

Abstract
Summary: Pathway Commons is a resource permitting simultaneous queries of multiple pathway databases. However, there is no standard mechanism for using these data (stored in BioPAX format) to annotate and build quantitative mathematical models. Therefore, we developed a new module within the virtual cell modeling and simulation software. It provides pathway data retrieval and visualization and enables automatic creation of executable network models directly from qualitative connections between pathway nodes.
Availability and implementation: Available at Virtual Cell (http://vcell.org/). Application runs on all major platforms and does not require registration for use on the user’s computer. Tutorials and video are available at user guide page.
Contact: vcell_support@uchc.edu
1 INTRODUCTION
The Pathway Commons collection of databases (Cerami et al., 2011) provides access to >442 000 interactions among 86 000 physical entities. The data are retrieved in the form of Biological PAthway eXchange, BioPAX (Demir et al., 2010) that can be converted to the Systems Biology Markup Language, SBML (Hucka et al., 2003)—the gold standard for exchanging mathematical models and the native standard for the Biomodels model database (Le Novère et al., 2006). However, while converting to SBML, the wealth of information in annotations is often lost because the semantics of general biological knowledge differs from that of mathematical models (Ruebenacker et al., 2009).
Here we describe a tool that allows users to access the rich trove of pathway data available through Pathway Commons and use it to build well-annotated mathematical models. It was implemented in the Virtual Cell (VCell) modeling and simulation framework (Cowan et al., 2012; Moraru et al., 2008), a platform designed for building and simulating compartmental and spatial models and analysis of simulation results. The VCell database contains >54 000 user models of which ∼500 are public. However, most of these models were created ‘ad-hoc’, using multiple iterations. In doing so, users rarely used detailed names to identify variables and functions in the mathematical model, but most often used simplified abbreviations like R, ATP or even arbitrary symbols such as S1, P1 and so forth. As a result, in the absence of extensive accompanying documentation, a model is often difficult or even impossible to understand by anyone except for the model author(s). As models increase in complexity, sharing and reusing models and components become a necessity, and creating a well-annotated model is essential. For this purpose, Minimal Information Required In the Annotation of Models (MIRIAM) community standard was developed (Le Novère et al., 2005). Our software facilitates this by linking every model element to extensive documentation coming from pathway databases, helping researchers to produce models that are easier to understand and shared with the community.
The tool stores connectivity information derived from pathway interactions and can convert it into a reaction network in VCell, which can then be populated with rate constants and initial concentrations to generate kinetic simulations, which is a useful feature for researchers learning how to model. Online tutorials describe simple ways to create a simulation-ready qualitative model of a complete pathway. We also expect the tool to be appealing to bioinformaticians as a browser and editor for data in Pathway Commons. We hope that the ease of creating computational models to use pathway data can bring together researchers doing mathematical modeling, biologists working with pathway data and bioinformaticians querying and visualizing it.
2 LINKING PATHWAYS AND MODELS
2.1 Accessing and linking pathway data to existing models
The new Pathway Commons at VCell interface allows users to search pathway data from inside VCell (Fig. 1, panel A). The entries of interest can be imported and stored alongside a VCell model as a Pathway Model to be available whenever necessary (panel B). The pathway data can be searched, filtered and viewed as a diagram or as a list of objects (panel C). Each entry in a Pathway Model includes a list of identifiers from different databases with clickable web links that lead to relevant entries in any of these databases (panel D). While working on an existing VCell model, a user can create a relevant Pathway Model and link it to entities in the VCell Reaction Diagram (panel F) to fully annotate the appropriate elements of the mathematical system (panel E). All such linked elements are marked with the letter L in both the Pathway Diagram and the Reaction Diagram (panel A).
Fig. 1.
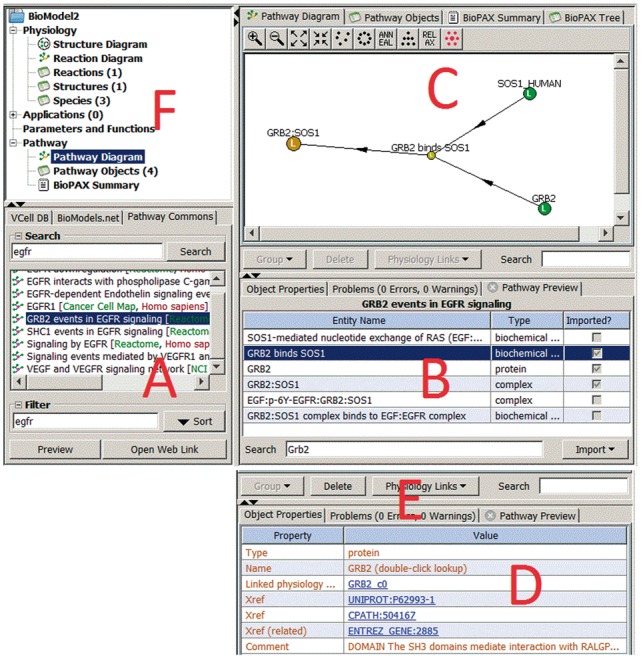
Pathway Commons at VCell. The Pathway database panel (A) allows the user to search Pathway Commons and select pathways for bringing into VCell. The Pathway preview panel (B) allows the user to import elements of the selected pathway into VCell. The Pathway view panel (C) allows the user to see multiple views of imported pathway data, to select elements to inspect in the Object properties panel (D) and to convert into VCell model elements (E). Selecting an item under Pathway in the navigation panel (F) will open the corresponding tab in the view panel (C)
2.2 Creating new models from pathway data
Selected physical entities and physical interactions from pathways can be automatically converted into VCell species and reactions in a Reaction Diagram. The default location of newly created species is in a single compartment and the default stoichiometries in newly created reactions are set to 1, as these types of information are often missing in the pathway databases. Users can then define multiple compartments and change reaction properties. Reaction kinetics defaults to mass action, but any kinetic formalism and rate laws can be specified. The new VCell model is fully annotated providing all available information about this element coming from multiple databases (Fig. 1, bottom right panel). Once all the initial concentrations (or copy numbers for stochastic simulations), rate constants and cellular geometry parameters are specified, the model can be sent to the appropriate VCell simulator to provide time courses computation.
3 IMPLEMENTATION
The format for VCell model representation is the VCell Markup Language (VCML), an XML dialect that has some similarities to SBML. Pathway Commons provides data in BioPAX format (Demir et al., 2010), which is an Resource Description Framework (RDF)-based ontology. As we pointed out earlier (Ruebenacker et al., 2009), BioPAX data are not fully compatible with XML-based modeling formats, and converting BioPAX data to annotated SBML or VCML will lead to data loss and distortion (Ruebenacker et al., 2009). Previously, we developed an intermediate representation in the form of the SBPAX ontology (Ruebenacker et al., 2009), which is an RDF/OWL schema that provides mapping of pathway entities to model elements. The first version of a BioPAX to model converter was implemented as a standalone software tool, SyBil (Ruebenacker and Blinov, 2011). This was then ported and integrated into core functionality of the client-server VCell platform.
We introduced two new components to VCML—the Pathway Model and the Relationship Model. These permit unambiguous storage of the complete extracted BioPAX data, without any loss that may be caused by translation, and the mapping of the pathway elements to VCell Model entities.
The Pathway Model contains all data that was extracted from Pathway Commons during model generation. Some of it can be linked to BioModel elements, but the user may keep unlinked data in the Pathway Model for future use. Pathway Commons currently supplies the data in BioPAX Level 2 format, but we already support importing data in Level 3 format. Sesame open-source framework is used for querying and analyzing these RDF data. It allows for generating Java source files from ontologies and use of SPARQL to query RDF. Java objects were created for all BioPAX Level 3 classes, with conversion from Level 2 done internally. The Pathway model is stored inside VCML as RDF annotations under the top level element. Use of RDF allows for seamless incorporation of pathway data coming from multiple sources. To operate on Pathway Model components, we implemented BioPAX I/O operations as new Java classes in VCell.
The Relationship Model is a new element of the VCML schema that links elements of VCell BioModel (species and reactions) to elements in the Pathway Model (physical entities and interactions). The mapping is many-to-many, so several pathway objects can be linked to a single model element and vice versa. Linked pathway entities have unique identities through the CPATH ID (the identifier assigned by Pathway Commons), and we store and display all UnificationXref IDs for reuse in different models.
ACKNOWLEDGEMENT
The authors thank Emek Demir, Igor Rodchenkov, Garry Bader and the BioPAX team for their valuable help.
Funding: National Institutes of Health (P41-GM103313, U54-RR022232, R01-GM095485).
Conflict of Interest: none declared.
REFERENCES
- Cerami EG, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AE, et al. Spatial modeling of cell signaling networks. Methods Cell Biol. 2012;110:195–221. doi: 10.1016/B978-0-12-388403-9.00008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir E, et al. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010;28:935–942. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- Le Novère N, et al. Minimum information requested in the annotation of biochemical models (MIRIAM) Nat. Biotechnol. 2005;23:1509–1515. doi: 10.1038/nbt1156. [DOI] [PubMed] [Google Scholar]
- Le Novère N, et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006;34:D689–D691. doi: 10.1093/nar/gkj092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru II, et al. The Virtual Cell modeling and simulation software environment. IET Syst. Biol. 2008;2:352–362. doi: 10.1049/iet-syb:20080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruebenacker O, Blinov ML. Using views of Systems Biology Cloud: application for model building. Theory Biosci. 2011;130:45–54. doi: 10.1007/s12064-010-0108-6. [DOI] [PubMed] [Google Scholar]
- Ruebenacker O, et al. Integrating BioPAX pathway knowledge with SBML models. IET Syst. Biol. 2009;3:317–328. doi: 10.1049/iet-syb.2009.0007. [DOI] [PubMed] [Google Scholar]