4306. [Google Scholar]; Angew Chem Int Ed. 2004;43:4210. [Google Scholar]
- 13.Variations in reaction conditions (e.g., employing a variety of bases, saturating with O2) did not overcome the formation of 13.
- 14.CCDC 960226 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 15.Citterio A, Finzi C, Santi R, Strology S. J Chem Res, Synop. 1988:156. [Google Scholar]
- 16.For recent alkaloid syntheses that feature the strategic use of indole oxidation chemistry, see: Han S, Movassaghi M. J Am Chem Soc. 2011;133:10768. doi: 10.1021/ja204597k.Qi X, Bao H, Tambar UK. J Am Chem Soc. 2011;133:10050. doi: 10.1021/ja203960b.
- 17.Huters AD, Quasdorf KW, Garg NK. Unpublished work. University of California; Los Angeles, CA: 2010. [Google Scholar]
- 18.Buckley BR, Fernández B, D-R Tetrahedron Lett. 2013;54:843. [Google Scholar]
- 19.Treatment of 4 with 3.0 equiv TBAF and 50.0 equiv MeOD in CH 3CN under an atmosphere of N2 at ambient temperature gave 40% deuterium incorporation at C3 after 10 min, whereas treatment under the same conditions for 1 h gave 50% deuterium incorporation at C3 and 25% deuterium incorporation at C14.
-
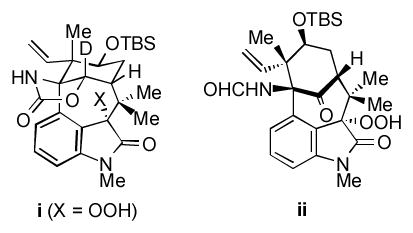
20.Efforts to isolate the putative peroxy species (Scheme 5) have been unsuccessful; however, we have isolated several related compounds, such as i and ii, by oxidation of the corresponding oxindoles.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.