

Abstract
The intra-host evolutionary and population dynamics of the human immunodeficiency virus type 1 (HIV-1), the cause of the acquired immunodeficiency syndrome, have been the focus of one of the most extensive study efforts in the field of molecular evolution over the past three decades. As HIV-1 is among the fastest mutating organisms known, viral sequence data sampled over time from infected patients can provide, through phylogenetic analysis, significant insights about the tempo and mode of evolutionary processes shaped by complex interaction with the host milieu. Five main aspects are discussed: the patterns of HIV-1 intra-host diversity and divergence over time in relation to different phases of disease progression; the impact of selection on the temporal structure of HIV-1 intra-host genealogies inferred from longitudinally sampled viral sequences; HIV-1 intra-host sub-population structure; the potential relationship between viral evolutionary rate and disease progression and the central evolutionary role played by recombination occurring in super-infected cells.
Key words: HIV-1, phylogenetic analysis, intrahost evolution, population dynamics
Introduction
The human immunodeficiency virus type 1 (HIV-1), a small diploid RNA retrovirus with a genome size of approximately 10,000 nucleotide bases, was isolated for the first time about three decades ago and recognized as the etiologic agent of the acquired immunodeficiency syndrome (AIDS).1 Early study soon demonstrated that HIV-1 displayed remarkable inter- and intra-host genetic heterogeneity,2-4 as well as specific evolutionary patterns within the infected host.5 Three main factors contribute to the fast evolution of the virus. First, the low-fidelity of the reverse transcriptase characterized by an error rate of 3.4×10–5 mutations per nucleotide per cycleresulting in the introduction of one nucleotide substitution every second to third newly synthesized genome.6,7 Second, the rapid viral generation time – defined as the time from release of a virion until it infects another cell and causes the release of a new generation of viral particles – estimated in 1.5-2.6 days with a production of ~1010–1012 new virions each day.8-11 Third, the elevated recombination rate with a number of crossovers between two RNA templates estimated to range from three to nine per genome per round of replication.12-14 These factors make HIV-1 one of the fastest evolving organisms known and the heterogeneous viral swarm that infects most HIV-1seropositive individuals fits the definition of a measurably evolving population, i.e. a population from which molecular sequences taken at different points in time will display a statistically significant number of genetic differences.15 In other words, HIV-1 evolution in vivo is an observable microevolutionary process that can be investigated through the analysis of viral sequences sampled longitudinally (heterochronous sequences) from an infected host.
HIV-1 intra-host evolutionary and population dynamics have been under extensive investigation,16,17 especially for their potential role in pathogenic processes,18 and because they constitute a major challenge for the development of an effective vaccine.19-21 Nevertheless, two fundamental questions – what are the intra-host ecological factors shaping viral diversity over time and what is the potential relationship between viral evolution and pathogenesis – are still presently debated.16
In the next sections, the current knowledge on HIV-1 intra-host evolutionary patterns in the absence of antiretroviral treatment (ART) will briefly be reviewed with special emphasis on results obtained through the development and application of phylogenetic techniques. I will show how phylogenetic analysis has been able to shed light on the complex interplay between viral evolution and immune selection that can ultimately result in disease progression.
Patterns of HIV-1 intra-host diversity and divergence over time
HIV-1 genetic variability at the nucleotide level can reach up to 5% within an infected subject and provides the viral population with the ability to adapt rapidly to changes in its environment.16 The infection is typically characterized by three phases – acute infection, clinical latency and progression to AIDS – during which viral genetic divergence and diversity display consistent patterns that have been associated with disease progression. 22,23 HIV-1 intra-host diversity and divergence over time can be defined according to specific phylogenetic criteria.24 Given a data set of HIV-1 sequences sampled longitudinally from an infected patient, diversity is the average pairwise genetic (nucleotide or amino acid) distance within the sequences sampled at a given time point, while divergence is their average genetic distance from the most recent common ancestor (MRCA), i.e. the root of the viral genealogy (phylogeny).
When an individual becomes infected with HIV-1, a relatively homogenous population of the virus is initially harbored because transmission is usually associated with a significant population bottleneck.25-27 A new infection can be established by just one single virion,28,29 although the number of variants transmitted seem to be dependent on the transmission route and bottlenecks associated with sexual transmission may be greater than with vertical transmission (e.g. mother to child or injecting drug use).30 It has been shown that in men who have sex with men (MSM) the minimum number of transmitted viruses can range from 2 to 10 with subsequent viral recombination leading to rapid and extensive genetic shuffling among viral lineages.31 A combined analysis also revealed a significantly higher frequency of multivariant transmission in MSM than in heterosexuals.31 The severity of transmission bottlenecks could be the result of anatomical barriers (e.g. mucosal barrier in heterosexual transmissions) and by immune selection mediated by early cytotoxic T cell responses.32 However, in an animal model, infection with a highly heterogeneous SIV viral swarm of CD8+ depleted Rhesus macaques, where immune selection is expected to be weak or inexistent, low frequency SIV variants characterized by specific amino acid motif in gp120 were still transmitted more efficiently than higher frequency ones in six different primates.33 Overall, it is important to keep in mind that these studies used, by necessity, indirect methods to quantify the amount of transmitted variants, because it is obviously very difficult to measure HIV-1 genetic diversity at the exact moment of transmission. Indeed, it has rightly been observed that homogeneity of the viral population during primary infection does not necessarily imply homogeneity of the transmitted variants.34
The clinical latency phase of HIV-1 infection is characterized by a progressive increase in both viral divergence and diversity, while during the last phase, when the immune system collapses and progression to AIDS begins, viral divergence stabilizes and viral diversity declines.23 This observation has been explained as a consequence of CD4+ T cell depletion, which likely results in less effective selection pressure on the virus, as well as significant decrease in target cells capable of sustaining viral replication.35
Temporal structure of HIV-1 intra-host genealogies
Phylogenetic studies have clearly shown that, in the absence of ART, the central paradigm governing HIV-1 intra-host evolutionary dynamics following primary infection is the turnover of genetic variants over time through sequential population bottlenecks.5,23 HIV-1 intra-host genealogies of heterochronous sequences display strong temporal structure, where sequences from the same sampling time tend to cluster together and are the direct ancestors of sequences from the following time point. A typical example, using a data set of HIV-1 env gp120 V1V3 sequences from a pediatric patient infected via mother to child transmission,36 is given by the maximum likelihood (ML) tree in Figure 1A. Increasing branch lengths from root to the latest sampled tips also reveal the increase in both viral diversity and divergence over time. The key point is that topology and branch length patterns of HIV-1 intra-host genealogies are the result of specific phylodynamic processes, defined as the interaction between evolutionary (i.e. mutation, genetic drift, selection) and ecological (population dynamics and environmental stochasticity) factors.37 In particular, the striking similarity between HIV-1 intra-host and influenza A inter-host genealogies, both characterized by strong temporal structure, seem to indicate that HIV-1 population bottlenecks may be driven by continual immune selection.37 Studies based on pairwise sequence comparisons of synonymous (dN) and nonsynonymous (dS) substitutions have suggested that neutral genetic drift is the dominant force shaping the intra-host evolution of the virus.38,39 However, such methods have the tendency to underestimate the strength of selection. 40 Analyses of dN/dS ratios using an ML framework that takes into account the phylogenetic tree relating the sequences under study, have shown that both purifying and positive selection play a substantial role in the continuous emergence in vivo of new variants and the ability of the virus to evade immune response.36,41,42 HIV intra-host adaptation rate in the env gene has, indeed, been estimated to the astounding value of one adaptive fixation event every ~2.5 months.43 Moreover, the comparison of HIV-1 replication rate of virus genomes isolated at early times following infection with that of later viruses has demonstrated an increase of fitness during chronic infection,44 and escapes from both CD8+ T-cell responses and neutralizing antibodies – major driving forces of rapid lineage turnover – are well documented.45-47
Figure 1.
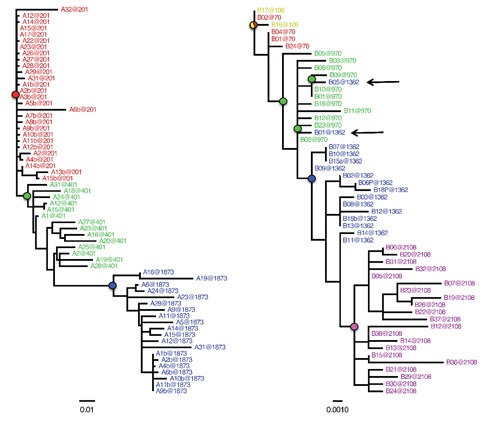
Maximum likelihood genealogies of HIV-1 intra-host sequences sampled over time from two different subjects. The trees were obtained with the HKY+G model. Branch lengths are scaled in nucleotide substitutions per site according to the bar at the bottom of each tree. The number after the @ sign in the sequence label indicates the sampling time in days post infection. Sequence names from different time points are displayed in different colors for clarity. The colored circles highlights the internal nodes representing the most recent common ancestor of the viral population sampled at a given time point. The root was inferred by choosing the rooted topology that resulted in the best linear regression between branch lengths and sampling times. The tree for subject S4 (left) includes a subset of env gp120 V1V3 sequences described in Salemi et al.36 (2007) from a pediatric patient infected through mother to child transmission. The tree for subject P5 (right) includes a subset of gag p24 sequences described in Norstrom et al.44 (2012) from a patient carrying the HLA-B*5701 allele with low risk of disease progression. Sequences sampled at a later time point clustering with sequences sampled earlier are indicated by arrows and may reveal archival genomes from viral reservoirs. The alignments used in the analysis are available from the author upon request.
The degree of temporal structure exhibited by HIV-1 genealogies of longitudinally sampled sequences can be affected by additional factors that can alter the ideal ladder-like topology. The ML tree in Figure 1B is an example obtained by analyzing gag p24 viral sequences from a subject carrying the HLA class I allele B*5701,48 which is associated with slower disease progression.49 By applying a nonparametric test for population structure, it has been shown that a complete within host viral population turnover may take up to 22 months.50 Therefore, the intermix of viral sequences at 70
and 106 days post infection shown in Figure 1B is expected and typical of genealogies inferred from longitudinal samples with relatively short time intervals. Other deviations, however, can provide critical insights on factors driving HIV-1 intra-host population dynamics. For example, the clustering of sequences sampled at more distant time points, like the one indicated by the arrows in Figure 1B, within a monophyletic clade of earlier sequences may indicate the activation (or re-activation) of archival genomes, possibly from a viral reservoir. Peripheral blood mononuclear cells have been noted to harbor small latent reservoirs of infectious virus,51,52 which seem to provide one of the mechanisms for lifelong persistence of HIV-1 under ART.53 Although phylogeny-based criteria have been proposed to detect reservoirs where viral replication is restricted,54 a full characterization of HIV-1 reservoirs is still lacking. From a phylogenetic perspective, a limiting factor is that topological differences among genealogies can intuitively be assessed by visual inspection (Figure 1A and B), but are actually difficult to quantify. A new statistic, Temporal Clustering (TC), has recently been developed to provide a quantitative measure of the degree of topological temporal structure in a serially sampled genealogy.55,56 TC values represent the expected deviation of an observed genealogy from the null hypothesis of no temporal structure (TC=0) and can be used to compare trees of different size. The tree with perfect temporal structure in Figure 1A has by definition TC=1, while the tree in Figure 1B has TC=0.72.56 In future studies, comparison of HIV-1 intra-host genealogies from patients sampled under different conditions could provide important insight on the dynamic of viral reservoirs. In addition, the ability to quantify and compare topological deviations from perfect temporal (ladder-like) structure among different trees may shed light on qualitative and quantitative differences in the evolutionary and ecological processes shaping each phylogeny.
HIV-1 intra-host effective population size and metapopulation structure
Strong evidence for the importance of selection on HIV-1 intra-host evolution is also provided by studies on the total viral population size within an infected host, estimated on the order of 1010 individual viruses.57,58 According to the general prediction of evolutionary theory, mutations in small size populations are produced rarely and their fixation will essentially depend on random genetic drift. On the other hand, in populations large enough that can be considered for any practical purpose of infinite size, mutations are produced more frequently and their fixation will eventually be decided by the deterministic action of natural selection. It has been suggested that it would be appropriate to consider the HIV-1 population within an infected host as if its size were infinite and, therefore, evolutionary driven by selection.22 On the other hand, several studies have calculated that HIV-1 intra-host effective population size (Ne), corresponding to the size of a hypothetical neutral idealized population (i.e. described by the standard Wright-Fisher model), is several orders of magnitude smaller (103-104) than the total population size.10,39,50,59 Such estimates would be more consistent with an evolutionary process driven by random genetic drift, despite the strong evidence of selection provided by dN/dS analyses (see previous section). At least two alternative explanations have been proposed to reconcile the discrepancy. First, Ne estimates in these studies were derived from methods implicitly assuming neutrality, making it impossible to assess whether low values reflect true stochastic processes or continual selective sweeps.34 Second, HIV-1 populations do not evolve under panmixia (one single population), but rather with some metapopulation structure (i.e. division in sub-populations) that causes a reduction in Ne.50 Indeed, several studies have shown the existence of diverse HIV-1 subpopulations infecting different body tissues, for example semen,60,61 breast milk,62,63 genital tract,64 and brain.65-68 The term compartments has been introduced to indicate tissues or cell types were viral replication is ongoing but viral gene-flow (in and out) is restricted because of anatomical barriers.24,54 Compartmentalization is shown, phylogenetically, by the existence of supported monophyletic clades including sequences isolated from a specific tissue, as displayed in Figure 2. The ML tree was inferred from HIV-1 sequences isolated from necropsy tissues of an infected subject who died with widespread atherosclerosis.69 Additional studies have also demonstrated that different brain regions harbor distinct viral populations characterized by specific evolutionary dynamics70,71 that may be associated with pathogenic processes like the onset of HIV-1 associated dementia.72,73
Figure 2.
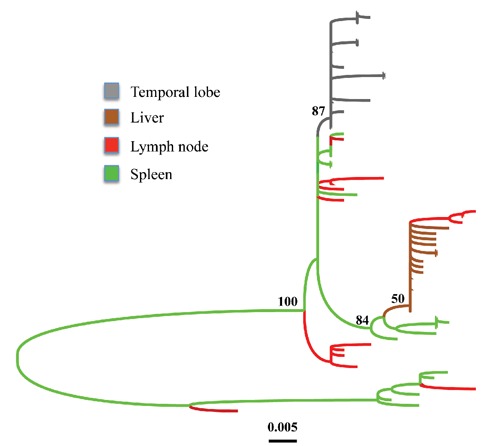
Maximum likelihood genealogy of HIV-1 gp120 V3C4 sequences amplified from necropsy tissues of a patient with widespread atherosclerosis. The tree includes a subset of the sequences obtained from subject AZ described in Lamers et al.65 (2011), and was inferred using the HKY+G model. Branch lengths are scaled in nucleotide substitutions per site according to the bar at the bottom and colored to highlight the tissue of origin according to the legend in the figure. The tissue of origin of the internal branches was inferred by maximum parsimonious reconstruction of ancestral states. Numbers along the branches represent percent bootstrap values (1000 replicates). The alignment used in the analysis is available from the author upon request.
Viral evolutionary rate and disease progression
Estimates of HIV-1 intra-host evolutionary rate range from 10-3 to 10-4 nucleotide substitutions per site per year depending on specific patients and gene region analyzed.16,74 Several studies have shown an inverse relationship between rate of viral evolution and disease progression,75-80 but contrary evidence has also been presented.81,82 A sophisticated molecular clock analysis of gp120 C2V5 sequences from nine subjects followed from seroconversion up to 11 years found that HIV-1 disease progression seems to be predicted by synonymous substitution rates, which are expected to be selectively neutral and, therefore, proportional to the underlying viral replication rate.83 Specifically, lower substitution rates were found in patients with slow disease progression. The authors speculated that such lower rates might reflect lower levels of persistent immune activation, which in turn impose significant constraints on viral generation times and viral evolution. T cell activation is the strongest predictor of progression to AIDS,84,85 and can determine the rate and continuity of viral replication.86
In the attempt to unify all these observations, Lee et al.35 in 2008 developed an HIV-1 intra-host evolution model that simulated the effects of mutation and fitness of sequence variants. The model indicated that the decrease of diversity and stabilization of divergence in the last phase of the infection are the result of a decrease in the proportion of offspring that are mutants as the distance from the founder strain increases, rather than of an increase in viral fitness. In other words, HIV-1 intra-host evolutionary rate is expected to decrease towards the end of the infection in correlation with the rate of CD4+ T cell decline because of the progressive disappearance of target cells that can sustain viral replication. Phylogenetic and molecular clock analysis of empirical data (15 patients followed for 3-12 years) supported the prediction of the model.35
Interestingly, a small proportion of HIV-1 infected patients (<1%), known as elite controllers or long-term nonprogressors (LTNPs), exhibit viral load (VL) below the detection limit of conventional assays (<50 copies/mL plasma) and can maintain CD4+ T cell counts of >500 cells/mm3 for more than 10 years without any therapy.87 Most LTNPs carry the HLAB* 5701 allele and their ability to control the infection have intensively been studied for its obvious clinical implications.49,87,88 From the standpoint of virus evolution, little work has been done since intra-host genetic variation is often minimal and difficult to evaluate given the undetectable VL. However, some subjects expressing HLA-B*5701 do show low levels of viremia (usually ranging between 1000 and 10,000 copies/mL) and can still remain clinically and/or immunologically stable for years without therapy.89,90 CD8+ T cell responses in these patients target several epitopes in the gag p24 viral gene,91-93 and an interesting association between HIV-1 intra-host evolutionary rate and immune response has recently been found.48 Heterochronous p24 sequence data were employed to estimate with a Bayesian molecular clock method the posterior distribution of viral evolutionary rates within subjects classified as either high-risk progressor (HRP) or low-risk progressor (LRP) on the basis of their CD4+ T cell count in early (10-13 weeks) infection.48 Lower evolutionary rates were found in LRPs who also exhibited higher polyfunctional responses than HRPs in terms of higher proportion of CD8+ T cells producing a combination of interferon-γ, inteleukin-2, MIP-1 and perforin when induced by wild-type HLA-B*5701-restricted epitopes. Further investigation of the interplay between specific CD8+ T cell responses and intra-host viral rate of evolution in these subjects may help defining correlates of protection, one of the greatest challenges in HIV-1 vaccine design.
The phylogenetic challenge of HIV-1 recombination
Due to super-infection and reverse transcriptase switches between alternative genomic templates, that are an integral part of HIV lifecycle, HIV-1 recombination rate per replication is likely exceeding the mutation rate.94,95 The impact of recombination on viral genetic diversity is evident at the molecular epidemiological scale. The majority of HIV-1 strains circulating worldwide cluster within a large group called M (for Main) that includes, besides ten phylogenetically distinct subtypes and two subsubtypes, several inter-subtype recombinants, known as circulating recombinant forms (CRFs).96,97 Recombination can affect HIV-1 evolutionary dynamics by providing a mechanism even more efficient than fast mutation rate for the virus to escape from the accumulation of deleterious mutations or to jump between adaptive peaks.16 It might accelerate progression to AIDS and can foster the emergence of viral variants able to evade immune pressure or with increased resistance to drugs.98-100 Analysis of HIV-1 sequences amplified from different tissues, sampled post mortem from patients with a number of AIDSrelated malignancies, have shown that recombination is common and more extensive in tissues with abnormal histopathology compared to normal tissues.101
As shown in Figure 3, intra-host recombination could be detected, in principle, by moving a sliding window along a multiple sequence alignment and calculating the bootstrap support for the phylogenetic clustering of a query sequence, the potential recombinant, with different reference sequences in different genomic regions. This analysis is an example of bootscanning; a method specifically developed to investigate recombination in HIV.102 Recombination violates the basic assumption of phylogeny inference of descent from a common ancestor. Recombinant sequences cannot be clustered correctly within one single phylogenetic tree but can be displayed within network-like graphs able to represent simultaneously different tree topologies (Figure 3).
Figure 3.
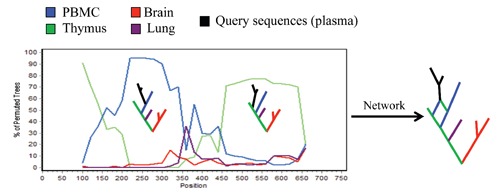
Putative recombination analysis of HIV-1 intra-host sequences sampled from different tissues as indicated by the color legend in the figure. A query sequence is compared to a set of reference sequences by a sliding window analysis, which calculates the bootstrap support for the clustering of the query sequence with a specific reference sequence along the genome. In this cartoon example, a query sequence from plasma clusters with high bootstrap support with a reference sequence from peripheral blood mononuclear cells (PBMCs) in the 5’ end of the genome, while with a sequence from thymus in the 3’ end. The two trees are schematically illustrated within the frame and the network shows how they could be represented by a cyclic graph where the recombinant sequence is simultaneously connected to the putative parental strains.
Using algorithms that do not explicitly model recombination can bias molecular clock as well as population genetic estimates.103,104 HIV-1 evolutionary dynamics should therefore be studied with methods that take recombination into account. Unfortunately, this is not the case for most of the work discussed in the previous sections, where authors have chosen either to ignore recombination or the common approach to identify recombinants and exclude them from the analysis. The latter strategy has two important caveats. First, because recombination is so frequent and plays such a central role, inferences on the evolutionary dynamics of the virus after exclusion of recombinants may be meaningless or, at the very least, incomplete. Second, despite the plethora of methods developed to detect recombination, each algorithm has limitations resulting, in general, in an under estimation of recombination, especially in data sets of closely related sequences.103,104 For example, while inter-subtype recombination is relatively straightforward to detect,105 bootscanning performs very poorly with HIV-1 sequences from individuals infected by the same subtype. Network-based methods have been more successful and have shown that the number of recombinant sequences detected by classic phylogenetic analysis can lead to a significant under estimation of the actual number of recombinants within an intra-host data set.106,107 However, it is clear that novel techniques and additional studies are still needed to assess the full impact of recombination on HIV-1 intra-host evolutionary and population dynamics.
Final remarks
This review briefly highlights some of the major findings that have contributed to our current understanding of HIV-1 intra-host phylodynamics. There is general agreement on deterministic selection processes as the main driving force of viral evolution within the infected host. The study of the temporal structure of genealogies inferred from heterochronous sequences is one of the key techniques that can be used to assess the impact of continual immune selection on viral population turnover and a promising tool to investigate further the activation or re-activation of viral reservoirs. In addition, the recent application of high-resolution deep sequencing to study HIV-1 population complexity and structure (biodiversity) within an infected host can allow the characterization of low frequency variants responsible for significant changes in the viral evolutionary landscape that would be impossible to detect by conventional methods. 108 On the other hand, a thorough understanding of early evolutionary events at the moment of transmission and during primary infection, as well as a full account of the role played by recombination is still lacking and among the future challenges of HIV molecular evolutionary research.
References
- 1.Barré-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for the acquired immunodeficiency syndrome (AIDS). Science 1983;292:1102-05 [DOI] [PubMed] [Google Scholar]
- 2.Hahn BH, Shaw GM, Taylor ME, et al. Genetic variation in HTLV-III/LAV over time in patients with AIDS or at risk for AIDS. Science 1986;232:1548-53 [DOI] [PubMed] [Google Scholar]
- 3.Balfe P, Simmonds P, Ludlam CA, et al. Concurrent evolution of human immunodeficiency virus type 1 in patients infected from the same source: rate of sequence change and low frequency of inactivating mutations. J Virol 1990;64:6221-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korber BT, Allen EE, Farmer AD, Myers GL. Heterogeneity of HIV-1 and HIV-2. AIDS 1995;9:S5-S18 [PubMed] [Google Scholar]
- 5.Holmes EC, Zhang LQ, Simmonds P, et al. Convergent and divergent sequence evolution in the surface glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA 1992;89:4835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preston BD, Poiesz BJ, Loeb LA. Fidelity of HIV-1 reverse transcriptase. Science 1988;242:1168-71 [DOI] [PubMed] [Google Scholar]
- 7.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 1995;69:5087-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995;373: 117-22 [DOI] [PubMed] [Google Scholar]
- 9.Perelson AS, Neumann AU, Markowitz M, et al. HIV-1 dynamics in vivo virion clearance rate, infected cell life-span, viral generation time. Science 1996;271:1582-6 [DOI] [PubMed] [Google Scholar]
- 10.Rodrigo AG, Shpaer EG, Delwart EL, et al. Coalescent estimates of HIV-1 generation time in vivo. Proc Natl Acad Sci USA 1999;96:2187-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu YX. Estimating mutation rate and generation time from longitudinal samples of DNA sequences. Mol Biol Evol 2001;18:620-6 [DOI] [PubMed] [Google Scholar]
- 12.Zhuang J, Jetzt AE, Sun G, et al. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J Virol 2002;76:11273-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jetzt AE, Yu H, Klarmann GJ, et al. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J Virol 2000;74:1234-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy DN, Aldrovandi GM, Kutsch O, Shaw G.M. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci USA 2004;101:4204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond AJ, Pybus OG, Rambaut A, et al. Measurably evolving populations. Trends Ecol Evol 2003;18:481-8 [Google Scholar]
- 16.Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nat Rev Genet 2004;5:52-61 [DOI] [PubMed] [Google Scholar]
- 17.Castro-Nallar E, Pérez-Losada M, Burton GF, Crandall KA. The evolution of HIV: inferences using phylogenetics. Mol Phylogenet Evol 2012;62:777-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowak M, Anderson RM, McLean AR, et al. Antigenic diversity threshold and the development of AIDS. Science 1991;254:963-9 [DOI] [PubMed] [Google Scholar]
- 19.Carr JK. Viral diversity as a challenge to HIV-1 vaccine development. Curr Opin HIV AIDS 2006;1:294-300 [DOI] [PubMed] [Google Scholar]
- 20.Nickle DC, Rolland M, Jensen MA, et al. Coping with viral diversity in HIV vaccine design. PLoS Comput Biol 2007;3:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salemi M. Toward a robust monitoring of HIV subtypes distribution worldwide. AIDS 2011;25:713-4 [DOI] [PubMed] [Google Scholar]
- 22.Coffin JM. Molecular biology of HIV. Crandall KA, The evolution of HIV. Baltimore, MD: John Hopkins University Press; 1999. pp. 3-40 [Google Scholar]
- 23.Shankarappa R, Margolick JB, Gange SJ, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 1999;73:10489-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nickle DC, Jensen MA, Shriner D, et al. Evolutionary indicators of human immunodeficiency virus type 1 reservoirs and compartments. J Virol 2003;77:5540-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards CTT, Holmes EC, Wilson DJ, et al. Population genetic estimation of the loss of genetic diversity during horizontal transmission of HIV-1. BMC Evol Biol 2006;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang LQ, MacKenzie P, Cleland A, et al. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol 1993;67:3345-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu T, Mo H, Wang N, et al. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 1993;261:1179-81 [DOI] [PubMed] [Google Scholar]
- 28.Keele BF, Giorgi EE, Salazar-Gonzalez JF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA 2008;105:7552-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salazar-Gonzalez JF, Bailes E, Pham KT, et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 2008; 82:3952-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scarlatti G. Mother-to-child transmission in HIV-1: advances and controversies of the twentieth century. AIDS Rev 2004;6:67-8 [PubMed] [Google Scholar]
- 31.Li H, Bar KJ, Wang S, Decker JM, et al. High Multiplicity Infection by HIV-1 in Men Who Have Sex with Men. PLoS Pathog 2010;6:e1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salazar-Gonzalez JF, Salazar MG, Keele BF, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/ founder viruses in acute and early HIV-1 infection. J Exp Med 2009;206:1273-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strickland SL, Gray RR, Lamers SL. Efficient transmission and persistence of low-frequency SIVmac251 variants in CD8-depleted rhesus macaques with different neuropathology. J Gen Virol 2012;93:925-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes EC. The evolution and emergence of RNA viruses. Oxford, UK: Oxford University Press; 2009 [Google Scholar]
- 35.Lee HY, Perelson AS, Park SC, Leitner T. Dynamic correlation between intrahost HIV-1 quasispecies evolution and disease progression. PLoS Comput Biol 2008;4:e1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salemi M, Burkhardt BR, Gray RR, et al. Phylodynamics of HIV-1 in lymphoid and non-lymphoid tissues reveals a central role for the thymus in emergence of CXCR4-using quasispecies. PLoS One 2007;2:e950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004;303:327-32 [DOI] [PubMed] [Google Scholar]
- 38.Plikat U, Nieselt-Struwe K, Meyerhans A. Genetic drift can dominate short-term human immunodeficiency virus type 1 nef quasispecies in vivo. J Virol 1997;71:4233-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shriner D, Shankarappa R, Jensen MA, et al. Influence of random genetic drift on human immunodeficiency virus type 1 env evolution during chronic infection. Genetics 2004;166:1155-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crandall KA, Kelsey CR, Imamichi H, et al. Parallel evolution of drug resistance in HIV: failure of nonsynonymous/synonymous substitution rate ratio to detect selection. Mol Biol Evol 1999;16:372-82 [DOI] [PubMed] [Google Scholar]
- 41.Edwards CT, Holmes EC, Pybus OG, et al. Evolution of the human immunodeficiency virus envelope gene is dominated by purifying selection. Genetics 2006;174:1441-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998;148:929-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williamson S. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol Biol Evol 2003;20:1318-25 [DOI] [PubMed] [Google Scholar]
- 44.Troyer RM, Collins KR, Abraha A, et al. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol 2005; 79:9006-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips RE, Rowland-Jones S, Nixon DF, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 1991;354:453-9 [DOI] [PubMed] [Google Scholar]
- 46.Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci USA 2003;100:4144-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frost SD, Wrin T, Smith DM, et al. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc Natl Acad Sci USA 2005; 102:18514-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norström NM, Buggert M, Tauriainen J, et al. Combination of immune and viral factors distinguishes low-risk versus highrisk HIV-1 disease progression in HLA-B* 5701 subjects. J Virol 2012;86:9802-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Migueles SA, Sabbaghian MS, Shupert WL, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA 2000; 97:2709-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Achaz G, Palmer S, Kearney M, et al. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol Biol Evol 2004;21:1902-12 [DOI] [PubMed] [Google Scholar]
- 51.Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med 2002;53:557-93 [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Chung C, Hu BS, et al. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J Clin Investig 2000;106:839-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finzi D, Blankson J, Siliciano JD, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 1999;5:512-17 [DOI] [PubMed] [Google Scholar]
- 54.Nickle DC, Shriner D, Mittler , et al. Importance and detection of virus reservoirs and compartments of HIV infection. Curr Opin Microbiol 2003;6:410-6 [DOI] [PubMed] [Google Scholar]
- 55.Gray RR, Pybus OG, Salemi M. Measuring the temporal structure in serially sampled phylogenies. Methods Ecol Evol 2011;2:437-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Norström MM, Prosperi MC, Gray RR, et al. PhyloTempo: a set of r scripts for assessing and visualizing temporal clustering in genealogies inferred from serially sampled viral sequences. Evol Bioinform Online 2012;8:261-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piatak M Jr, Saag MS, Yang LC, et al. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 1993;259:1749-54 [DOI] [PubMed] [Google Scholar]
- 58.Haase AT, Henry K, Zupancic M, et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 1996;274:985-9 [DOI] [PubMed] [Google Scholar]
- 59.Leigh Brown AJ. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc Natl Acad Sci USA 1997;94:1862-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coombs RW, Speck CE, Hughes JP, et al. Association between culturable human immunodeficiency virus type 1 (HIV-1) in semen and HIV-1 RNA levels in semen and blood: evidence for compartmentalization of HIV-1 between semen and blood. J Infect Dis 1998;177:320-30 [DOI] [PubMed] [Google Scholar]
- 61.Kiessling AA, Fitzgerald LM, Zhang D, et al. Human immunodeficiency virus in semen arises from a genetically distinct virus reservoir. AIDS Res Hum Retroviruses 1998;14:S33-41 [PubMed] [Google Scholar]
- 62.Becquart P, Courgnaud V, Willumsen J, Van de Perre P. Diversity of HIV-1 RNA and DNA in breast milk from HIV-1 infected mothers. Virology 2007;363:256-60 [DOI] [PubMed] [Google Scholar]
- 63.Gray RR, Salemi M, Lowe A, et al. Multiple independent lineages of HIV-1 persist in breast milk and plasma. AIDS 2011;25:143-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poss M, Rodrigo AG, Gosink JJ, et al. Evolution of envelope sequences from the genital tract and peripheral blood of women infected with clade A human immunodeficiency virus type 1. J Virol 1998; 72:8240-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korber BT, Kunstman KJ, Patterson BK, et al. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J Virol 199468:7467-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hughes ES, Bell JE, Simmonds P. Investigation of the dynamics of the spread of human immunodeficiency virus to brain and other tissues by evolutionary analysis of sequences from the p17gag and env genes. J Virol 1997;71:1272-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris A, Marsden M, Halcrow K, et al. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J Virol 1999;73:8720-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohagen A, Devitt A, Kunstman KJ, et al. Genetic and functional analysis of full-length human immunodeficiency virus type 1 env genes derived from brain and blood of patients with AIDS. J Virol 2003;77;12336-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamers SL, Gray RR, Salemi M, et al. HIV-1 Phylogenetic analysis shows HIV-1 transits through the meninges to brain and peripheral tissues. Infect Genet Evol 2011;11:31-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang J, Jozwiak R, Wang B, et al. 1998. Unique HIV type 1 V3 region sequences derived from six different regions of brain: region-specific evolution within host-determined quasispecies. AIDS Res Hum Retroviruses 1998;14:25-30 [DOI] [PubMed] [Google Scholar]
- 71.Shapshak P, Segal DM, Crandall KA, et al. Independent evolution of HIV type 1 in different brain regions. AIDS Res Hum Retroviruses 1999;15:811-20 [DOI] [PubMed] [Google Scholar]
- 72.Salemi M, Lamers SL, Yu S, et al. Phylodynamic analysis of human immunodeficiency virus type 1 in distinct brain compartments provides a model for the neuropathogenesis of AIDS. J Virol 2005;79:11343-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lamers SL, Salemi M, Galligan DC, et al. HIV-1 Evolutionary patterns associated with pathogenic processes in the brain. J Neurovir 2010;16:230-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lemey P, Rambaut A, Pybus OG. HIV dynamics within and among hosts. AIDS Rev 2006;8:125-40 [PubMed] [Google Scholar]
- 75.Ganeshan S, Dickover RE, Korber BT, et al. Human immunodeficiency virus type 1 genetic evolution in children with different rates of development of disease. J Virol 1997;71:663-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Halapi E, Leitner T, Jansson M, et al. Correlation between HIV sequence evolution, specific immune response and clinical outcome in vertically infected infants. AIDS 1997;11:1709-17 [DOI] [PubMed] [Google Scholar]
- 77.Williamson S, Perry SM, Bustamante CD, et al. A statistical characterization of consistent patterns of human immunodeficiency virus evolution within infected patients. Mol Biol Evol 2005;22:456-68 [DOI] [PubMed] [Google Scholar]
- 78.Delwart EL, Pan H, Sheppard HW, et al. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J Virol 1997;71:7498-508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shioda T, Levy JA, Cheng-Mayer C.Small amino acid changes in the V3 hypervariable region of gp120 can affect the T-cellline and macrophage tropism of human immunodeficiency virus type 1. Proc Natl Acad Sci USA 1992;89:9434-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolinsky SM, Korber BTM, Neumann AU, et al. Adaptive evolution of human immunodeficiency virus type1 during the natural course of infection. Science 1996;272:53742. [DOI] [PubMed] [Google Scholar]
- 81.Markham RB, Wang WC, Weisstein AE, et al. Patterns of HIV-1 evolution in individuals with differing rates of CD4 T cell decline. Proc Natl Acad Sci USA 1998;95:12568-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McNearney T, Hornickova Z, Markham R, et al. Relationship of human immunodeficiency virus type 1 sequence heterogeneity to stage of disease. Proc Natl Acad Sci USA 1992;89:10247-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lemey P, Kosakovsky Pond SL, Drummond AJ, et al. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput Biol 2007;3:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 1999;179:859-70 [DOI] [PubMed] [Google Scholar]
- 85.Sousa AE, Carneiro J, Meier-Schellersheim M, et al. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol 2002;169:3400-6 [DOI] [PubMed] [Google Scholar]
- 86.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med 2006;12:289-95 [DOI] [PubMed] [Google Scholar]
- 87.Migueles SA, Connors M. Long-term non-progressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA 2010;304:194-201 [DOI] [PubMed] [Google Scholar]
- 88.Makedonas G, Betts MR. Living in a house of cards: re-evaluating CD8+ T-cell immune correlates against HIV. Immunol Rev 2011;239:109-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sheppard HW, Lang W, Ascher MS, et al. The characterization of non-progressors: long-term HIV-1 infection with stable CD4+ T-cell levels. AIDS 1993;7:1159-66 [PubMed] [Google Scholar]
- 90.Cao Y, Qin L, Zhang L, et al. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med 1995;332:201-8 [DOI] [PubMed] [Google Scholar]
- 91.Brockman MA, Schneidewind A, Lahaie M, et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 2007;81:12608-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 2009;206:1253-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dyer WB, Zaunders JJ, Yuan FF, Wang B, et al. Mechanisms of HIV non-progression; robust and sustained CD4+ T-cell proliferative responses to p24 antigen correlate with control of viraemia and lack of disease progression after long-term transfusion-acquired HIV-1 infection. Retrovirology 2008;5:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jung A, Maier R, Vartanian JP, et al. Recombination: multiply infected spleen cells in HIV patients. Nature 2002;418:144. [DOI] [PubMed] [Google Scholar]
- 95.Jost S, Bernard MC, Kaiser L, et al. A patient with HIV-1 superinfection. N Engl J Med 2002;347:731-6 [DOI] [PubMed] [Google Scholar]
- 96.Robertson DL, Anderson JP, Bradac JA.HIV-1 nomenclature proposal. Science 2000;288:55-6 [DOI] [PubMed] [Google Scholar]
- 97.McCutchan FE. Global epidemiology of HIV. J Med Virol 2006;78:S7-S12 [DOI] [PubMed] [Google Scholar]
- 98.Liu SL, Mittler JE, Nickle DC, et al. Selection for human immunodeficiency virus type 1 recombinants in a patient with rapid progression to AIDS. J Virol 2002;76:10674-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morris A, Marsden M, Halcrow K, et al. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J Virol 1999;73:8720-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kellam P, Larder BA. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J Virol 1995;69:669-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lamers SL, Salemi M, Galligan DC, et al. Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS One 2009;4:e5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses 1995;11:1423-5 [DOI] [PubMed] [Google Scholar]
- 103.Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. PNAS 2001;98:13757-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Posada D. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol Biol Evol 2002;19:708-17 [DOI] [PubMed] [Google Scholar]
- 105.Lole KS, Bollinger RC, Paranjape RS, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999;73:152-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salemi M, Gray RR, Goodenow MM. An exploratory algorithm to identify intra-host recombinant viral sequences. Mol Phylogenet Evol 2008;49:618-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mild M, Esbjornsson J, Fenyo E, Medstrand P. Frequent intrapatient recombination between human immunodeficiency virus type 1 R5 and X4 envelopes: implications for coreceptor switch. J Virol 2007;81:3369-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yin L, Liu L, Sun Y, Hou W, et al. High-resolution deep sequencing reveals biodiversity, population structure, and persistence of HIV-1 quasispecies within host ecosystems. Retrovirology 2012;9:108. [DOI] [PMC free article] [PubMed] [Google Scholar]