

Abstract
G-protein coupled receptors (GPCRs) are the primary target class of currently marketed drugs, accounting for about a quarter of all drug targets of approved medicines. However, almost all the screening efforts for novel ligand discovery rely exclusively on cellular systems overexpressing the receptors. An alternative ligand discovery strategy is a fragment-based drug discovery, where low molecular weight compounds, known as fragments, are screened as initial starting points for optimization. However, the screening of fragment libraries usually employs biophysical screening methods, and as such, it has not been routinely applied to membrane proteins. We present here a surface plasmon resonance biosensor approach that enables, cell-free, label-free, fragment screening that directly measures fragment interactions with wild-type GPCRs. We exemplify the method by the discovery of novel, selective, high affinity antagonists of human β2 adrenoceptor.
Keywords: Fragment screening, G-protein coupled receptors, surface plasmon resonance, β2 adrenoceptor
G-protein coupled receptors (GPCRs) are the principal class of drug targets. The conventional approach to the discovery of GPCR ligands has focused on the high-throughput screening (HTS) of cellular systems overexpressing the receptors against very large collections of compounds (ten to hundreds of thousands of compounds), with typical molecular weights ranging from 350 to 500 Da (27 to 38 non-hydrogen atoms). In contrast, fragment screening is now established as an important new approach to drug discovery.1 The principle of fragment-based drug discovery is that a relatively small number of low molecular weight fragments can represent large areas of chemical space. However, fragment screening has not been routinely applied to membrane proteins. This is particularly limiting, as around a quarter of current drug targets of approved medicines are GPCRs. Fragment-based drug discovery starts with the screening of a small library, consisting often of only several hundred to a couple of thousand low molecular weight compounds, called fragments. Fragments are usually in the molecular weight range of 100 to 300 Da (8 to 23 non-hydrogen atoms). The low molecular weights of fragments usually result in significantly lower affinity hits, compared to HTS hits; however, the interactions can be very ligand efficient2 as the fragments can adopt optimum orientations in the active site. Conventional ligand displacement assay methods measuring the displacement of radiolabeled and fluorescent-labeled ligands have been used to fragment screening of histamine-H43 and adenosine-A3 receptors,4 respectively, in cell-based assays. However, for fragment screening there are significant advantages to exploiting highly sensitive label-free, biophysical screening techniques in order to detect potential low affinity fragment hits. Currently, surface plasmon resonance (SPR) has become a dominant technology for fragment screening.5 The advantage of SPR based screening is that it has unparalleled potential not only to design screening procedures to identify both orthosteric and allosteric ligands6 but also more importantly to use more sophisticated targets such as protein complexes, which are otherwise not feasible in cellular systems.7
In recent years, biophysical techniques have advanced to face the challenge of studying membrane bound GPCRs. Thermostabilized mutated GPCRs, developed for crystallization, have been shown to be suitable for biophysical fragment screening, by SPR and nuclear magnetic resonance (NMR).8−10 However, the process of engineering conformational stability results in GPCR constructs that have been shown to lack the full range of wild-type pharmacology.11−13 Ideally, biophysical fragment screening of the wild-type sequence GPCRs, which retain the full range of pharmacology,7,14,15 may have advantages for ligand discovery over mutated, thermostabilized GPCRs. We have developed a biosensor fragment screening protocol that enables cell-free, label-free fragment screening that directly measures fragment interactions with the nonstabilized, purified wild-type GPCR. We exemplify the method by the discovery of novel antagonists of the human β2 adrenoceptor.
The β2 adrenoceptor has been the prototypical GPCR for the development of new methods. The β2 adrenoceptor was the first ligand-binding GPCR to be purified from cell membranes; the first to be cloned and sequenced;16 provided the first high-resolution crystal structures;17−19 provided the first example of structure-based discovery of novel GPCR ligands by virtual screening;20 and was the first receptor to be crystallized in complex with its G protein, Gs.21 Hence, the β2 adrenoceptor was selected as the ideal candidate GPCR for novel biophysical methods development.
A human β2 adrenoceptor construct containing a FLAG tag at the N-terminus and histidine 10 (His-10) tag at the C-terminus was generated for baculovirus expression in Sf9 cells (Supporting Information Figure S1). The receptor was solubilized and purified as described before.22 The β2 adrenoceptor was captured via His-10 tag on NTA sensor chip.
To establish whether the captured β2 adrenoceptor is pharmacologically active on the surface, binding of an agonist (fenoterol) and an antagonist (alprenolol) was measured (Figure 1a). The measured affinity of alprenolol (KD = 790 pM) corresponds very well to the binding affinity measured in native membrane using radioligand binding (ki = 1 nM).23 Fenoterol exhibited affinity KD = 139 nM, which is also in very good agreement with measured affinity by radioligand binding (ki = 126 nM).24 These data suggest that immobilization of the purified β2 adrenoceptor on the SPR surface does not alter its pharmacological properties and therefore represents a viable approach for in vitro drug screening.
Figure 1.

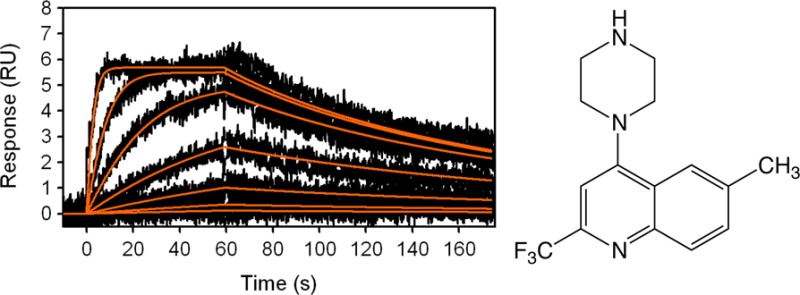
SPR sensorgrams of (a) an antagonist alprenolol and an agonist fenoterol, and (b) fragments A to E interacting with β2 adrenoceptor. Red lines represent kinetic fit. The right-side inserted graphs for fragments D and E represent equilibrium fits.
As a proof of principle, a library consisting of 656 fragments5 was screened against the immobilized β2 adrenoceptor at one concentration. The average molecular weight (MW) of the fragment library is MW = 187 (equating to 13 non-hydrogen (heavy) atoms) with fragment sizes varying from 94 Da (7 heavy atoms) to 341 Da (24 heavy atoms). To reduce the nonspecific binding, especially at high concentrations that could be a source of false positives when screening fragment libraries, it is important to find a suitable reference surface. The reference surface should ideally be captured in the same manner as the target protein but in an inactive state of ligand binding. Receptors with pharmacologically blocked binding sites have previously been demonstrated as highly suitable reference surfaces for GPCR analysis by SPR.6 The β2 receptor was preincubated with a slow-off-rate agonist compound BI-16710725 to create a control/reference target receptor (i.e., ligand binding site-blocked state), which was immobilized by the method described above. A third channel was used as a blank reference surface. To evaluate screening data, single-point concentrations were read for each of the fragments before the end of the injection. Examples of collected data are shown in Supporting Information Figure S2a,b. Each sensorgram was then inspected against all three surfaces (target, reference target, and blank). Examples for two of the hit compounds are shown in Supporting Information Figure S2c,d. Eighty-one compounds were then further selected and screened at six concentrations, ranging from 300 to 1.2 μM. A total of five fragment hits were confirmed: fragments A to E with affinities ranging from KD = 17 nM to KD = 22 μM (Table 1). Fragments A to E were screened at concentrations adjusted to the affinity of each fragment in duplicate (Figure 1b).
Table 1. Kinetic Parameters (ka, kd), Affinity (KD), and Ligand Efficiency (LE) (kcal/mol/non-hydrogen atom) Values for Alprenolol, Fenoterol, and Fragments A to E Binding to β2 Adrenoceptor, Collected in the DDM (Dodecylmaltoside) Detergent Measured Using Surface Plasmon Resonance (SPR) at 10 °C, and Radioligand Competition (ki) for β1 and β2 Adrenoceptors for Fragments A to E at Room Temperaturea.
| SPR |
competition
binding (ki) |
|||||
|---|---|---|---|---|---|---|
| compd | ka (M–1 s–1) | kd (s–1) | KD | LE | β2 | β1 |
| alprenolol | 8.11(±0.02) × 105 | 6.4(±0.05) × 10–4 | 790 (±60) pM | 0.65 | ||
| fenoterol | 5.45(±0.09) × 105 | 7.6(±0. 1) × 10–2 | 139.0 (±0.9) nM | 0.40 | ||
| A | 5.06(±0.03) × 105 | 8.94(±0.04) × 10–3 | 17.6 (±0.1) nM | 0.48 | 177.8 (±16.8) nM | 1467 (±75.9) nM |
| B | 1.16(±0.02) × 106 | 1.08(±0.02) × 10–1 | 93.4 (±0.7) nM | 0.54 | 342.8 (±22.6) nM | 963.4 (±64.7) nM |
| C | 6.62(±0.07) × 105 | 5.47(±0.05) × 10–2 | 82.5 (±0.7) nM | 0.54 | 191.4 (±14.2) nM | 152.0 (±9.7) nM |
| D | NA | NA | 22.2 (±0.7) μM | 0.40 | 14.1(±1.2) μM | 9.9 (±0.7) μM |
| E | NA | NA | 3.5 (±0.1) μM | 0.50 | 15.0 (±1.4) μM | 12.9 (±0.8) μM |
The errors reported for the SPR data represent the SD from duplicates and for radioligand binding are the mean ± SE obtained from three independent experiments done in duplicates.
Fragment hits A to E were further tested for binding to the β2 adrenoceptor in the presence of MNG detergent. Minimal differences in the affinities were observed compared to data obtained using DDM detergent suggesting a very limited influence of the different detergents on binding activity of the compounds. (Supporting Information Table S1 and Figure S3). In addition, fragments A and B were resynthesized (Supporting Information) and their high affinities reconfirmed by SPR against the β2 adrenoceptor in the presence of the MNG detergent (Supporting Information Table S2 and Figure S4). The structures of the confirmed hits are shown in Figure 2.
Figure 2.

Chemical structures of the validated β2 adrenoceptor fragment hits A to E and the synthesized fragment analogues F to L.
In order to further confirm these potential hits, we measured the affinities of the fragments A to E in a radioligand competition binding assay. Moreover, the radioligand competition binding assay also tests whether these fragments occupy the orthosteric ligand binding pocket of the receptor. All five fragments showed specific competitive inhibition of [125I]-cyanopindolol (CYP) binding with ki values, which are close to the affinities observed by SPR (Table 1 and Figure 3a). This data suggest that these fragments occupy the classical orthosteric binding pocket of the β2 adrenoceptor and that they are bonafide receptor ligands capable of binding to the receptor in native membranes. As the ligand binding pocket of the β1 adrenoceptor and the β2 adrenoceptor are highly conserved and there are already a series of nonselective ligands described in the literature, we tested whether these fragments also bind to the β1 adrenoceptor (Figure 3b) in a competition binding assay with radioligands. Interestingly, fragment A exhibited about 10-fold selectivity for the β2 adrenoceptor compared to the β1 adrenoceptor. However, the other four fragments exhibited nonselective binding to the β2 adrenoceptor and the β1 adrenoceptor (Table 1). In addition, the selectivity of fragments A to E were further tested by screening the compounds against a panel of 27 GPCRs consisting of the α1 and α2 adrenoceptors, the serotonin receptors, the dopamine receptors, and the histamine receptors (Supporting Information Table S3). Fragment A is relatively selective for the β2-adrenoceptor with off-target affinities against only three other receptors ki < 1 μM (5-HT2Bki = 407 nM; 5-HT2Cki = 965 nM; histamine H1 ki = 399 nM) and measured binding activity against a further three receptors with affinities within 10-fold of the radioligand displacement ki (α2A adrenoceptor ki = 1202 nM; 5HT6 ki = 1262 nM; β1 adrenoceptor ki = 1467 nM).
Figure 3.

Radioligand competition binding and functional assays for the β2 adrenoceptor with fragments A to E. (a,b) Dose-dependent competition binding curves of the fragments for the β2 adrenoceptor (a) and β1 adrenoceptor (b). (c,d) Stimulatory dose response curves of the fragments obtained from cAMP production (c) and β-arrestin recruitment (d) assays. (e,f) Inhibitory effects of the fragments at 10 μM on isoproterenol-stimulated dose responses in cAMP production (e) and β-arrestin recruitment (f) assays. All of the data points represent mean ± SE obtained from three independent experiments done in duplicate. Dose response curves for each compound were obtained using the nonlinear iterative curve-fitting computer program Prism. ISO, isoproterenol; CYP, cyanopindolol; ICI, ICI-118,551 (a β2 adrenoceptor-specific antagonist); CGP, CGP-20712A (a β1 adrenoceptor-specific antagonist).
Next, we tested the functional activity of these fragments in cell-based signaling assays to investigate whether these fragments are blockers or agonists for the β2 adrenoceptor. Interestingly, none of these fragments activated either the G protein coupling, monitored through elevation of the cAMP level or β-arrestin recruitment to the β2 adrenoceptor (Figure 3c,d). Rather, all of these fragments inhibited isoproterenol-induced responses in both the cAMP production and β-arrestin recruitment assays (Figure 3e,f). For the β-arrestin recruitment assay, the C-terminus of the β2 adrenoceptor is replaced with the C-terminus of the V2 vasopressin receptor in order to increase the signal-to-noise ratio while retaining the ligand binding properties of the native β2 adrenoceptor. The relative inhibition for these fragments corresponded well with their affinities for the β2 adrenoceptor. Fragments D and E led to modest inhibition of isoproterenol-induced cAMP production and β-arrestin recruitment, and only at high concentrations, reflective of their low affinity for the receptor (Supporting Information Figure S5a,b). In order to further confirm the specificity of these ligands for the β adrenoceptors, we tested these fragments on another Gs coupled receptor, the arginine-vasopressin type 2 receptor (AVPR2). However, we did not observe any detectable agonistic or blocking activity of these fragments on either cAMP production or β-arrestin recruitment in the AVPR2 system, suggesting that these are β adrenoceptor selective ligands (Supporting Information Figure S6a–c). Interestingly, none of the fragments from this small library exhibited agonistic activity although our SPR assay described here does have the capability of identifying classical small molecule agonists as demonstrated by the measurement of fenoterol binding.
To elucidate the features responsible for the high affinity of fragment A, a limited number of analogues were prepared and tested using the SPR β2 adrenoceptor binding assay in the MNG detergent (Table 2; Supporting Information Figure S7 and Table S4), to give initial data about binding requirements. 4-Piperazine-quinoline was prepared as an undecorated core for comparison (fragment F). The undecorated F is also a high affinity fragment with a high ligand efficiency (KD = 348 nM ; LE = 0.52 kcal/mol/non-hydrogen atom). The following initial structure–activity relationships are observed: (i) addition of a substituent at the R2 position (B, Me; G, CF3) gave increases in potency, while maintaining high ligand efficiency; (ii) a substitution in the 6-position (compare A, R6 = CH3, with G, R6 = H) loses 2-fold in potency but retains ligand efficiency, suggesting no significant interaction at this vector; (iii) a chloro substitution in the 7-position (compare C, R7 = Cl, with F, R7 = H) gave a 4-fold increase in potency and retained ligand efficiency, while the larger CF3 substituent (H) gave a significant reduction in both potency and ligand efficiency, possibly indicating limited scope for substitution at this position; (iv) the 4-position NH group of the piperazine ring (substituent A) is a major contributor to affinity. Replacement of the 4-position NH of fragment A with NMe (fragment I; KD = 1800 nM) or a CF2 (fragment J; KD = 67 000 nM) leads to a loss in activity of 50-fold and 2000-fold, respectively. The replace with the secondary amine group (A) with an aliphatic ether oxygen (K) leads to ablation of activity. Removal of the benzene ring entirely to change the 4-piperazine-quinoline (F) to a 4-piperazine-pyridine (L) reduces the affinity by 400-fold, but L retains a high ligand efficiency (KD = 141 600 nM; LE = 0.44).
Table 2. Structure–Activity Relationship Table for the Fragments A, B, C, and F to L Interacting with the β2 Adrenoceptor, Collected in the MNG Detergent, Measured Using Surface Plasmon Resonance (SPR).
| fragment | A | R2 | R6 | R7 | KD | LE |
|---|---|---|---|---|---|---|
| A | NH | CF3 | CH3 | H | 39.0 (±0.1) nM | 0.45 |
| B | NH | CH3 | H | H | 72.5 (±0.4) nM | 0.54 |
| C | NH | H | H | Cl | 76.5 (±0.7) nM | 0.54 |
| F | NH | H | H | H | 348.3 (±0.6) nM | 0.52 |
| G | NH | CF3 | H | H | 84.2 (±0.2) nM | 0.46 |
| H | NH | H | H | CF3 | 8 (±0.1) μM | 0.33 |
| I | NCH3 | CF3 | CH3 | H | 1.8 (±0.1) μM | 0.34 |
| J | CF2 | CF3 | CH3 | H | 67.1 (±0.5) μM | 0.22 |
| K | O | CF3 | CH3 | H | no binding | NA |
| L | NH | 141.6 (±0.7) μM | 0.42 |
In summary, we have established the utility of biosensor-based fragment screening of wild-type GPCRs by the discovery of novel, high affinity, antagonists of the β2 adrenoceptor. We have demonstrated that fragment screening by SPR can be undertaken on tagged, native GPCRs without the need for extensive protein engineering. The advantage of the method is that by screening tagged native receptors, without the need for introducing stabilizing mutations, the pharmacology of the wild-type receptor can be maintained. The process of engineering conformational thermostability of a GPCR has been shown to markedly affect the pharmacology of mutant receptors compared to wild-type receptors.11,12 Engineering conformational stability requires the receptor to be trapped in a conformation induced by a test ligand to produce either an agonist or inverse agonist/antagonist conformation.12,13 For example, antagonist-conformation stabilized adenosine A2A receptors are incapable of binding agonists with an appreciable affinity, vice versa antagonist binding to agonist-conformation stabilized adenosine A2A receptor display weaker affinity.11,12 In contrast, biophysical fragment screening of wild-type GPCRs retain the full breadth of pharmacology in measuring agonists and antagonists7,14,15 and thus provide the opportunity for discovering compounds with diverse mechanisms and potentially novel binding sites.6
The SPR fragment screening method not only provides a useful new approach to the discovery of novel GPCR ligands, but it also presents unique opportunities to screen for ligands against biased signaling conformations of the receptors as well as receptor signaling complexes.
Supporting Information Available
Details of the experimental procedures, author contributions, and the acknowledgements. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
○ T.A., S.A., and A.K.S. contributed equally to this work.
This work is funded by the MSD Scottish Life Sciences Fund (to I.N. and I.H.G), in part by grants HL16037 and HL70631 from the National Institutes of Health (to R.J.L.) and NIH contracts and grants supporting drug discovery and receptor pharmacology (to B.L.R.). R.J.L. is an Investigator of the Howard Hughes Medical Institute. B.L.R. also received support from the Michael Hooker Chair of Pharmacology. The work is funded in part by the Innovative Medicines Initiative’s K4DD consortium under grant agreement no 115366 (I.N. and A.L.H.) and benefits from the Wellcome Trust Strategic Awards to the University of Dundee (WT083481 and 100476/Z/12/Z).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Murray C.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Verheij M. H.; de Graaf C.; de Kloe G. E.; Nijmeijer S.; Vischer H. F.; Smits R. A.; Zuiderveld O. P.; Hulscher S.; Silvestri L.; Thompson A. J.; van Muijlwijk-Koezen J. E.; Lummis S. C.; Leurs R.; de Esch I. J. Fragment library screening reveals remarkable similarities between the G protein-coupled receptor histamine H4 and the ion channel serotonin 5-HT3A. Bioorg. Med. Chem. Lett. 2011, 21, 5460–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L. A.; Vernall A. J.; Denman J. L.; Briddon S. J.; Kellam B.; Hill S. J. Fragment screening at adenosine-A3 receptors in living cells using a fluorescence-based binding assay. Chem. Biol. 2012, 19, 1105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova I.; Hopkins A. L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova I.; Besnard J.; Hopkins A. L. Screening for GPCR ligands using surface plasmon resonance. ACS Med. Chem. Lett. 2011, 2, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova I.; Pancera M.; Wyatt R. T.; Myszka D. G. A biosensor-based approach toward purification and crystallization of G protein-coupled receptors. Anal. Biochem. 2006, 353, 278–283. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Rich R. L.; Myszka D. G.; Figaroa F.; Siegal G.; Marshall F. H. Fragment screening of stabilized G-protein-coupled receptors using biophysical methods. Methods Enzymol. 2011, 493, 115–136. [DOI] [PubMed] [Google Scholar]
- Chen D.; Errey J. C.; Heitman L. H.; Marshall F. H.; Ijzerman A. P.; Siegal G. Fragment screening of GPCRs using biophysical methods: Identification of ligands of the adenosine A(2A) receptor with novel biological activity. ACS Chem. Biol. 2012, 7, 2064–2073. [DOI] [PubMed] [Google Scholar]
- Christopher J. A.; Brown J.; Doré A. S.; Errey J. C.; Koglin M.; Marshall F. H.; Myszka D. G.; Rich R. L.; Tate C. G.; Tehan B.; Warne T.; Congreve M. Biophysical fragment screening of the β1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design. J. Med. Chem. 2013, 56, 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson N.; Jazayeri A.; Errey J.; Baig A.; Hurrell E.; Zhukov A.; Langmead C. J.; Weir M.; Marshall F. H. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 2011, 60, 36–44. [DOI] [PubMed] [Google Scholar]
- Magnani F.; Shibata Y.; Serrano-Vega M. J.; Tate C. G. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Vega M. J.; Magnani F.; Shibata Y.; Tate C. G. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova I.; Sodroski J.; Myszka D. G. Solubilization, stabilization, and purification of chemokine receptors using biosensor technology. Anal. Biochem. 2005, 339, 271–281. [DOI] [PubMed] [Google Scholar]
- Navratilova I.; Dioszegi M.; Myszka D. G. Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology. Anal. Biochem. 2006, 355, 132–139. [DOI] [PubMed] [Google Scholar]
- Dixon R. A.; Kobilka B. K.; Strader D. J.; Benovic J. L.; Dohlman H. G.; Frielle T.; Bolanowski M. A.; Bennett C. D.; Rands E.; Diehl R. E.; Mumford R. A.; Slater E. E.; Sigal I. S.; Caron M. G.; Lefkowitz R. J.; Strader C. D. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature 1986, 321, 75–79. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Rosenbaum D. M.; Kobilka T. S.; Thian F. S.; Edwards P. C.; Burghammer M.; Ratnala V. R.; Sanishvili R.; Fischetti R. F.; Schertler G. F.; Weis W. I.; Kobilka B. K. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [DOI] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H. J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Cherezov V.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H. J.; Yao X. J.; Weis W. I.; Stevens R. C.; Kobilka B. K. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 2007, 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B. K. Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal. Biochem. 1996, 231, 269–71. [DOI] [PubMed] [Google Scholar]
- Baker J. G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharmacol. 2005, 144, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January B.; Seibold A.; Whaley B.; Hipkin R. W.; Lin D.; Schonbrunn A.; Barber R.; Clark R. B. Beta2-adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J. Biol. Chem. 1997, 272, 23871–23879. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Fung J. J.; Pardon E.; Casarosa P.; Chae P. S.; Devree B. T.; Rosenbaum D. M.; Thian F. S.; Kobilka T. S.; Schnapp A.; Konetzki I.; Sunahara R. K.; Gellman S. H.; Pautsch A.; Steyaert J.; Weis W. I.; Kobilka B. K. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.