

Abstract
Purpose
To investigate the genotype and phenotype in families with adenosine triphosphate–binding cassette, sub-family A, member 4 (ABCA4)–associated retinal degeneration.
Methods
Three families with at least one family member with known homozygous or compound heterozygote mutations in the ABCA4 gene were studied. The investigations included full field electroretinography (ff-ERG), multifocal ERG (mERG), Goldmann visual fields, optical coherence tomography (OCT), and standard ophthalmological examination. Microarray (Asper) was used for ABCA4 genotyping.
Results
In family 1, the proband (age 23) was homozygote for the c768 G>T mutation. She was diagnosed with cone rod dystrophy (CRD) while her aunt (age 69) was compound heterozygote for the c768 G>T and c2894 A>G mutations and had autosomal recessive retinitis pigmentosa (arRP). The father (age 61) and the mother (age 60) of the proband were asymptomatic carriers of the c768 G>T mutation. In family 2, the proband (age 25) was homozygote for the c5917del. She was diagnosed with CRD. Her father and two sisters were compound heterozygote for the c5917del and c5882 G>A mutations. The eldest sister (age 23) suffered from Stargardt disease (STGD) while the youngest sister (age 12) and their father (age 48) had no visual complaints. Anyhow, their ERG measurements indicated changes corresponding to STGD. The mother (age 42), (heterozygote for the c5917 delG mutation) and the youngest child (age 9; heterozygote for the c5882 G>A mutation) had a normal phenotype. In family 3, the proband (age 43) was compound heterozygote for c768 G>T and c3113 C>T and had been diagnosed with STGD. Her son (age 12), who was homozygote for the c768 G>T mutation, had wider scotomas with earlier onset (age 6), ff-ERG cone responses in the lower range of normality, and reduced mERG. At the moment, he is classified as having STGD but may progress to CRD. The father (age 45) was asymptomatic and heterozygote for the c768 G>T mutation. The patients with progressive disorders (CRD or arRP) had prolonged implicit times for the 30 Hz flicker ff-ERG and the mERG. All patients with two mutations in the ABCA4 gene demonstrated attenuation of retinal thickness on the OCT macular map.
Conclusions
This study confirms that ABCA4 mutations lead to a spectrum of retinal degenerations ranging from STGD to CRD or arRP. At the time of diagnosis, it is not possible to predict the severity of the condition only from genotyping. Our results suggest that prolongation of implicit times for the ff-ERG and/or mERG seem to be associated with progressive conditions such as CRD and arRP. Since ABCA4 mutations are common in the general population, different family members can harbor various combinations of mutations resulting in diverse phenotype and prognosis in the same family, further emphasizing the importance of a combination of genetic and electrophysiological tests at the first visit and follow-up.
Introduction
Mutations in the adenosine triphosphate–binding cassette, sub-family A, member 4 (ABCA4) gene lead to a wide spectrum of autosomal recessive (ar) retinal degeneration, including Stargardt disease (STGD) [1-4], cone-rod dystrophy (CRD) [5-11], retinitis pigmentosa (RP) [5,6,8-10,12], and probably some forms of age-related macular degeneration (AMD) [13]. The ABCA4 gene encodes an adenosine triphosphate (ATP)-binding cassette (ABC) transporter protein, ABCA4, specifically located in the rim of the photoreceptor discs in the outer segments of rods and cones [14,15]. The ABC transporter proteins comprise a superfamily of proteins that actively moves a wide range of compounds, for example, vitamins, lipids, amino acids, and polypeptides, across cell membranes [16]. In photoreceptors, the ABCA4 transporter has the important function of actively transferring N-retinylidene-phosphatidylethanolamine (NRPE) and phosphatidylethanolamine (PE) from the lumen of the outer segment disc membranes to the cytoplasmic leaflet following photobleaching of opsins [17,18]. NRPE is formed by the binding of all-trans retinal to the phospholipid PE. The ABCA4 rim protein thus facilitates the removal of potentially toxic retinoid compounds from photoreceptors. Retinal degenerations associated with mutations in the ABCA4 gene are consequently proposed to be caused by the accumulation of a lipofuscin fluorophore, N-retinylidene-N-retinylethanolamine (A2E), that is formed when the ABCA4 rim protein is missing or dysfunctional and therefore cannot clear the photoreceptor outer segment from NRPE. A2E is eventually accumulated in the cells of the retinal pigment epithelium (RPE) resulting in RPE cell death and probably secondary loss of photoreceptors [18,19]. However, other authors have suggested that degeneration of photoreceptors might precede changes in the RPE [20,21].
In STGD, patients suffer from juvenile-onset macular dystrophy typically leading to rapid impairment of central vision during the first or second decades of life. Most patients retain intact peripheral visual function despite having central scotomas of various sizes. The disorder is characterized by progressive bilateral atrophy of the foveal RPE and deep orange-yellow retinal flecks around the macula and/or in the midperiphery [22]. Electrophysiological examinations typically show normal or only mildly reduced full-field electroretinography (ff-ERG) but reduced multifocal electroretinography (mERG) [23,24]. In other patients, ABCA4 mutations are instead associated with CRD resulting in early loss of visual acuity, reduced color vision, and progressive constriction of visual fields in combination with central scotomas. In the fundus, pigment changes in the macula and midperiphery eventually develop [25]. The mERG is typically significantly reduced, and the photopic ff-ERG is more distinctively and earlier reduced than the scotopic ff-ERG [11,22,24-26]. In arRP, the first symptoms of the disease are night blindness and progressive loss of peripheral vision followed by reduced visual acuity [11,22]. Typical fundoscopic changes include narrowed retinal vessels, pale optic nerve head, and bone spicule-like pigmentation in the periphery [11]. The scotopic ff-ERG is reduced more gravely and before the photopic ff-ERG [11,22,24].
Thus, there is marked variability in ABCA4 retinal phenotypes. Some authors have proposed a model that correlates severity of disease with residual function of the ABCA4 rim protein based on the effect of the individual mutation on gene transcription and protein translation [6,8,11,27-30]. Other authors suggest that zero or low activity of ABCA4 can be associated with early onset of disease rather than a specific form of retinal degeneration (STGD, CRD, or arRP) [31]. The purpose of our study was to investigate the genotype in three families with ABCA4-associated retinal degeneration and to explore the phenotype among different individuals and generations.
Methods
Patients
Three families with mutations in the ABCA4 gene were studied (Table 1). Family 1 consists of a girl (1IIa) with CRD, her parents (1Ib and c), and her aunt (1Ia), who has arRP (Figure 1). No consanguinity is known in this family. Family 2 comprises four siblings (2IIa–d) and their parents (2Ia and b). The family is of Hungarian origin, and the parents are second cousins. The eldest sister (2IIa) has been diagnosed with CRD and the second eldest (2IIb) with STGD. In family 3, the mother (3Ia) has STGD, and her son (3IIa) has visual problems as well. The parents are not related. This study was conducted in accordance with the tenets of the Declaration of Helsinki and was approved by the Ethical Committee for Medical Research at Lund University. All subjects gave informed consent to participate in the study.
Table 1. Patient demographics.
| Subject | Age (years) | Sex | Age at onset (years) | VA OD | VA OS | ff-ERG cone | ff-ERG rod | mERG | Visual fields |
|---|---|---|---|---|---|---|---|---|---|
| 1 Ia |
69 |
F |
early childhood |
CF |
CF |
↓↓ |
↓↓ |
↓↓ |
Minimal temporal crescent-shaped remnants |
| 1 Ib |
61 |
M |
NVP |
1.0 |
1.0 |
N |
N |
- |
Normal |
| 1 Ic |
60 |
F |
NVP |
0.8* |
0.4** |
N |
N |
- |
Normal |
| 1 IIa |
23 |
F |
9 |
0.04 |
0.05 |
↓↓ |
N |
↓↓ |
Large nasal paracentral/central scotomas |
| 2 Ia |
42 |
F |
NVP |
0.7 |
1.0 |
N |
N |
- |
Normal |
| 2 Ib |
48 |
M |
40 |
0.5 |
0.3 |
N |
N |
↓ |
Small nasal central scotomas for 04:e |
| 2 IIa |
25 |
F |
7 |
0.03 |
0.04 |
↓↓ |
↓↓ |
↓↓ |
Large central/para-central scotomas and peripheral constriction |
| 2 IIb |
23 |
F |
20 |
0.16 |
0.13 |
N |
N |
↓ |
Small central scotomas for 03:e OD and 04:e OS within the central 5° |
| 2 IIc |
12 |
F |
NVP |
1.0 |
1,0 |
N |
N |
↓ |
Normal |
| 2 IId |
9 |
M |
NVP |
1.0 |
1.0 |
N |
N |
- |
Normal |
| 3 Ia |
41 |
F |
15 |
0.2 |
0.1 |
N |
N |
↓ |
Small central scotomas within 5–10°, concentric reduction of II4:e |
| 3 Ib |
43 |
M |
NVP |
- |
- |
- |
- |
- |
- |
| 3 IIa | 12 | M | 6–7 | 0.09 | 0.16 | ↓ | N | ↓ | Central scotomas within 20–30° |
Abbreviations: VA visual acuity, OD right eye, OS left eye, F female, M male, CF count fingers, NVP no visual problems, *clinical presentation at last visit reduced vision due to capsular opacities after cataract surgery, **reduced vision due to cataract Ff-ERG recordings: ↓reduced, ↓↓severely reduced, N normal
Figure 1.
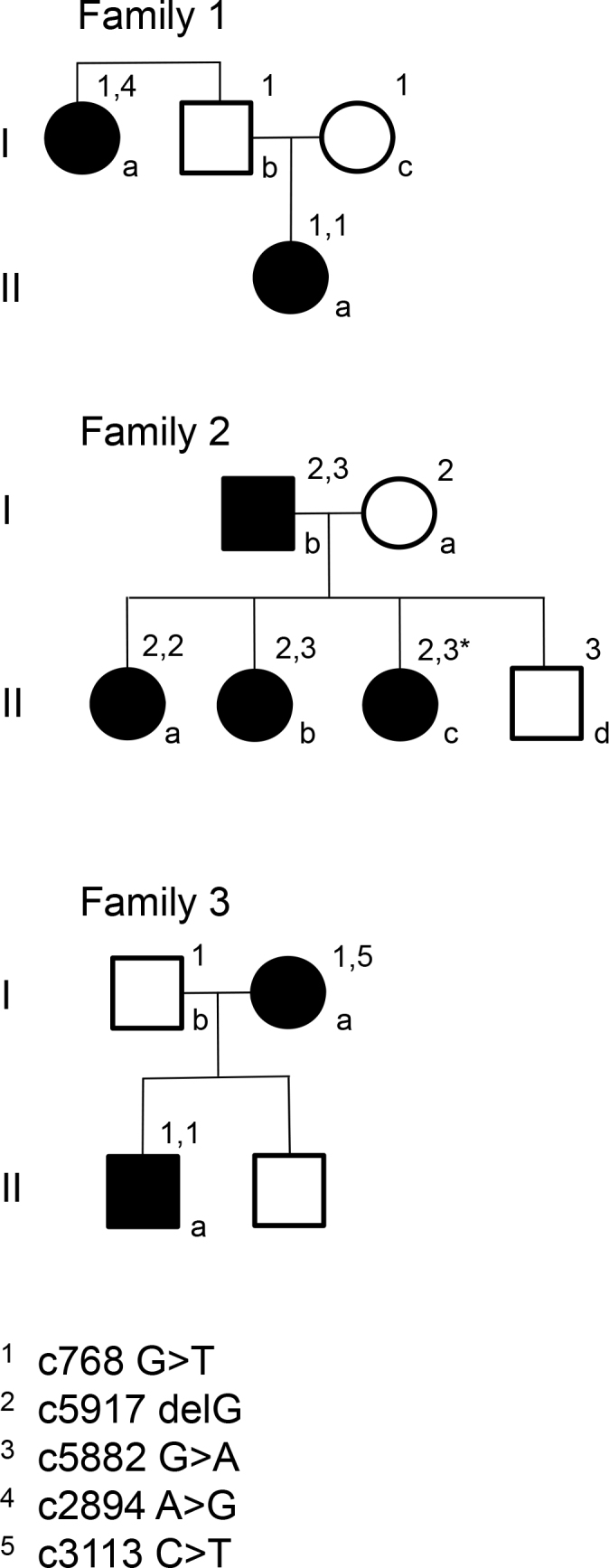
Pedigrees for the families. *indicates a carrier of a double mutation. She is asymptomatic, although the multifocal electroretinography (mERG) shows reduced amplitudes.
Ophthalmologic examination
The ophthalmologic examination included best-corrected visual acuity (VA), assessed on the Snellen chart, slit-lamp biomicroscopy, and dilated fundus examination.
Fundus photography
Fundus photographs of the right or both eyes were obtained with a Topcon fundus camera (Topcon, Inc., Oakland, NJ) focusing on the optic disc, the macula, and the periphery.
Visual field testing
Kinetic perimetry was performed with a Goldmann perimeter using standardized objects V4e and I4e. In some cases, additional objects were used (e.g., II4e, I2e, and 02e).
Full-field electroretinography
Ff-ERGs were recorded either with an Espion E2 analysis system (Diagnosys, Lowell, MA) or with a Nicolet Viking analysis system (Nicolet Biomedical Instruments, Madison, WI). The latter had previously been used in the department, and some of the older investigations in patients followed over a long period of time were performed with this equipment. The procedure adhered to the standardized protocol for clinical electroretinography recommended by the International Society for Clinical Electrophysiology of Vision (ISCEV) [32,33] in addition to slight modifications of flash intensities and the recording of 30 Hz flicker under dark-adapted condition. The modifications were made for longitudinal comparison of results in the same individual, since ff-ERGs have been recorded according to the same protocol for many years in our department. In patients with low amplitudes (e.g., combined rod-cone response <10 µV), special recordings were obtained using computer averaging (30 flashes), a bipolar artifact rejecter, and a line frequency notch filter (50 Hz). To register weak cone responses, stimulation included 200 flashes of 30 Hz flickering white light and a digital, narrow band pass filter added to the ff-ERG equipment. The narrow bandpass filter was tuned at 30 Hz (12 dB at 29 Hz and 31 Hz) to measure signals as low as 0.1 µV [34]. To ensure reproducibility, the recordings were repeated for all stimulus intensities until two successive identical curves were obtained.
Multifocal electroretinography
MERGs were recorded with a Visual Evoked Response Imaging System (VERIS 4; EDI, San Mateo, CA) according to the ISCEV guidelines [35,36]. The pupils were maximally dilated (cyclopentolate 1% and 10% phenylephrine hydrochloride). The first-order component of the mERG was analyzed regarding the amplitudes and implicit times of P1 (first positive peak); within the four innermost of the six concentric rings, the central (innermost) ring was the summed responses from the first and second rings.
Optical coherence tomography
Retinal thickness was measured with a Topcon 3D OCT-1000 (Topcon, Inc., Paramus, NJ). This is a Fourier domain-based optical coherence tomography (OCT), which captures 3D scans with scan size 6×6 mm and scan density 512×128. In most patients, the internal fixation target (preset for macular imaging) was used. In patients with poor central vision, however, the external fixation target had to be applied. Further analyses were performed by using the standard retinal thickness map of the OCT software (version 4.00). The retinal thickness map consists of three concentric circles, each with a diameter of 1, 3, and 6 mm, respectively. The two outer rings are further divided into four segments, corresponding to a superior, nasal, inferior, and temporal segment. For each segment of the retinal map, the mean thickness of the retina, from the RPE to the inner limiting membrane (ILM), is measured. The mean thickness of each segment is also automatically compared to an age-matched normative database. Measurements outside normal limits are highlighted in specific colors depending on whether they are thinner than normal, outside the first (red) or fifth (yellow) percentile, or thicker than normal, outside the 95th (orange) or 99th (purple) percentile.
Genetic analysis
Screening for all known mutations in the ABCA4 gene was performed at Asper Biotech using the ABCA4 genotyping microarray (Asper Ophthalmics, Tartu, Estonia). The test currently detects 632 mutations in the ABCA4 gene. Testing for known mutations concerning RP was also performed.
Results
Family 1
Patients 1Ia and 1IIa harbored two different combinations of mutations in the ABCA4 gene, (Figure 1, Table 2) with diverse phenotypes. Patient 1Ia had severely reduced retinal function centrally (mERG) and in the periphery (ff-ERG; Table 1 and Table 3) and small temporal remnants of the visual fields (Figure 2). She was diagnosed with arRP many years ago. Fundoscopic changes were typical, and OCT showed attenuation (Figure 2). Patient 1IIa (followed for 14 years) demonstrated a typical CRD phenotype (Table 1), with large scotomas that over time deepened but did not grow (Figure 3). Ff-ERG was reduced; it was most marked for the cones, and the IT for the 30 Hz flicker was delayed (Table 3). MERG was severely reduced with prolonged implicit times (ITs). Fundoscopic changes were typical for CRD, and OCT showed attenuation (Figure 2). Her parents (1Ib and c) were asymptomatic besides from that the mother (1Ic) recently had cataract surgery in her right eye and still suffered from cataract in the left eye.
Table 2. Genotype and phenotype for the subjects.
| Subject |
ABCA4 allele 1 |
ABCA4 allele 2 |
Phenotype* | ||
|---|---|---|---|---|---|
| Nucleotide change | Effect | Nucleotide change | Effect | ||
| 1 Ia |
c768 G>T |
V256V/splice |
c2894 A>G |
N965S/missense |
arRP |
| 1 Ib |
c768 G>T |
V256V/splice |
─ |
─ |
NVP |
| 1 Ic |
c768 G>T |
V256V/splice |
─ |
─ |
NVP |
| 1 IIa |
c768 G>T |
V256V/splice |
c768 G>T |
V256V/splice |
CRD |
| 2 Ia |
c5917 delG |
V1973X/frameshift |
─ |
─ |
NVP |
| 2 Ib |
c5917 delG |
V1973X/frameshift |
c5882 G>A |
G1961E/missense |
STGD |
| 2 IIa |
c5917 delG |
V1973X/frameshift |
c5917 delG |
V1973X/frameshift |
CRD |
| 2 IIb |
c5917 delG |
V1973X/frameshift |
c5882 G>A |
G1961E/missense |
STGD |
| 2 IIc |
c5917 delG |
V1973X/frameshift |
c5882 G>A |
G1961E/missense |
STGD |
| 2 II d |
c5882 G>A |
G1961E/missense |
─ |
─ |
NVP |
| 3 Ia |
c768 G>T |
V256V/splice |
c3113 C>T |
A1038V/missense |
STGD |
| 3 Ib |
c768 G>T |
V256V/splice |
─ |
─ |
NVP |
| 3 IIa | c768 G>T | V256V/splice | c768 G>T | V256V/splice | STGD |
Abbreviations: arRP; autosomal recessive retinitis pigmentosa, CRD; cone rod dystrophy, STGD; Stargardt disease, NVP; no visual problems *clinical presentation at last visit
Table 3. Ff-ERG results for the right eye of the patients.
| Patient and age at examination | Rod response; dark-adapted blue single flash b-wave Amplitude. (µV) | Combined response; dark-adapted white single flash b-wave Amplitude. (µV) | Combined response; dark-adapted white single flash a-wave Amplitude. (µV) | Cone response; dark-adapted 30 Hz flickering white light b-wave Amplitude. (µV) | Cone response; dark-adapted 30 Hz flickering white light b-wave IT (ms) |
|---|---|---|---|---|---|
| 1Ia (63)
(69) |
1
10 |
40
24 |
23
20 |
10
7 |
40.1
41.0 |
| 1Ib (61) |
132 |
256 |
124 |
38 |
29.5 |
| 1Ic (60) |
183 |
296 |
145 |
39 |
32 |
| 1IIa (9)
(16)
(20
(23) |
55
65
60
82 |
100
89
83
110 |
60
45
48
51 |
20
6
7
5 |
37.2
40.8
42.7
41.0 |
| 2Ia (41) |
170 |
250 |
92 |
41 |
29.5 |
| 2Ib (48) |
140 |
330 |
200 |
55 |
28.5 |
| 2IIa (10)
(15)
(25) |
a
20
11 |
29
39
23 |
14
7
2 |
11
5
7 |
40.0
43.5
45.0 |
| 2IIb (22) |
280 |
370 |
250 |
75 |
27.5 |
| 2IIc (12) |
320 |
420 |
190 |
74 |
27.0 |
| 2IId (9) |
160 |
360 |
120 |
39 |
28.5 |
| 3Ia (21)
(28) |
190
130 |
300
270 |
100
100 |
60
53 |
29.2
31.0 |
| 3Ib |
b |
b |
b |
b |
b |
| 3IIa (10) | 120 | 200 | 110 | 29 | 30.0 |
a: no value recorded due to failure of ff-ERG registration. b: ff-ERG not recorded
Figure 2.
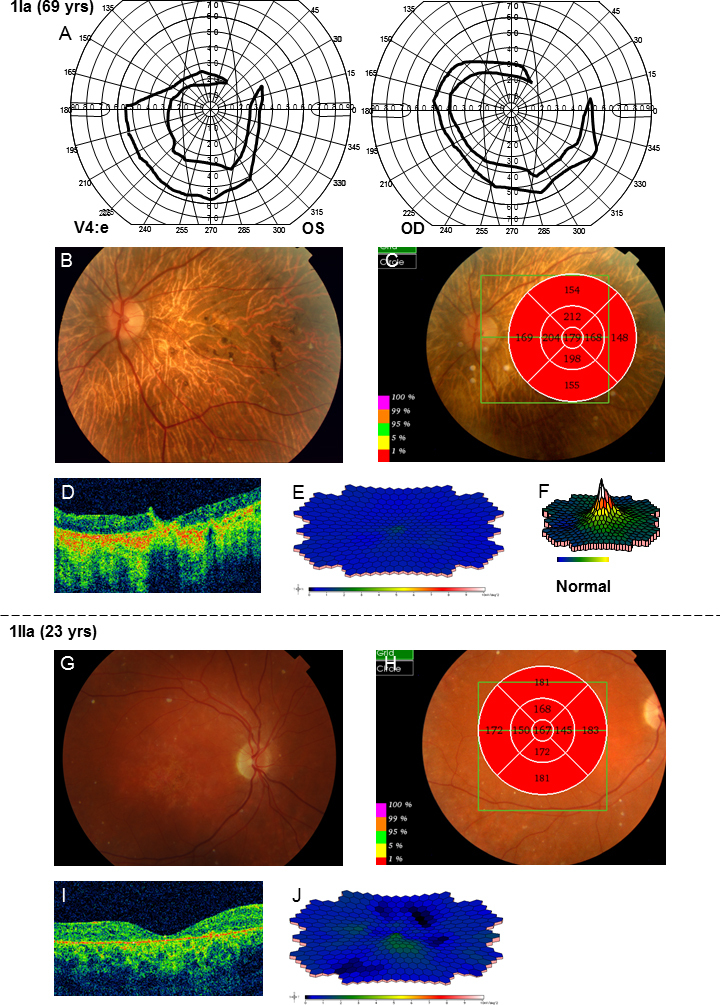
Results of examinations for patients 1Ia and 1IIa. A: The latest Goldmann perimetry shows only small crescent-shaped temporal remnants. B: The fundus photograph shows changes typical for autosomal recessive retinitis pigmentosa (arRP), including pale optic nerve head, narrowed retinal vessels, bone spicule-like pigmentations in the periphery but also atrophy and pigmentations in the macula. C–D: Optical coherence tomography (OCT) measurement of macular thickness demonstrates attenuation in all segments of the retinal thickness map. E: Multifocal electroretinography (mERG) shows severely reduced amplitudes and delayed implicit times compared to a normal registration (F). G: The fundus photograph from patient 1IIa reveals pigment changes in the macula and midperiphery, deep orange-yellow retinal flecks in the posterior pole and midperiphery, and a slightly pale optic nerve head. H–I: OCT measurement of retinal thickness shows an attenuated retina in all segments. J: mERG demonstrates severely reduced central and paracentral amplitudes and delayed implicit times.
Figure 3.
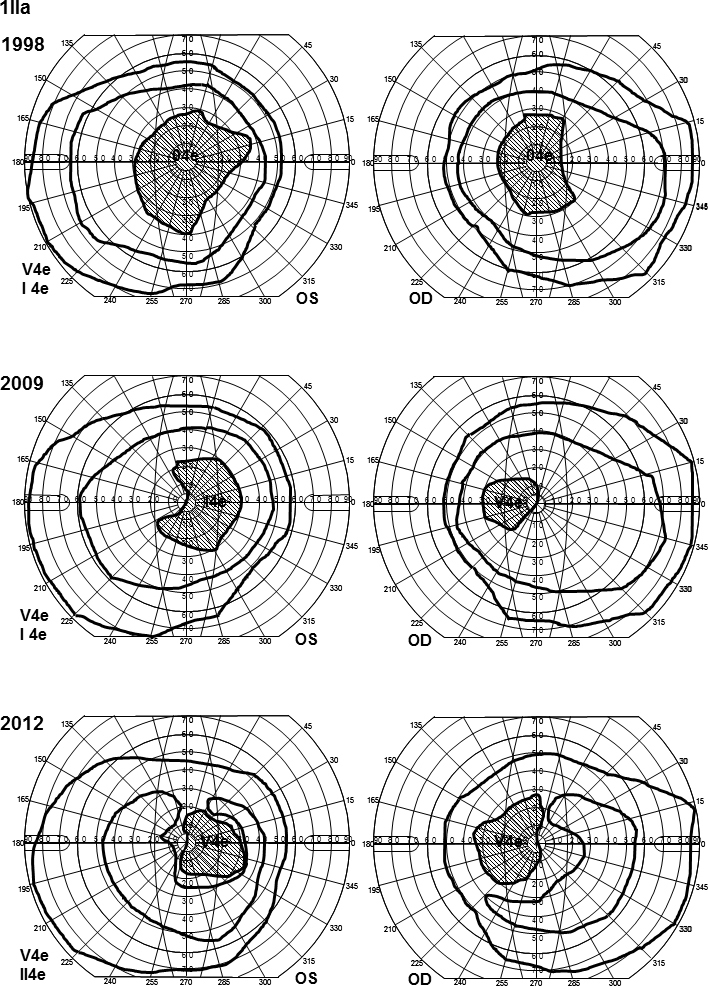
Goldmann perimetry for patient 1IIa. Visual fields show large scotomas with deepening over time. The V4e isopter is almost normal, but the I4e isopter shows slight constriction. At the last examination, the patient could not detect the I4e object, and the II4 e was used instead showing constriction in both eyes.
Family 2
All family members harbored one or two mutations in the ABCA4 gene (Table 2). Patient 2IIa initially had almost normal visual fields for her age, but over time, they became increasingly constricted with large scotomas (Figure 4). The ff-ERG was severely reduced, and the 30 Hz flicker IT was prolonged, showing further delay over time (Table 3). The mERG was severely reduced, and the registration was of bad quality due to poor fixation. Fundoscopic changes were mostly consistent with arRP, but due to the character of her visual fields and the ff-ERG, patient 2IIa was classified as having CRD. Patient 2IIb was diagnosed with late onset STGD (Table 1). She had small central scotomas and reduced mERG amplitudes within the central 15° (Figure 5). The ff-ERG parameters were normal. Subtle fundoscopic changes, typical of STGD, were found as well as an attenuated retina on OCT. The youngest sister, patient 2IIc, who has the same mutations as patient 2IIb (Table 2) has no symptoms yet, but the mERG was reduced (Figure 6), and OCT demonstrated attenuated retina in the innermost five segments of the macular map as well as intraretinal fluid in the center. Only subtle changes were found in the fundus (Figure 6). The youngest of the siblings, patient 2IId, and the mother, patient 2Ia, harbored only one ABCA4 mutation and accordingly are carriers without symptoms and with normal examination results (Table 1 and Table 3). The father, patient 2Ib, had been diagnosed with age-related macular degeneration at another eye clinic 4 years before the study, due to yellow flecks around the macula (Figure 6). Our investigations revealed a condition consistent with late onset STGD, and he harbored the same mutations as his two daughters, patients 2IIb and 2IIc. He had small scotomas within the most central 5° and corresponding reduction in the paracentral mERG (Figure 6) but normal implicit times. OCT showed attenuated retina in the five innermost segments (Figure 6).
Figure 4.
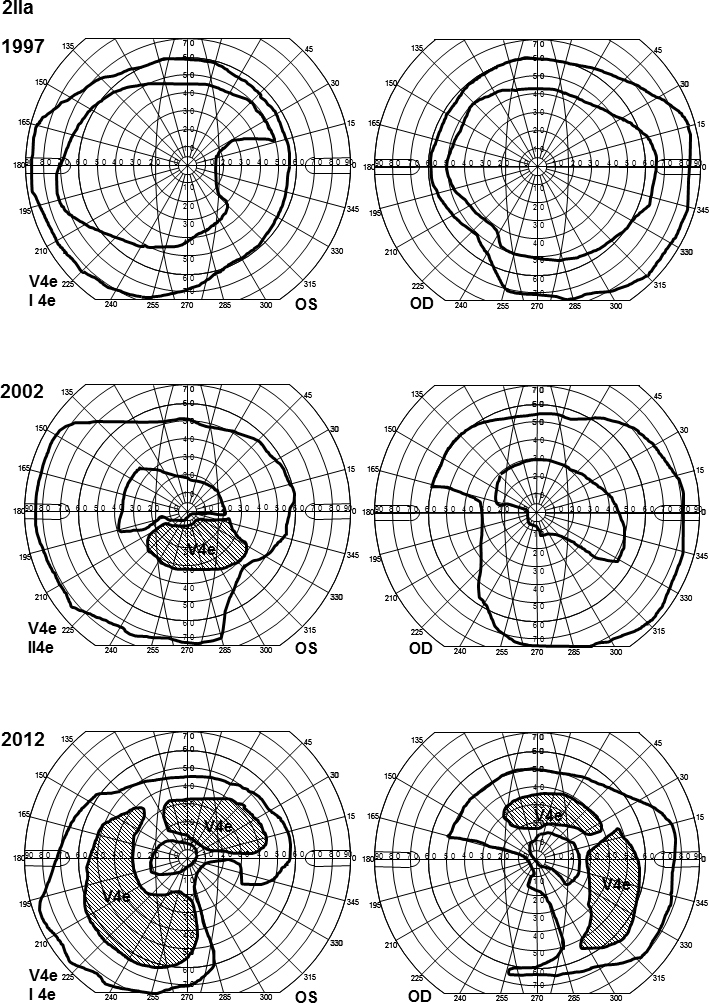
Goldmann perimetry for patient 2IIa. The outer borders of the visual fields were initially quite normal for the V4e isopter but showed nasal and inferior constriction for the I4e object in the left eye. Over the years, the visual fields have become increasingly constricted concerning the V4e isopter inferiorly and nasally and the I4e isopter concentrically. Moreover, she has developed large temporal and upper scotomas for V4e.
Figure 5.
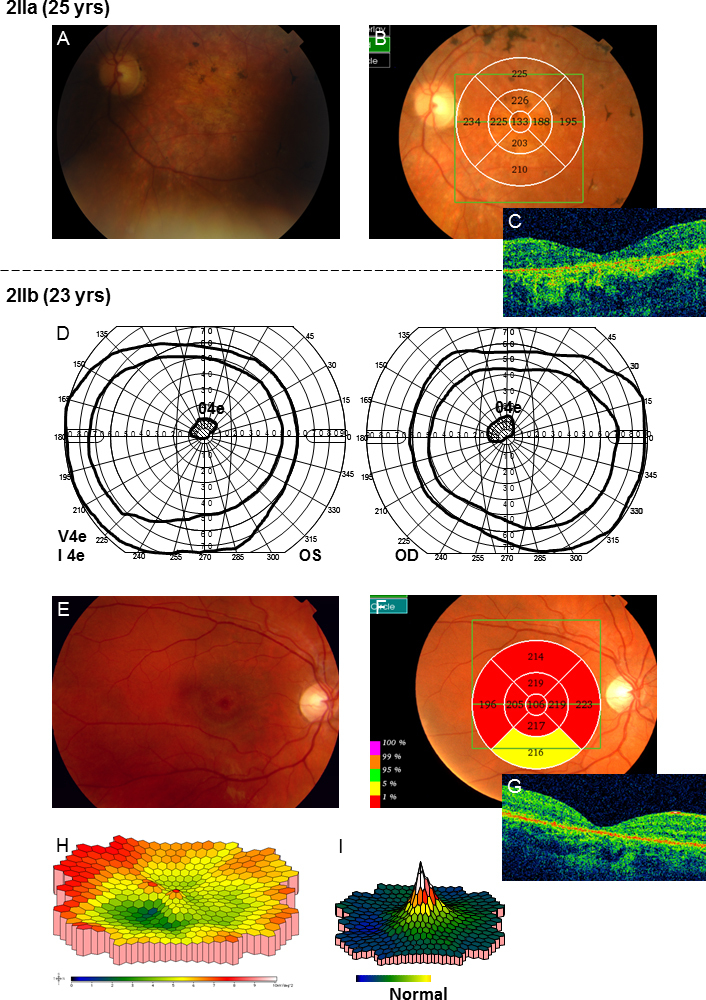
Results of the examinations for patients 2IIa and 2IIb in family 2. A: Fundoscopic changes including slightly pale optic nerve heads, attenuated vessels, a few pigmentations and atrophies in the maculas and bone spicule-like pigmentations in the midperiphery were found. B–C: Optical coherence tomography (OCT) measurement of retinal thickness showed attenuation in all segments of the retinal map. D: Goldmann visual field plots for patient 2IIb showed normal outer borders for the V4e and I4e but small scotomas for the 04e object. E: Around the maculas, discrete yellow flecks and irregular pigmentation was found. F–G: OCT showed attenuation of retinal thickness in all segments of the macular map (H) multifocal electroretinography (mERG) showed reduced amplitudes within the central 15° compared to the normal condition (I). The implicit times of the mERG were normal though.
Figure 6.
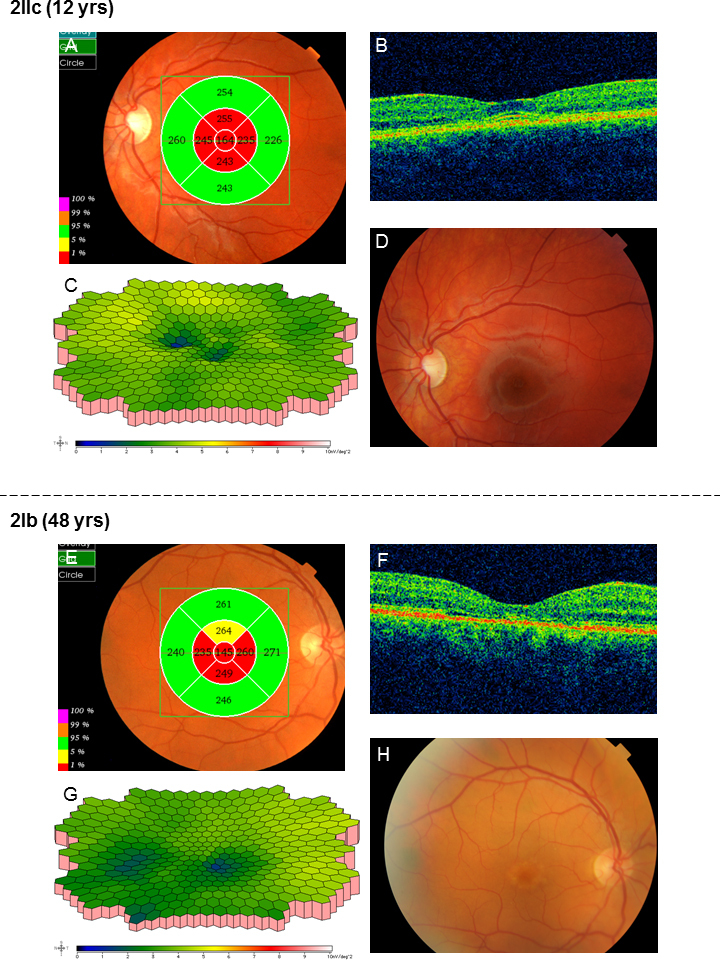
Results of the examinations for patients 2IIc and 2Ib in family 2. A: In patient 2IIc, optical coherence tomography (OCT) showed attenuated retina in the innermost five segments of the macular map even before visual acuity (VA) was reduced. B: Moreover, intraretinal fluid was found in the center of the macula. C: The amplitudes of the multifocal electroretinography (mERG) were reduced especially in the center compared to the normal condition (Figure 5I), but implicit times were normal. D: Only subtle unspecific changes with discrete irregular macular pigmentation were found in the fundus. E: OCT of the father in family 2, patient 2Ib, also showed attenuated retina in the five innermost segments of the macular map although the macular contour was normal (F). G: The mERG showed reduced paracentral amplitudes. Implicit times were normal. H: This patient had previously, at another clinic, been diagnosed with age-related macular degeneration due to yellow flecks around the macula.
Family 3
Two different ABCA4 mutations were found in family 3 (Table 2). The mother, patient 3Ia, was diagnosed with STGD at the age of 15 with a history of reading problems and photophobia. She had small central scotomas that have grown slightly deeper and wider over the years (Figure 7). The mERG was reduced in the paracentral and central areas (Figure 8), and the ITs for the latest mERG (LE) were slightly prolonged. OCT showed macular attenuation, and the fundoscopic changes are typical for STGD (Figure 8). Her son, patient 3IIa, was younger (age 6) at the onset of symptoms (photophobia and reading problems), and his VA was worse at first visit (Table 1). Goldmann perimetry showed large and deep central scotomas (Figure 8). The ff-ERG parameters were normal, but the amplitudes of the 30 Hz flicker were in the lower range of the reference interval (Table 3). MERG indicated reduced paracentral amplitudes and slightly prolonged ITs for the outermost ring. OCT showed attenuated retina in all segments of the macular map, although fundoscopy revealed only slightly irregular pigmentation (Figure 8). The father, patient 3Ib, who harbors only one mutation, had no symptoms (Table 1 and Table 3). Genetic testing for RP were also performed in the patients and showed no mutations.
Figure 7.
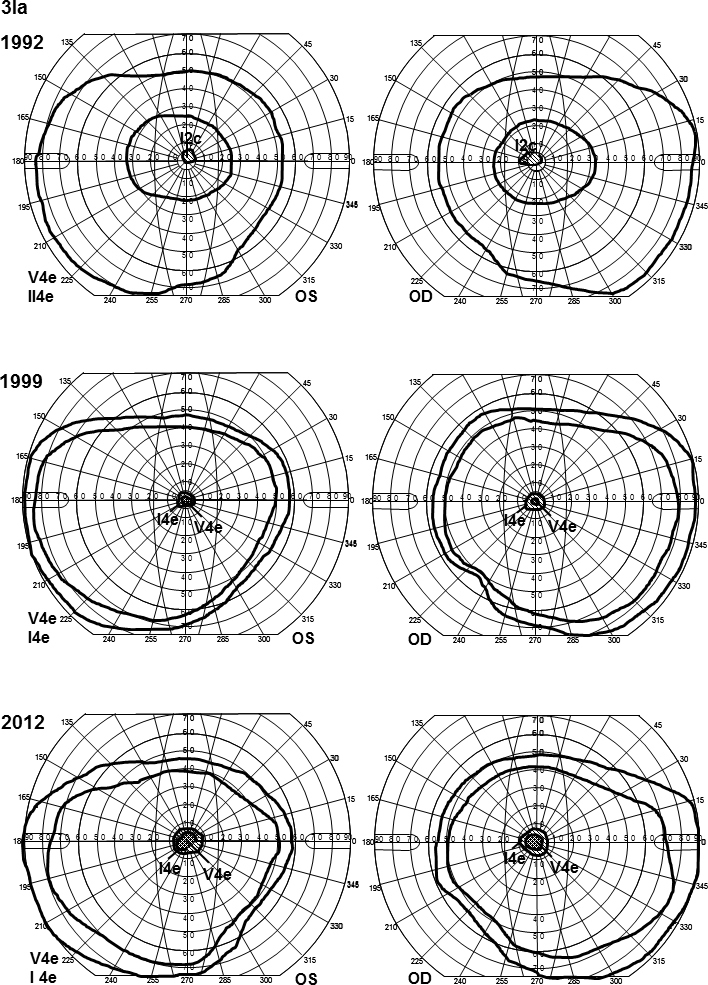
Goldmann visual field plots for patient 3Ia showing normal outer borders for the V4e and I4e isopters, along with small central scotomas. The scotomas have grown deeper (from I2c to V4e) and slightly wider over the years (from 1992 to 2012). Notice that the II4e object and not the I4e was used in 1992.
Figure 8.
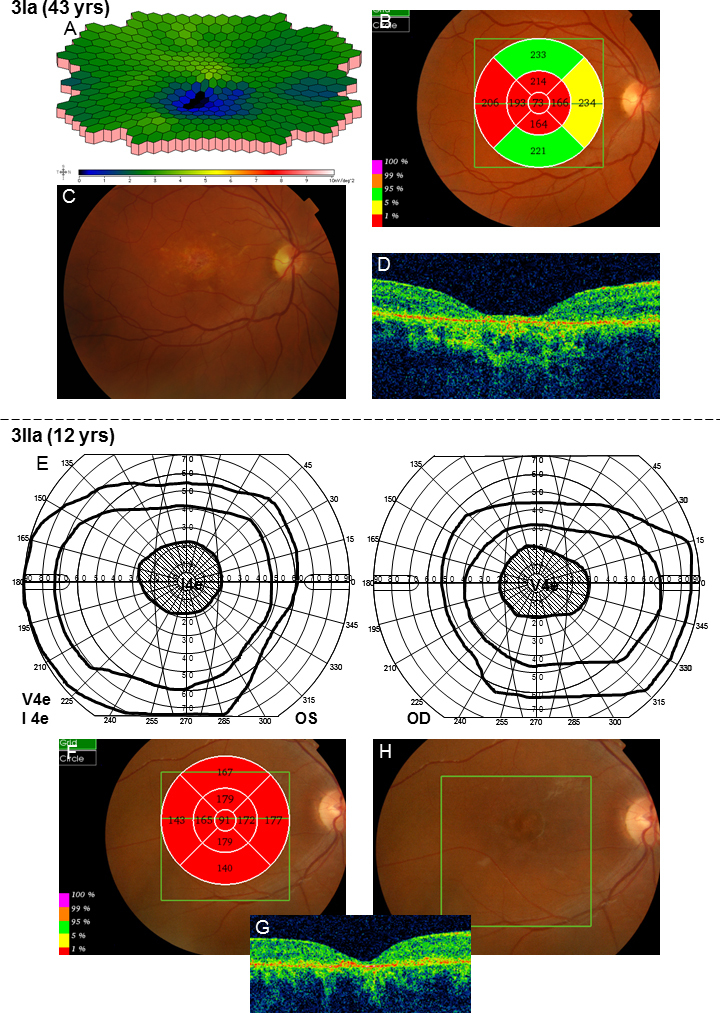
Results of the examinations for patients 3Ia and 3IIa in family 3. A: Multifocal electroretinography (mERG) showed reduced central and paracentral amplitudes. The implicit times for the latest mERG were normal in the right eye but slightly prolonged in the left eye. B: The optical coherence tomography (OCT) macular thickness map showed attenuation in all segments except the outer superior and inferior ones. C: Fundoscopic changes include yellow flecks, atrophies, and irregular pigmentation around the maculas. D: In addition, the contour of the macula appeared attenuated. E: Goldmann visual field plots from patient 3IIa showed large central scotomas for the V4e object, including the most central 20–25°. Moreover, the V4e isopters were normal but the I4e isopters seemed slightly constricted. F–G: Retinal thickness measured with OCT is attenuated in all segments of the macular map. H: Fundoscopy showed slightly irregular pigmentation in the maculas.
Discussion
The fact that ABCA4 mutations are associated with a wide spectrum of retinal degenerations regarding age of onset, visual problems, and course of disease leads to great challenges concerning prognostic counseling. Many investigators have tried to correlate the type of gene defects to a specific phenotype [6,11,28,29], but this hypothesis is not always reliable [8,27,30]. The great number of mutations in the ABCA4 gene (more than 630 known at the moment) also makes it difficult to make predictions only by identifying the genotype, since not all combinations of mutations have even been described yet. To widen the knowledge in this field, we have studied three families with known ABCA4 mutations.
Five patients from two of the families demonstrated clinical and electrophysiological signs of STGD. Three of them, patients 2Ib, 2IIb, and 2IIc, were from family 2 and harbored the same mutations (c5917 delG and c5882 G>A). The c5882 G>A mutation is mild [37] and has previously been associated with retinal changes restricted to the parafoveal area [8,23,31,38] and late onset [8,23,27,31,37], findings that correlate well with the observations in our patients (onset at the age of 48 and 23). The youngest patient, 2IIc, was, at the time of the study, still asymptomatic, which may indicate later onset for her. Family 2 is from Hungary, and the c5882 G>A mutation is common in Hungarian patients with STGD [39]. In this and other [23,38] studies, the c5882 G>A mutation has been associated with reduced retinal thickness on OCT. Interestingly, the patient with worse VA (patient 2IIb) showed more widespread retinal attenuation (Figure 5F) than the patients with better VA (Figure 6A,E). Even more interesting, retinal thickness was reduced in patient 2IIc despite full VA and almost normal fundus appearance. Thus, morphological retinal changes seem to precede visual deterioration suggesting that OCT may be useful in investigating children with reduced VA of unknown cause. Moreover, our patients showed normal ff-ERG results but reduced mERGs, indicating functional changes restricted to the macular region. Similar electrophysiological findings have been reported by other authors [8,23,38] in association with c5882 G>A. In one study [38], however, a combination of c5882 G>A and mutations other than c5917 delG gave ff-ERG changes consistent with CRD or arRP. This, again, highlights the difficulties in predicting the severity of a condition from mutational analysis alone. Gerth et al. [8] and Rivera et.al [40]. have described mother/daughter pairs with the same mutations as patients 2Ib and 2IIa. The elder generation (with the c5882 G>A and c5917 delG mutations) showed similar findings with late onset, moderately reduced VA, normal ff-ERG, and reduced mERG. The younger generation, including patient 2IIa, with homozygote c5917 delG mutations, instead demonstrated signs of progressive CRD with early onset, much reduced ff-ERG responses for cones and rods, and prolonged 30 Hz flicker IT (Table 3). Similar electrophysiological findings and attenuated retina on OCT were found by Testa et al. [38] in patients with the same mutations. In view of this, homozygote c5917 delG mutations seem to cause serious and progressive forms of retinal degeneration such as CRD and arRP [8,38,40].
The remaining two patients with STGD were a mother (patient 3Ia) and her son (patient 3IIa). Patient 3Ia was compound heterozygote for the c3113 C>T and c768 G>T mutations while her son, patient 3IIa, was homozygote for c768 G>T. Patient 3Ia seemed to have a milder condition with later onset, small, quite stable scotomas (Figure 7) and normal ff-ERG (Table 3) while her son was worse afflicted, showing early onset, larger scotomas (Figure 8), ff-ERG cone responses on the lower margin, and more widely spread retinal attenuation on OCT (Figure 8F compared to Figure 8B). The c3113 C>T mutation has previously most often been described as a component of the complex allele c1622 T>C;c3113 C>T (L541P/A;A1038V) [11,25,38], but it is also known to be pathogenic as a separate mutation [15,40]. This mutation has most frequently been found in late onset STGD [30,37,41], but association with onset in the early teens, as in patient 3Ia, has also been described [8]. The c3113 C>T mutation is a mild or moderate mutation [37] usually associated with normal ff-ERG and reduced mERG [8,41] findings that correspond well to the observations in patient 3Ia.
The c768G>T mutation, however, is severe [11,25,37,42,43], leading to serious and progressive retinal degenerations, such as CRD. Patients 1IIa and 3IIa were homozygote for the c768G>T mutation. Patient 1IIa, accordingly, had large central scotomas (Figure 3) and progressive deterioration of VA and cone function. For patient 3IIa, the follow-up period was quite short, but until now, he has demonstrated a similar clinical picture with deteriorating VA and large scotomas that do not match the small scotomas of his mother, patient 3Ia. Thus, the phenotype of 3IIa more resembles the phenotype of 1IIa, besides from that the ff-ERG has not shown that much cone reduction yet. Future follow-up will reveal whether his condition will progress to a CRD. If not, other genes or environmental factors, not yet found, may modify the phenotype.
Patients with homozygote c768G>T and c5917 delG mutations seem to have worse retinal degeneration. This might be explained by the theory of residual function of the ABCA4 protein [11,29]. c768G>T is a splice site mutation leading to loss of transcripts for the ABCA4 gene [43], in turn leading to absent translation of the ABCA4 protein. The lack of ABCA4 leads to accumulation of toxic retinoid compounds that damage the photoreceptors. The c5917 delG is a frameshift mutation probably likewise resulting in an absent or seriously defective ABCA4 protein with consequent photoreceptor damage.
Patient 1Ia, who also harbored the c768G>, but in combination with c2894A>G, had the most serious form of ABCA4 associated retinal degeneration, a form of arRP. The c2894A>G also previously has been described in patients with severely reduced ff- and mERG parameters [38] and seems to be a common mutation in the Scandinavian population [44].
Interestingly, all our patients (1Ia, 1IIa, and 2IIa) with progressive forms of retinal degeneration had delayed ITs for the ff- and/or mERG (Table 3), and delayed ITs have been suggested as a risk factor for progressive disease in other studies [45-48] as well. One possible drawback of this study is the method of gene testing since microarray analysis may miss significant mutations that could have been identified with gene sequencing. Thus, differences in phenotype between similarly genotyped patients may be caused by undetected mutations within the ABCA4 gene or in other potentially modulating genes.
In conclusion, this study confirms that ABCA4 mutations lead to a wide spectrum of retinal degenerations with various levels of visual deficit ranging from STGD to CRD and arRP. Since these mutations occur frequently in the general population (4.5%–10%) [30,43,49-51], different family members can harbor various combinations of mutations leading to diverse phenotypes and prognosis within the same family. Although the same combination of mutations often gives a similar clinical picture, this is not always the case, emphasizing the importance of electrophysiological and genetic testing. In this study, delayed ITs seem to indicate progressive conditions such as CRD or arRP, something that may be useful in prognostic counseling and when it comes to choosing cases for future gene therapy.
Acknowledgments
The author thanks Ing-Marie Holst and Boel Nilsson for skillful technical assistance and Sten Andréasson for fruitful collaboration. This study was supported by grants from Skåne County Council Research and Development Foundation, and Stiftelsen Synfrämjandets Forskningsfond. The author has no conflicts of interest to disclose.
References
- 1.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR). FEBS Lett. 1997;409:247–52. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 3.Illing M, Molday LL, Molday RS. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem. 1997;272:10303–10. doi: 10.1074/jbc.272.15.10303. [DOI] [PubMed] [Google Scholar]
- 4.Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–34. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res. 2001;73:877–86. doi: 10.1006/exer.2001.1093. [DOI] [PubMed] [Google Scholar]
- 6.Cremers FP, Van De Pol DJ, Van Driel M, Den Hollander AI, Van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 7.Fishman GA, Stone EM, Eliason DA, Taylor CM, Lindeman M, Derlacki DJ. ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol. 2003;121:851–5. doi: 10.1001/archopht.121.6.851. [DOI] [PubMed] [Google Scholar]
- 8.Gerth C, Andrassi-Darida M, Bock M, Preising MN, Weber BH, Lorenz B. Phenotypes of 16 Stargardt macular dystrophy/fundus flavimaculatus patients with known ABCA4 mutations and evaluation of genotype-phenotype correlation. Graefes Arch Clin Exp Ophthalmol. 2002;240:628–38. doi: 10.1007/s00417-002-0502-y. [DOI] [PubMed] [Google Scholar]
- 9.Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 10.Klevering BJ, Van Driel M, Van De Pol DJ, Pinckers AJ, Cremers FP, Hoyng CB. Phenotypic variations in a family with retinal dystrophy as result of different mutations in the ABCR gene. Br J Ophthalmol. 1999;83:914–8. doi: 10.1136/bjo.83.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maugeri A, Klevering BJ, Rohrschneider K, Blankenagel A, Brunner HG, Deutman AF, Hoyng CB, Cremers FP. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67:960–6. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, Del Rio T, Dean M, Vilageliu L, Gonzalez-Duarte R, Balcells S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–2. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 13.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–91. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–8. doi: 10.1038/77004. [DOI] [PubMed] [Google Scholar]
- 15.Sun H, Nathans J. Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet. 1997;17:15–6. doi: 10.1038/ng0997-15. [DOI] [PubMed] [Google Scholar]
- 16.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–8. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 19.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–9. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomes NL, Greenstein VC, Carlson JN, Tsang SH, Smith RT, Carr RE, Hood DC, Chang S. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009;50:3953–9. doi: 10.1167/iovs.08-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullins RF, Kuehn MH, Radu RA, Enriquez GS, East JS, Schindler EI, Travis GH, Stone EM. Autosomal recessive retinitis pigmentosa due to ABCA4 mutations: clinical, pathologic, and molecular characterization. Invest Ophthalmol Vis Sci. 2012;53:1883–94. doi: 10.1167/iovs.12-9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allikmets R. Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet. 2000;67:793–9. doi: 10.1086/303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cella W, Greenstein VC, Zernant-Rajang J, Smith TR, Barile G, Allikmets R, Tsang SHG. 1961E mutant allele in the Stargardt disease gene ABCA4 causes bull's eye maculopathy. Exp Eye Res. 2009;89:16–24. doi: 10.1016/j.exer.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119:359–69. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- 25.Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci. 2002;43:1980–5. [PubMed] [Google Scholar]
- 26.Szlyk JP, Fishman GA, Alexander KR, Peachey NS, Derlacki DJ. Clinical subtypes of cone-rod dystrophy. Arch Ophthalmol. 1993;111:781–8. doi: 10.1001/archopht.1993.01090060069025. [DOI] [PubMed] [Google Scholar]
- 27.Burke TR, Tsang SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 2011;32:165–74. doi: 10.3109/13816810.2011.565397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shroyer NF, Lewis RA, Allikmets R, Singh N, Dean M, Leppert M, Lupski JR. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res. 1999;39:2537–44. doi: 10.1016/s0042-6989(99)00037-1. [DOI] [PubMed] [Google Scholar]
- 29.van Driel MA, Maugeri A, Klevering BJ, Hoyng CB, Cremers FP. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 1998;19:117–22. doi: 10.1076/opge.19.3.117.2187. [DOI] [PubMed] [Google Scholar]
- 30.Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet. 2001;108:346–55. doi: 10.1007/s004390100493. [DOI] [PubMed] [Google Scholar]
- 31.Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, Lupski JR. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005;14:2769–78. doi: 10.1093/hmg/ddi310. [DOI] [PubMed] [Google Scholar]
- 32.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 33.Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography (2004 update). Doc Ophthalmol. 2004;108:107–14. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- 34.Andréasson SO, Sandberg MA, Berson EL. Narrow-band filtering for monitoring low-amplitude cone electroretinograms in retinitis pigmentosa. Am J Ophthalmol. 1988;105:500–3. doi: 10.1016/0002-9394(88)90241-3. [DOI] [PubMed] [Google Scholar]
- 35.Hood DC, Bach M, Brigell M, Keating D, Kondo M, Lyons JS, Marmor MF, Mcculloch DL, Palmowski-Wolfe AM. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc Ophthalmol. 2012;124:1–13. doi: 10.1007/s10633-011-9296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hood DC, Bach M, Brigell M, Keating D, Kondo M, Lyons JS, Palmowski-Wolfe AM. ISCEV guidelines for clinical multifocal electroretinography (2007 edition). Doc Ophthalmol. 2008;116:1–11. doi: 10.1007/s10633-007-9089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, Den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology. 2012;119:1199–210. doi: 10.1016/j.ophtha.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Testa F, Rossi S, Sodi A, Passerini I, Di Iorio V, Della Corte M, Banfi S, Surace EM, Menchini U, Auricchio A, Simonelli F. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: implications for gene therapy. Invest Ophthalmol Vis Sci. 2012;53:4409–15. doi: 10.1167/iovs.11-8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hargitai J, Zernant J, Somfai GM, Vamos R, Farkas A, Salacz G, Allikmets R. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2005;46:4402–8. doi: 10.1167/iovs.05-0504. [DOI] [PubMed] [Google Scholar]
- 40.Rivera A, White K, Stohr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, Weber BH. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. 2000;67:800–13. doi: 10.1086/303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burke TR, Tsang SH, Zernant J, Smith RT, Allikmets R. Familial discordance in Stargardt disease. Mol Vis. 2012;18:227–33. [PMC free article] [PubMed] [Google Scholar]
- 42.Klevering BJ, Yzer S, Rohrschneider K, Zonneveld M, Allikmets R, Van Den Born LI, Maugeri A, Hoyng CB, Cremers FP. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur J Hum Genet. 2004;12:1024–32. doi: 10.1038/sj.ejhg.5201258. [DOI] [PubMed] [Google Scholar]
- 43.Maugeri A, Van Driel MA, Van De Pol DJ, Klevering BJ, Van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Dahl N, Brunner HG, Deutman AF, Hoyng CB, Cremers FP. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet. 1999;64:1024–35. doi: 10.1086/302323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenberg T, Klie F, Garred P, Schwartz M. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol Vis. 2007;13:1962–9. [PubMed] [Google Scholar]
- 45.Berson EL, Gouras P, Hoff M. Temporal aspects of the electroretinogram. Arch Ophthalmol. 1969;81:207–14. doi: 10.1001/archopht.1969.00990010209011. [DOI] [PubMed] [Google Scholar]
- 46.Eksandh L, Ekstrom U, Abrahamson M, Bauer B, Andreasson S. Different clinical expressions in two families with Stargardt's macular dystrophy (STGD1). Acta Ophthalmol Scand. 2001;79:524–30. doi: 10.1034/j.1600-0420.2001.790520.x. [DOI] [PubMed] [Google Scholar]
- 47.Hood DC, Holopigian K, Greenstein V, Seiple W, Li J, Sutter EE, Carr RE. Assessment of local retinal function in patients with retinitis pigmentosa using the multi-focal ERG technique. Vision Res. 1998;38:163–79. doi: 10.1016/s0042-6989(97)00143-0. [DOI] [PubMed] [Google Scholar]
- 48.Seeliger M, Kretschmann U, Apfelstedt-Sylla E, Ruther K, Zrenner E. Multifocal electroretinography in retinitis pigmentosa. Am J Ophthalmol. 1998;125:214–26. doi: 10.1016/s0002-9394(99)80094-4. [DOI] [PubMed] [Google Scholar]
- 49.Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 50.Maugeri A, Flothmann K, Hemmrich N, Ingvast S, Jorge P, Paloma E, Patel R, Rozet JM, Tammur J, Testa F, Balcells S, Bird AC, Brunner HG, Hoyng CB, Metspalu A, Simonelli F, Allikmets R, Bhattacharya SS, D'urso M, Gonzalez-Duarte R, Kaplan J, Te Meerman GJ, Santos R, Schwartz M, Van Camp G, Wadelius C, Weber BH, Cremers FP. The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur J Hum Genet. 2002;10:197–203. doi: 10.1038/sj.ejhg.5200784. [DOI] [PubMed] [Google Scholar]
- 51.Riveiro-Alvarez R, Aguirre-Lamban J, Lopez-Martinez MA, Trujillo-Tiebas MJ, Cantalapiedra D, Vallespin E, Avila-Fernandez A, Ramos C, Ayuso C. Frequency of ABCA4 mutations in 278 Spanish controls: an insight into the prevalence of autosomal recessive Stargardt disease. Br J Ophthalmol. 2009;93:1359–64. doi: 10.1136/bjo.2008.148155. [DOI] [PMC free article] [PubMed] [Google Scholar]