

Abstract
Creutzfeldt-Jakob disease (CJD) is a rapidly progressive and ultimately fatal disorder of the central nervous system. It occurs worldwide with an incidence of 0.5-1 new case per million population per year. No specific treatment is available and management is limited to supportive care. Autopsy or biopsy provides a definitive diagnosis. Because of the transmissible nature of the disease and hesitancy of patients/family members to give consent for biopsy, numerous challenges in confirming the clinical diagnosis are faced by healthcare professionals. We report a case of 66-year-old male who was hospitalized due to hip fracture following a fall. Acute mental status changes followed the surgical fixation of hip fracture which triggered neurologic work up. This finally revealed suspicion and confirmation of CJD. Patient had progressive cognitive decline with akinetic mutism during further hospital stay and was later discharged home with hospice. Shorter thereafter he died at home. This case demonstrates the importance of keeping an open mind towards possibility of CJD when faced with esoteric neurologic presentations. Also this case provides insight into challenges in quarantine and sterilization of surgical instruments when these patients go through major surgeries.
Key words: sCJD, Myoclonus, MRI brain, Diffusion weighted imaging, Quarantine
Introduction
Sporadic Creutzfeldt-Jakob disease (sCJD) is a fatal human transmissible spongiform encephalopathy. It occurs because of spontaneous transformation of normal prion protein to abnormal proteins. The sporadic form of CJD is the most common prion disorder in humans and accounts for ~85% of all cases of human prion disease, while inherited prion disease account for 10-15% of all cases. Iatrogenic CJD seems to be the result of the accidental inoculation of patients with prion. Variant CJD (vCJD) is the result of exposure to tainted beef from cattle with bovine spongiform encephalopathy (BSE). Hallmark of prion disease, whether sporadic, inherited or acquired by infection, is that it involves the aberrant metabolism of Prion related Protein (PrP). In humans, the PrP gene is designated as Prion related Nucleo-peptide(PRNP) and is located on the short arm of chromosome 20. Different clinical and pathological sub-types of sporadic CJD have been linked to polymorphism status at codon 129 of PRNP combined with the type of protease resistant prion protein found in the brain.1,2
Common findings of sCJD on neurological examination include, but are not limited to, rapidly progressive dementia, myoclonus, visual disturbances, cerebellar ataxia, and pyramidal/extra pyramidal signs. These signs are non-specific and can be caused by a variety of diseases.3 Different kinds of atypical presentations which lead to initial diagnosis of stroke, depression and supranuclear palsy are also known in literature.4
In conjunction with above mentioned clinical signs diagnostic tests like detection of 14-3-3 protein in CSF, characteristic MRI findings and typical EEG pattern are used to make a diagnosis of probable or possible CJD. The diagnosis of CJD is usually confirmed by the presence of spongiform degeneration with deposition of prion protein particularly in cerebral cortex and immunodiagnostic testing of brain tissue obtained at biopsy.
Case Report
A 66 yr old male was admitted to the hospital following a fall on 12/29/2011. The fall was under strange circumstances wherein he felt sudden vertigo sensation while shutting his car door. He sustained an intertrochanteric fracture of hip during the fall. His initial evaluation indicated that he had profound dysmmetria in both upper extremities and absent proprioception. Further discussion with patient’s wife, indicated he had problems with balance since 2 months prior to presentation. He saw an otorhinolaryngologist for the same and underwent canolith repositioning procedure with the presumed initial diagnosis of benign positional vertigo. She felt that his balance problems had gotten worse since then.
During post operative course of hip surgery patient spiked temperature and also had mental status changes. A lumbar puncture was done and meningeal infection was rule out. CSF was also sent for 14-3-3 assay along with complete paraneoplastic panel. He was then also noted to have slurred speech. An MRI of the brain was done which showed on FLAIR (fast fluid attenuated inversion recovery) images that there were T2 hyper intense signal changes and restricted diffusion in the bilateral basal ganglia(caudate and putamen nuclei) and right frontal cortex (Figure 1). Although the official radiology report stated the diagnosis as basal ganglia infarct, teleradiology service hinted at the possibility of CJD along with toxic/metabolic encephalopathy. EEG revealed diffuse slowing but no sharp wave activity. Patients fever subsided but he continued to have severe ataxia.
Figure 1.
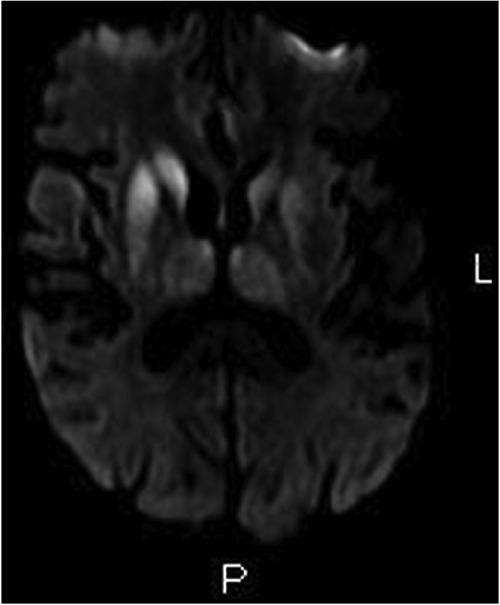
Magnetic resonance imaging of the brain: FLAIR image.
Based on the CDC criteria (Table 1), since patient had cerebellar problem with myoclonus and also MRI findings a diagnosis of probable CJD was made. Brain biopsy was suggested so that definitive diagnosis of CJD could be made but family refused. Patient was then send to inpatient rehab unit to work on balance and coordination while being closely followed by medicine service.
Table 1.
Centers for Disease Control and Prevention criteria for Sporadic Creutzfeldt-Jakob disease, 2010.
| Disease | Criteria |
|---|---|
| Definite | Detection of protease-resistant Prion Protein or scrapie-associated fibrils by neuropathology, immunochemical technique, and/or Western blot. |
| Probable | No findings indicating alternative diagnoses AND progressive dementia with at least 2 of (i)-(iv) AND at least one of (a)-(c). |
| Possible | No findings indicating alternative diagnoses AND progressive dementia with duration of less than 2 years AND with at least 2 of (i)-(iv) AND at least one of (a)-(c). |
Adapted from: WHO and Zerr et al.13,14 (i) Myoclonus; (ii) Visual or cerebellar problems; (iii) Pyramidal or extra pyramidal features; (iv) Akinetic mutism. (a) Periodic sharp wave complexes on electroencephalography; (b) Positive 14-3-3 protein in the cerebrospinal fluid with a disease duration of less than 2 years; (c) High signal abnormalities in caudate nucleus and/or putamen on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR) MRI.
In the Rehab Unit patient had more myoclonus, a kinetic mutism along with worsening cognitive decline. Repeat MRI was obtained which did reveal changes of CJD more clearly. By this time his CSF immunoassay was positive for 14-3-3 protein and a diagnosis of CJD was established. When the diagnosis was told to family they choose palliative care, patient was then discharged to home with hospice. Patient expired 5 days after discharge to home.
Autopsy an performed at University of Chicago Medical center revealed spongiform changes in basal ganglia, thalamus, substantia nigra and cerebellum with relative cortical sparing (Figure 2). The spongiform changes in cerebellum were associated with atrophic changes in internal granular layer and Purkinje cell layer (Figure 3). The morphologic changes fitted into one of the strains associated with relative cortical sparing and more prominent basal ganglia/cerebellar disease. Immunohistochemical exam done at National Prion Disease Pathology Surveillance Center, Cleveland (USA) confirmed sporadic CJD of VV2 type. The VV2 phenotype was based on molecular subtype determined by PRNP genotype and pathologic prion protein according to classification of sporadic CJD proposed by Parchi et al.5 VV2 is an ataxic variant which has ataxia at onset in 15% and has longer duration of illness. The PrP gene sequencing ruled out the presence of pathogenic mutation in the coding region of PrP gene and further abnormal prion protein type was characterized by Western Blot.
Figure 2.
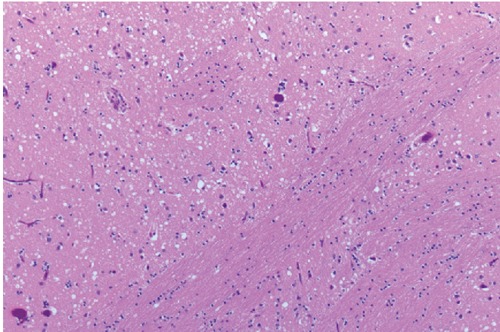
Tissue slide of basal ganglia.
Figure 3.
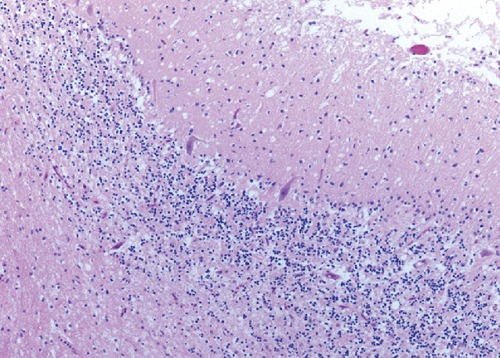
Tissue slide of cerebellum.
Discussion
Making a diagnosis of CJD is challenging since early clinical features resemble closely other neurological disorders like stroke, dementia and multiple sclerosis.6-8 Current diagnostic criteria for possible CJD vary based on clinical signs utilized.9 A first set of criteria was proposed by Masters et al. in 1979.10 These original criteria have been extensively used and adapted by different investigators. More recently, the EU collaborative group decided to consider the presence of the 14-3-3 brain protein in CSF as an additional diagnostic criterion for clinically probable CJD.11,12 In European MRI-CJD consortium proposed in 2009 MRI has gained significant diagnostic role in suspected cases of CJD. The frequency of diagnosing CJD increases with additional data but at the cost of potentially increasing the false positive rate.13 A CDC diagnostic criterion (Table 1) was adapted from Zerr et al.13 and Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation, Feb 9-11, 1998, Geneva, Switzerland.14
Although it’s important to keep CJD in mind when considering obscure neurological presentations, consideration of this diagnosis early on without excluding more common conditions would be unwise. Our patient had indolent onset of disease with otorhinolaryngological symptoms two months prior to hospitalization. Such presentation with otorhinolaryngological symptoms of hearing problem and ataxia has been earlier described in literature.15 By the time our patient had fall with altered mental status which prompted work up, his spongiform brain disease was quiet advanced leading to rapid cognitive decline and death within a month of diagnosis.
The definitive diagnosis is by histological exam of the brain once suspicion is established. Most of the time getting biopsy in a timely manner is not possible. This is true given the costly and aggressive nature of diagnostic test for a non treatable condition. Also the rigorous decontamination and sterilization techniques for handling tissue at biopsy may make it impractical in a community setting.16 Frankly biopsy is rarely undertaken and patients who die after CJD like disease have autopsy at specialized tertiary centers to confirm diagnosis. Lately MRI and immunoassay on the CSF in conjunction with periodic sharp complexes pattern on EEG have been useful tools for the ante mortem diagnosis of CJD. In our patient tip off was the finding on MRI of bilateral areas of increased signal intensity affecting the basal ganglion followed by positive 14-3-3 in CSF. The CSF 14-3-3 immunoassay has sensitivity and specificity of 95% and 93% respectively. MRI and CSF 14-3-3 protein have emerged as useful tools for the ante mortem diagnosis of CJD given challenges of obtaining tissue. High signal changes in the basal ganglia and cerebral cortex on FLAIR and DWI (Diffusion weighted imaging) are useful in the diagnosis of sCJD. DWI is the most sensitive MRI technique in early clinical diagnosis of CJD can detect characteristic lesions in the majority of patients regardless of the presence of Periodic Sharp Complexes.17 The characteristic MRI lesion pattern is bilateral symmetric hyper intense signal changes in the basal ganglia and cortical regions.18 In the appropriate clinical setting these MRI findings are quiet helpful to diagnose CJD although significant overlap with other conditions does exist.
Conclusions
Our patient underwent a hip replacement surgery at a time when his diagnosis was not made so standard sterilization method was used which typically isn’t adequate for eradication of CDJ prion protein virus. This presents a theoretical risk of prion protein transmission through surgical equipment. Although this risk is well recognized for neurosurgical procedures, there has been no report in literature of CJD transmission after orthopedic procedure. Both laboratory and clinical research has determined that human-to-human transmission of CJD can occur as the result of tissue implant, use of contaminated instruments, or administration of human hormones extracted from the organs of human cadavers. Recently, three cases of vCJD acquired via blood transfusion were reported in the UK.19 In all these cases, the blood was extracted from persons with vCJD prior to their diagnosis. The third recipient had abnormal prion protein isolated in lymphoid tissue at post-mortem.
It is important to have strict quarantine and sterilization of struments for any surgery for a patient with suspicion for CJD. In our patient the diagnosis of Probable CJD was only after MRI brain done several days after hip surgery so special decontamination of surgical instruments was not possible. Since the ante mortem diagnosis is rare and disease runs short course before death, it presents a challenge for implementing control and quarantine measures. It’s therefore prudent to implement the highest level of sterilization and quarantine measures for anyone presenting with progressive dementia along with any 2 of 4 clinical features (as mentioned above in Table 1. CDC’s Diagnostic criteria, 2010) when alternative diagnosis is ruled out with routine investigations.
The role of MRI and CSF 14-3-3 assay protein early on in disease is well justified, in the face of importance of strict quarantine needed early on in the disease especially since tissue diagnosis is often not available until after the death of patient. Lastly it’s important for physicians to keep the possibility of CJD in mind when evaluating patients with altered mental status when no distinct etiology is immediately apparent.
References
- 1.Campbell S, Warner M, Margaret E, et al. CJD - a case of mistaken identity. Lancet 2004;364:2068. [DOI] [PubMed] [Google Scholar]
- 2.Collinge J. Human prion diseases and bovine spongiform encephalopathy. Hum Mol Genet 1997;6:1699-705 [DOI] [PubMed] [Google Scholar]
- 3.Kojima G, Tatsuno BK, Inaba M, et al. Creutzfeldt-Jacob disease: a case report and differential diagnosis. Hawaii J Med Public Health 2013;72:136-9 [PMC free article] [PubMed] [Google Scholar]
- 4.Panagariya A, Jain RS, Sharma AK. Stroke like presentation of Creutzfeldt-Jakob disease: an unusual variant. J Assoc Physicians India 1999;47:548-50 [PubMed] [Google Scholar]
- 5.Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathologica 2012;50:20-45 [PubMed] [Google Scholar]
- 6.Pachalska M, Kurzbauer H, Formińska-Kapuścik M, et al. Atypical features of dementia in a patient with Creutzfeldt-Jakob disease. Med Sci Monit 2007;13:9-19 [PubMed] [Google Scholar]
- 7.Marciani MG, Bernardi G, Sancesario G, et al. Rapidly progressive form of Creutzfeldt-Jakob disease without dementia: a case report. Int J Neurosci 1996;84:115-20 [DOI] [PubMed] [Google Scholar]
- 8.Vingerhoets FJ, Hegyi I, Aguzzi A, et al. An unusual case of Creutzfeldt-Jakob disease. Neurology 1998;51:617-9 [DOI] [PubMed] [Google Scholar]
- 9.Brandel JP, Delasnerie-Laupretre N, Laplanche JL, et al. Diagnosis of Creutzfeldt-Jakob disease: effect of clinical criteria on incidence estimates. Neurology 2000;54:1095-9 [DOI] [PubMed] [Google Scholar]
- 10.Masters CL, Harris JO, Gajdusek DC, et al. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol 1979;5:177-88 [DOI] [PubMed] [Google Scholar]
- 11.Hsich G, Kenney K, Gibbs CJ Jr, et al. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. New Engl J Med 2006; 335:924-30 [DOI] [PubMed] [Google Scholar]
- 12.Zerr I, Bodemer M, Gefeller O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jacob disease. Ann Neurol 1998;43:32-40 [DOI] [PubMed] [Google Scholar]
- 13.Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jacob disease. Brain 2009;132:2659-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WHO Global surveillance, diagnosis, and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. Available from: http://www.who.int/csr/resources/publications/bse/whoemczdi989.pdf Accessed: July 2013 [Google Scholar]
- 15.Bigelow DC, Eisen MD, Yen DM, et al. Otolaryngological manifestations of Creutzfeldt-Jakob disease. Arch Otolaryngol Head Neck Surg 1998;124:707-10 [DOI] [PubMed] [Google Scholar]
- 16.Tanwani LK, Furman CD, Ritchie CS. Diagnostic challenges in Creutzfeldt-Jacob disease: case report. South Med J 2003; 96:832-5 [DOI] [PubMed] [Google Scholar]
- 17.Pereira E. Diffusion weighted sequence on MRI for the diagnosis of Creutzfeldt-Jakob disease. Arq Neuropsiquiatr 2002;60:906-8 [DOI] [PubMed] [Google Scholar]
- 18.Onofrj M, Fulgente T, Gambi D, Macchi G. Early MRI findings in Creutzfeldt-Jakob disease. J Neurol 1993;240:423-6 [DOI] [PubMed] [Google Scholar]
- 19.Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang 2006;91:221-30 [DOI] [PubMed] [Google Scholar]