

Summary
BRAF is an oncogenic protein kinase that drives cell growth and proliferation through the MEK-ERK signaling pathway. BRAF inhibitors have demonstrated anti-tumor efficacy in melanoma therapy, but have also found to be associated with the development of cutaneous squamous cell carcinomas (cSCC) in certain patients. Here, we report that BRAF is phosphorylated at Ser729 by AMP-activated protein kinase (AMPK), a critical energy sensor. This phosphorylation promotes the association of BRAF with 14-3-3 proteins and disrupts its interaction with the KSR1 scaffolding protein, leading to attenuation of the MEK-ERK signaling. We also show that phosphorylation of BRAF by AMPK impairs keratinocyte cell proliferation and cell cycle progression. Furthermore, AMPK activation attenuates BRAF inhibitor-induced ERK hyperactivation in keratinocytes and epidermal hyperplasia in mouse skin. Our findings reveal a mechanism for regulating BRAF signaling in response to energy stress and suggest a strategy for preventing the development of cSCC associated with BRAF-targeted therapy.
Introduction
The complex regulation of metabolic activity within cancer cells is critical to their survival and proliferation. AMP-activated protein kinase (AMPK) plays a central role in maintaining energy homeostasis as a sensor of cellular energy levels in both normal and transformed cells (Hardie et al., 2012; Mihaylova and Shaw, 2011; Steinberg and Kemp, 2009). AMPK exists as a heterotrimeric complex comprising a catalytic kinase subunit (α) and two regulatory subunits, β and γ. The activity of AMPK is regulated through binding of AMP and/or ADP to its γ regulatory subunit, followed by its obligatory phosphorylation by upstream activating kinases, including the tumor suppressor LKB1 and Ca2+/CaM-dependent kinase kinase (CAMKK). In this way, it is thought that AMPK is able to integrate different signaling events with the metabolic state of the cell. The activation of AMPK can be triggered by metabolic stress, such as hypoxia, ischemia, glucose deprivation and reactive oxygen species (ROS), or by physiological stimuli such as skeletal muscle contraction, adipokines and cytokines. Additionally, its activation can be pharmacologically manipulated by various drugs and xenobiotics, such as the antidiabetic drugs metformin and phenformin as well as aspirin, resveratrol and berberine (Hardie, 2012).
Upon its activation, AMPK phosphorylates downstream effectors to stimulate ATP-producing catabolic pathways while suppressing ATP-consuming biosynthetic pathways, thus maintaining an energy balance (Hardie et al., 2012; Mihaylova and Shaw, 2011; Steinberg and Kemp, 2009). In addition to its well-established roles in regulating metabolic processes, recent studies have revealed that AMPK also couples the cellular energy sensing to the regulation of cell growth and proliferation. AMPK has been shown to regulate mTOR (mammalian target of rapamycin)-mediated protein synthesis and cell growth through direct phosphorylation of both TSC2 and Raptor proteins in the mTOR signaling pathway (Gwinn et al., 2008; Inoki et al., 2003; Shaw et al., 2004). In addition, activation of AMPK has been reported to enhance phosphorylation of tumor suppressor p53 at Ser15 and p27Kip1 at its C-terminus (Jones et al., 2005) (Imamura et al., 2001) (Liang et al., 2007). These phosphorylation events may partially explain the cell cycle arrest caused by energy stress. More recently, a chemical genetics screen identified protein phosphatase 1 regulatory subunit 12C (PPP1R12C) as a direct substrate of AMPKα2 involved in mitosis regulation and the phosphorylation of PPP1R12C by AMPK was shown to be required for completion of mitosis (Banko et al., 2011). However, how AMPK coordinates cellular energy status and cell proliferative responses remains an intriguing question.
The RAF-MEK-ERK protein kinase cascade is a major signaling pathway that transmits extracellular mitogenic signals to cell proliferative responses among other cellular functions (Osborne et al., 2012; Udell et al., 2011). The RAF Ser/Thr kinase family is comprised of three members, A-, B- and C-RAF that share similar domain structures. Either homo- or hetero- dimerization of RAF family proteins, has been suggested to be a critical step in phosphorylation and activation of MEK (Mitogen-activated protein kinase/Extracellular signal-regulated kinase Kinase) and subsequently ERK (Extracellular signal-Regulated Kinase) in response to RAS activation. RAFs, MEKs and ERKs are tethered together by Kinase Suppressor of RAS (KSR) proteins (also homologs of RAF family members) that mainly serve as scaffolds of the signaling cascade. Mutations in the RAF-MEK-ERK signaling pathway are frequently found in human cancer. Among them, BRAF mutations are found in 50% of melanoma and 6% of human cancer overall (Gray-Schopfer et al., 2007). More than 90% of BRAF mutations involve a single base substitution in a codon in the kinase domain leading to V600E amino acid change and constitutive activation of BRAF protein kinase activity and consequently downstream MEK-ERK signaling (Gray-Schopfer et al., 2007). Small molecule inhibitors targeting the catalytic site of BRAF have shown significant anti-tumor activities in recent clinical trials, and one of them, Zelboraf (also known as Vemurafenib or PLX4032) has been approved for the treatment of metastatic melanomas harboring the BRAF V600E mutation (Perez-Lorenzo and Zheng, 2012; Ribas and Flaherty, 2011). However, side effects such as development of well-differentiated cutaneous squamous cell carcinomas (cSCC) and keratoacanthomas have been found in ∼ 15-30% of patients treated with the BRAF inhibitors (Robert et al., 2011). Although BRAF inhibitors block MEK-ERK signaling in cancer cells with BRAF mutations, at pharmacologically achievable doses they paradoxically activate MEK-ERK signaling in cells with wild-type BRAF by binding to one subunit of RAF family dimers and activating the adjacent subunit (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010). This paradoxical activation of MEK-ERK signaling and accordingly hyper-proliferation of keratinocytes may underlie the adverse effect of BRAF inhibitors (Downward, 2011; Su et al., 2012) .
Here we report that AMPK phosphorylates BRAF at Ser729, promotes its association with 14-3-3 signaling adaptor and disrupts the hetero-dimerization of BRAF with KSR1 and CRAF (also known as RAF1). Phosphorylation of BRAF by AMPK in response to energy stress plays a key role in attenuating BRAF-mediated mitogenic responses, including cell cycle progression and cell proliferation. More importantly, activation of AMPK prevents the paradoxical activation of MEK-ERK signaling in keratinocytes and epidermal hyperplasia in mouse skin induced by BRAF inhibitors.
Results
Activation of AMPK attenuates RAF-MEK-ERK Signaling
We have previously reported that while AICAR, an AMPK activator, induces robust activation of AMPK signaling in cells expressing wild type (WT) BRAF, it fails to do so in melanoma cells expressing V600E mutant BRAF (Zheng et al., 2009). Interestingly, we also found that AICAR treatment of C140 mouse melanocytes expressing WT BRAF attenuated the basal phosphorylation of ERK (Zheng et al., 2009). In Figures 1A and S1 we show that the AMPK activators AICAR, phenformin and A-769662 suppress ERK activation in the keratinocyte cell line CCD1106, in mouse embryonic fibroblasts (MEFs) and in WM3854 melanoma cells, all of which express wild-type BRAF. To determine whether the attenuation of ERK activation by A-769662 is dependent on AMPK, we compared the response of ERK signaling to A-769662 in immortalized MEFs derived from either AMPK α1/α2 double knockout (AMPK-null) mice or WT mice. While the direct AMPK activator, A-769662 attenuated phosphorylation at the activation sites of MEK and ERK in wild-type MEFs, it had little effect on the AMPK-null MEFs (Figure 1B). Similar results were obtained for AICAR in these cells (Figure S1B). Taken together, these results strongly suggest that AMPK regulates RAF-MEK-ERK signaling at the level of, or upstream of RAF kinases.
Figure 1. Activation of AMPK attenuates RAF-MEK-ERK signaling in various cell lines.
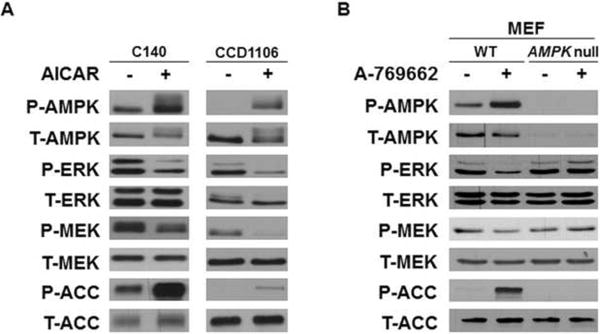
(A) C140 melanocytes or CCD1106 human keratinocytes were treated with or without 2 mM AICAR for 2 hr. (B) WT and AMPK-null MEFs were treated with or without 100 μM A-769662 in DMEM media containing 10% FBS for 2 hr. Cell lysates were used for western blotting with indicated antibodies. See also Figure S1.
AMPK Phosphorylates BRAF at Ser729
AMPK has been shown to phosphorylate CRAF on Ser621 in vitro (Sprenkle et al., 1997), but not in vivo (Noble et al., 2008). Since Ser621 of CRAF is well conserved among the three RAF kinase family members (Figure 2B), we therefore investigated whether BRAF is phosphorylated in vivo on the analogous site (Ser729) upon AMPK activation. Phosphorylation of Ser729 in BRAF has previously been identified through [32P]-labeled tryptic-phospho-peptide analysis (Ritt et al., 2010), but the kinase(s) mediating this phosphorylation in vivo is unknown. To test the possibility that AMPK regulates the phosphorylation of this residue in vivo, Cos-7 cells were transiently transfected with FLAG-tagged BRAF and then treated with or without 2 mM AICAR for 2 hr. The FLAG-BRAF was then recovered by immunoprecipitation using anti-FLAG M2 agarose beads, digested with trypsin or chymotrypsin and subjected to LC-MS/MS analysis to assess total BRAF phosphorylation under these two conditions. Among the various phospho-peptides identified, Ser729-containing peptides were the only species consistently found to be phosphorylated in the AICAR treated samples and they were rarely detected in samples from the untreated cells (Figure 2A). We further analyzed the relative ratios of phospho-Ser729 peptides versus peptides in the unphosphorylated states using total ion intensities over elution peaks (Zheng et al., 2009) and found that treatment with AICAR increased the fraction of BRAF phosphorylated at Ser729 from ~30% to ~70% based on this semi-quantitative analysis (Figure 2A).
Figure 2. AMPK phosphorylates BRAF at Ser 729 both in vitro and in vivo.
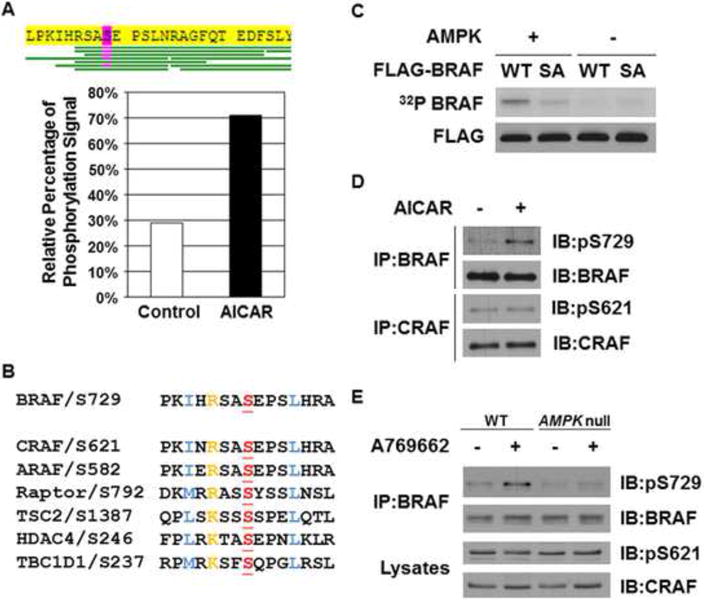
(A) Identification of phosphorylated BRAF peptides containing Ser729 by LC-MS/MS analysis. COS7 cells transfected with FLAG-BRAF were treated with or without 2 mM AICAR for 2 hr. FLAG-BRAF proteins were immunoprecipitated using anti-FLAG M2 agarose beads and subjected to trypsin or chymotrypsin digestion followed by the LC-MS/MS analysis. Percentages of phospho-Ser 729 peptides versus these peptides in the unphosphorylated states were calculated using total ion intensities over elution peaks.
(B) Alignment of the sequence surrounding Ser729 in BRAF with the analogous sites in CRAF and ARAF and other AMPK phosphorylation sites in selective proteins.
(C) AMPK directly phosphorylates BRAF in vitro. FLAG-BRAF WT or S729A (SA) mutant proteins immunoprecipitated from HEK293 cells were incubated with active recombinant AMPK proteins in the presence of γ-32P-ATP.
(D) Treatment with AICAR increases phosphorylation of BRAF Ser729, but not CRAF Ser621, in CCD1106 cells. Cells were treated with or without 2 mM AICAR for 2 hr.
(E) AICAR-induced BRAF Ser729 phosphorylation is dependent on the presence of AMPK. WT or AMPK-null MEFs were treated with or without 100 μM A-769662 for 2 hr.
See also Figure S2.
BRAF Ser729 is flanked by a sequence that conforms well to the optimum phosphorylation consensus motif of AMPK (Gwinn et al., 2008; Hardie, 2007) (Scott et al., 2002) and shows strong similarity with several other well-characterized AMPK substrate sites (Figure 2B), suggesting that it could be a direct AMPK substrate. To examine whether AMPK is capable of phosphorylating BRAF directly in vitro, FLAG-tagged BRAF was immuno-precipitated from 293 cells and incubated with recombinant AMPK protein (α1/β1/γ1 complex) and γ-32P-ATP. To avoid background 32P incorporation due to auto-phosphorylation of BRAF, a kinase-dead mutant of BRAF (K483M) was used. As shown in Figure 2C, FLAG-BRAF was phosphorylated by AMPK in vitro, and this phosphorylation was greatly attenuated by the mutation of Ser729 to Ala.
Using an antibody directed against a synthetic phosphopeptide based on the sequence surrounding pSer729 of BRAF, we found that A-769662 strongly induces phosphorylation of endogenous BRAF at Ser729 in CCD1106 keratinocytes (Figure 2D). Control experiments confirm that this antibody specifically recognized phosphorylated BRAF at Ser729 and does not recognize BRAF S729A mutant (Figure S2). A-769662 also induced phosphorylation of BRAF at Ser729 in wild type MEFs but not in AMPK-null MEFs, indicating that AMPK is required for optimal phosphorylation (Figure 2E). The basal Ser729 phosphorylation in the absence of A-769662 treatment could be mediated by another AMPK-related kinase. In agreement with previous findings that AMPK activation did not result in the phosphorylation of CRAF Ser621 (Noble et al., 2008), we were unable to detect any change in the phosphorylation of CRAF Ser621 in WT MEFs upon treatment with AICAR (Figure 2D). In addition, no difference in CRAF Ser621 phosphorylation was found between WT and AMPK-null MEFs. Collectively, our findings strongly indicate that BRAF is phosphorylated by AMPK at Ser729 following AICAR treatment.
AMPK associates with BRAF
To examine whether AMPK is able to physically interact with BRAF, we transiently transfected 293 cells with either FLAG-BRAF-expressing or empty vectors and subjected both to immunoprecipitation with anti-FLAG M2 agarose beads. After western blot analysis for AMPK on these immunoprecipitates, we found that endogenous AMPK specifically associated with the anti-FLAG immunoprecipitation complex from cells transfected with FLAG-BRAF (Figure 3A). In addition, we found that endogenous BRAF from 293 cells co-immunoprecipitated with both Myc-tagged WT and Myc-tagged K45R kinase-dead (KD) mutant of AMPKα2 (Figure 3B). Finally, we detected endogenous BRAF in anti-AMPKα precipitates from CCD1106 cells, but not from the control IgG precipitates (Figure 3C). These data demonstrate that AMPK associates with BRAF, further supporting that AMPK directly phosphorylates BRAF.
Figure 3. AMPK associates with BRAF.
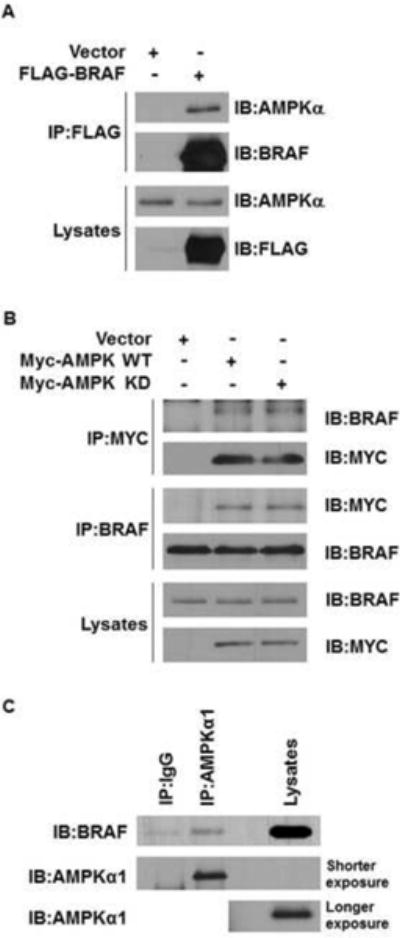
(A) FLAG-BRAF proteins coimmunoprecipitate with endogenous AMPKα proteins.
(B) Myc-AMPKα2 proteins coimmunoprecipitate with endogenous BRAF proteins.
(C) Endogenous BRAF coimmunoprecipitate with endogenous AMPKα1 proteins in CCD1106 melanocytes. HEK293 cells were transfected with FLAG-BRAF (A), Myc-AMPKα2 WT, or K45R (kinase-dead, KD) mutant (B) as indicated.
Phosphorylation of BRAF at Ser729 is critical for the attenuation of ERK signaling by AMPK
To assess whether attenuation of MEK-ERK signaling by AMPK activation is dependent on the phosphorylation of BRAF by AMPK at Ser729, we stably expressed FLAG-tagged WT or FLAG-tagged S729A phosphorylation-deficient mutant of BRAF in Braf-null MEFs via retroviral infection. Expression of either of these proteins increased the basal level of ERK activation (Figure 4A). However, while AICAR suppressed MEK and ERK activation in the cells expressing wild type BRAF, it had no effect on cells expressing the S729A mutant BRAF (Figure 4A). Similar results were observed in CCD1106 keratinocytes (Figure 4B) or C140 melanocytes (Figure S3) stably expressing either WT or an S729A mutant of BRAF as well. It is noted that there was still some background AICAR effect in the presence of the S729A mutant in these cells, presumably because they also have endogenous wild-type BRAF proteins. Taken all together, the experiments presented in Figures 1 through 4 indicate that AICAR inhibits activation of MEK and ERK by inducing phosphorylation of Ser729 of BRAF and that the primary kinase mediating this effect is AMPK.
Figure 4. Phosphorylation of BRAF Ser729 is critical for attenuation of ERK signaling by AMPK.
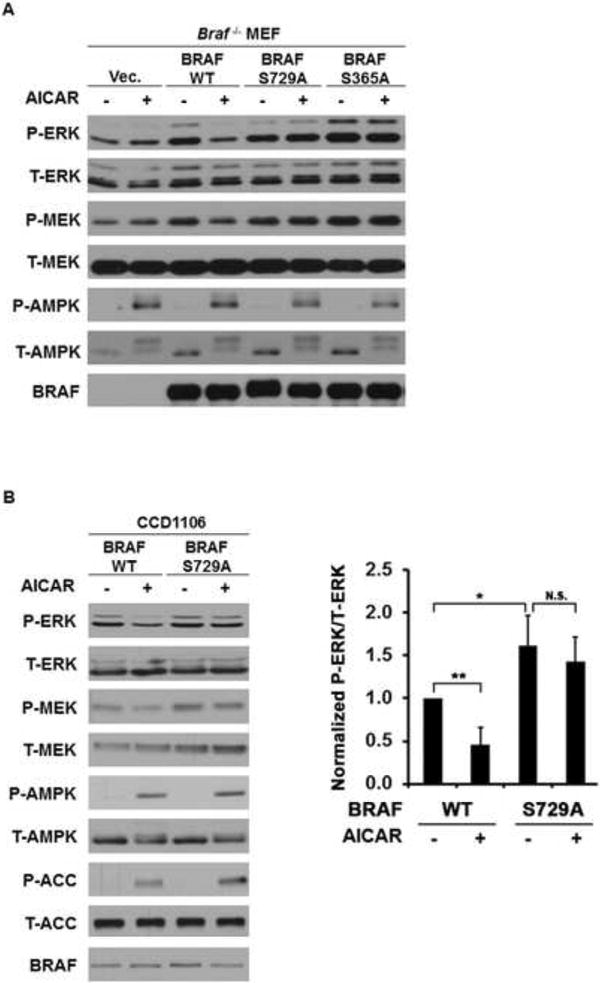
(A) Attenuation of MEK-ERK signaling by AMPK activation is mediated by BRAF Ser729 in MEFs. BRAF-null MEFs were stably transfected with control vector or various BRAF constructs as indicated. Cells were treated with 1 mM AICAR for 1 hr.
(B) Attenuation of MEK-ERK signaling by AMPK activation is mediated by BRAF S729 in CCD1106 cells. Cells stably expressing various BRAF constructs were treated with 2 mM AICAR for 2 hr. Quantification analysis was done for three independent experiments. Data are presented as mean +/- SEM; *P<0.05;**P<0.01; N.S., not significant.
See also Figure S3.
Phosphorylation of BRAF at Ser729 by AMPK enhances the association between BRAF and 14-3-3 and disrupts the BRAF-KSR1 interaction
To examine whether phosphorylation of BRAF by AMPK modulates the BRAF kinase activity, we performed in vitro cascade-kinase assays to measure the kinase activity of endogenous BRAF protein immunoprecipitated from C140 cells treated with AICAR or control. As shown in Figure 5A, we did not detect a significant direct effect of AICAR treatment on BRAF kinase activity. Consistent with this observation, it was previously shown that mutation of Ser729 to alanine does not affect the kinase activity of BRAF in vitro (Ritt et al., 2010). These results suggest that the relevant effect of Ser729 phosphorylation of BRAF by AMPK does not involve the inhibition of BRAF kinase catalytic activity per se. However, phosphorylated Ser729 has been previously shown as one of the two sites on BRAF that bind the signaling adaptor 14-3-3 and this association has been proposed to play an inhibitory role on BRAF signaling (Ritt et al., 2010) (MacNicol et al., 2000) (Brummer et al., 2006). To determine whether activation of AMPK drives the interaction between BRAF and 14-3-3, cell lysates from WT and Ampk-null MEFs that had been treated with AICAR were used for immunoprecipitation with anti-14-3-3 antibody, and the presence of BRAF in the immunocomplex were examined. As shown in Figure 5B, AICAR treatment promotes the association of 14-3-3 with BRAF, in WT MEFs, but not in Ampk-null MEFs. Importantly, this effect was not seen with either CRAF or ARAF suggesting that AMPK regulation is specific for BRAF. Similar results were observed in GST-14-3-3 pull-down experiments using lysates from CCD1106 cells treated with AICAR (Figure 5C). Moreover, we have confirmed that indeed Ser729 is critical for the association of BRAF with 14-3-3 in MEFs, since mutation of Ser729 to Ala abolished the association (Figure 5G). These findings together support that phosphorylation of BRAF by AMPK promotes its association with 14-3-3.
Figure 5. AMPK-dependent phosphorylation of BRAF Ser729 promotes its binding to 14-3-3 and disrupts its association with KSR1.
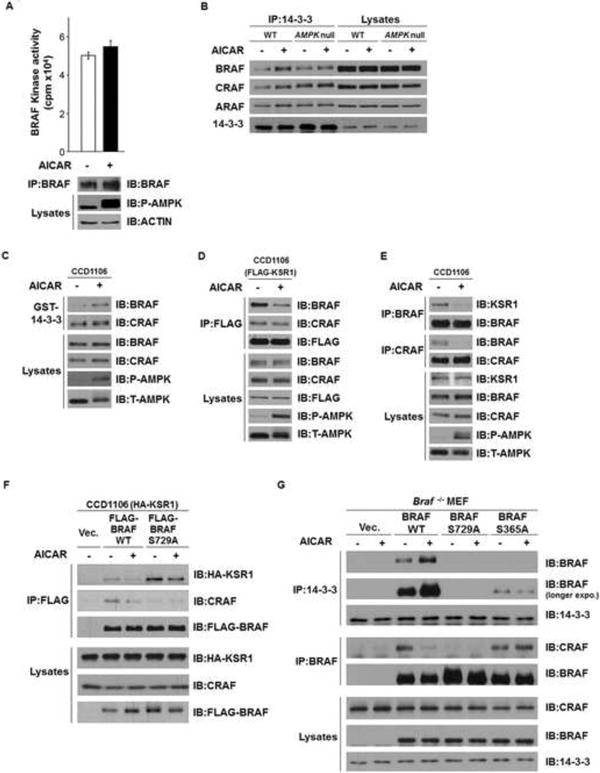
(A) Treatment with AICAR does not affect the kinase activity of BRAF. C140 cells were treated with or without 2 mM AICAR for 2 hr. Endogenous BRAF proteins were immunoprecipitated and the kinase activities of BRAF were measured using the BRAF kinase cascade assay. Data are represented as mean +/- SEM
(B) Activation of AMPK by AICAR promotes the BRAF/14-3-3 association in WT MEFs but not Ampk-null MEFs. Lysates from WT or Ampk-null MEFs were immunoprecipitated with anti-14-3-3 antibodies, followed by immunoblotting.
(C) Activation of AMPK by AICAR promotes the association between 14-3-3 and BRAF, but not CRAF. CCD1106 cells lysates were incubated with GST-14-3-3 in GST pulldown assays.
(D) Activation of AMPK by AICAR disrupts the association between FLAG-KSR1 and BRAF. CCD1106 cells were transiently infected with FLAG-KSR1 expressing retrovirus. Cells were treated with 2 mM AICAR for 2 hr.
(E) Activation of AMPK by AICAR disrupts the endogenous BRAF-KSR1 dimer formation.
(F) Ser729 of BRAF is critical for the disruption of BRAF-KSR1 association upon activation of AMPK. CCD1106 cells stably expressing HA-KSR1 were transiently infected by various FLAG-BRAF expressing retrovirus as indicated.
(G) Phosphorylation of BRAF Ser729 is critical for the association between BRAF and 14-3-3. Braf-null MEFs stably expressing various BRAF constructs were subjected to immunoprecipitation. Cells were treated with 1 mM AICAR for 1 hr as indicated.
See also Figure S4.
Because both the association between BRAF and the scaffold protein KSR1 and the heterodimerization between BRAF and CRAF have been shown to play important roles in driving the activation of RAF-MEK-ERK signaling pathway (Osborne et al., 2012), we next investigated whether phosphorylation of BRAF at Ser729 by AMPK modulates the BRAF/KSR1 and/or BRAF/CRAF associations. As shown in Figure 5D, treatment with AICAR in CCD1106 cells stably expressing FLAG-KSR1 led to decreased levels of BRAF, but not CRAF, in the anti-FLAG-KSR1 immunoprecipitates. AICAR also significantly disrupted the association of endogenous KSR1 proteins with BRAF in CCD1106 cells (Figure 5E). To further determine if this effect of AMPK on regulating BRAF-KSR1 association is dependent on the phosphorylation of BRAF by AMPK at Ser729, we carried out co-immunoprecipitation experiments using CCD1106 cells expressing HA-KSR1 together with either FLAG-tagged WT or S729A mutant of BRAF. We found that S729A BRAF showed stronger association with KSR1, as compared to WT BRAF (Figure 5F). Moreover, activation of AMPK by AICAR disrupts the association of KSR1 with FLAG-BRAF WT, but much less with the S729A mutant. These data strongly indicate that activation of AMPK disrupts the association between BRAF and KSR1 through phosphorylation of BRAF at Ser729.
We also tested whether AICAR treatment affected BRAF and CRAF heterodimerization. We found that, similar to its effect on the BRAF/KSR1 association, AICAR also disrupted the BRAF/CRAF association in various cell lines (Figures 5E, 5F and 5G). If this disruption is dependent on phosphorylation of BRAF Ser729, we would expect that the BRAF S729A mutant would bind more readily to CRAF. However, in agreement with a previous report (Ritt et al., 2010), mutation of Ser729 to Ala in BRAF reduced rather than increased the interaction between BRAF and CRAF in both CCD1106 cells and MEFs (Figures 5F and 5G). This could be because the hydroxy moiety of Ser729 either directly binds to CRAF to stabilize the BRAF-CRAF interaction or enhances a conformational state in BRAF that stabilizes the BRAF-CRAF heterodimer. Alternatively, AMPK might phosphorylate other targets to suppress BRAF-CRAF interactions. It is noteworthy that mutation of BRAF Ser365, another known 14-3-3 binding site, to Ala in the context of Ser729 being still present dramatically reduces both basal and AMPK stimulated 14-3-3 binding (Figure 5G) and allows BRAF-CRAF dimerization and ERK activation to be maintained in the presence of AICAR (Figures 5G and 4A). This result supports a model in which phosphorylation of both Ser365 and Ser729 of BRAF are required for high affinity binding to 14-3-3 (presumably due to binding to both pockets of the 14-3-3 homodimer) and that this allows 14-3-3 to block interactions between BRAF and CRAF.
Phosphorylation of BRAF by AMPK regulates keratinocyte cell proliferation and cell cycle progression
Because the RAF-MEK-ERK signaling pathway plays a major role in regulating cell proliferative responses, we next considered whether phosphorylation of BRAF by AMPK would play a role in regulating cell proliferation. We performed cell proliferation assays on CCD1106 cells stably expressing BRAF WT, S729A mutant or the vector control, and found that cells expressing the S729A mutant had significantly increased proliferation rates compared to cells expressing WT BRAF or the vector control (Figure 6A), suggesting that phosphorylation of BRAF at Ser729 negatively regulates cell proliferation. Next, we performed propidium iodide staining followed by fluorescence-activated cell sorting (FACS) to determine the effect of AICAR on the cell-cycle progression in these CCD1106 stable cells. In cells expressing WT BRAF, treatment of AICAR led to a reduced percentage of cells in G2/M phase and an increased percentage of cells in S phase (Figure 6B). In contrast, cells expressing BRAF S729A mutant were not sensitive to AICAR-induced decrease of G2/M cell fractions (Figure 6B), probably due to increased transit through the S phase. We further examined the effect of AICAR on these cells in cell proliferation assays. As shown in Figure 6C, AICAR exerted a stronger inhibitory effect on the proliferation of WT cells compared to S729A cells. Together, these results support the hypothesis that phosphorylation of BRAF Ser729 by AMPK in response to AICAR plays an inhibitory role on keratinocyte cell cycle progression and cell proliferation.
Figure 6. Phosphorylation of BRAF at Ser729 regulates keratinocyte cell proliferation and cell cycle progression.

(A) Expression of BRAF S729A mutant (SA) in CCD1106 keratinocytes promotes cell proliferation. Cells were seeded at 3X104 cells/well and cells were counted daily. Cell media were changed daily. Cell proliferation of CCD1106 cells stably expressing WT BRAF or S729A (SA) mutant was measured. One representative from three independent experiments is shown. Data are represented as mean +/- SEM
(B) Treatment of CCD1106 cells expressing BRAF SA mutant with AICAR leads to more dramatic decrease of cells in the G2 phase and increase of cells in S phase, compared to that of CCD1106 cells expressing WT BRAF. Cells were stained with propidium iodide and analyzed by FACS. Data are presented as mean +/- SEM (n=3); **P<0.01; N.S. indicates not significant. Histograms from one representative experiment are shown.
(C) CCD1106 cells expressing BRAF S729A mutant are less sensitive to the inhibitory effects of AICAR on cell proliferation than those expressing WT BRAF. Cells were treated with 0.3 mM AICAR for the indicated time. Cell numbers were counted daily and expressed as percentage of cell numbers of each stable line at Day 0 before AICAR treatment. Differences in cell numbers between cell lines at each time point were assessed by paired t test. Data are represented as mean +/- SEM. ANOVA: time X cell, F (2,48) =3.97, P<0.05.
See also Figure S5.
Activation of AMPK prevents BRAF inhibitor-induced hyper-activation of ERK and proliferative response in keratinocytes and mouse skin
BRAF kinase inhibitors, such as PLX4032 and GSK-2118436, have shown anti-tumor activities in melanoma patients with BRAF mutations (Chapman et al., 2011; Flaherty et al., 2010; Hauschild et al., 2012). However, ~15-30% of treated patients developed cutaneous squamous cell carcinomas and/or keratoacanthomas (Chapman et al., 2011; Flaherty et al., 2010; Ribas and Flaherty, 2011; Su et al., 2012). It has been suggested that the paradoxical activation of ERK signaling by BRAF kinase inhibitors in cells lacking BRAF mutation is involved in the development of these BRAF inhibitor-associated skin toxicities (Downward, 2011) (Su et al., 2012). Based on the inhibitory effect of AMPK activation on RAF-MEK-ERK signaling, we reasoned that activation of AMPK by AICAR might also attenuate BRAF inhibitor induced ERK activation in keratinocytes. To test this hypothesis, we pretreated CCD1106 human keratinocytes with 2 mM AICAR for 1 hr before subjecting them to treatment with the BRAF inhibitor PLX4032 for 1 hr. As shown in Figure 7A, PLX4032 indeed induced hyperactivation of ERK in these cells, and AICAR pretreatment induced AMPK activation and greatly attenuated the phosphorylation of ERK induced by PLX4032. Next, we further investigated the effect of phenformin, an AMPK activator, on PLX4720 BRAF inhibitor induced epidermal proliferation response in mouse skin. Phenformin was chosen because it has good bio-availability and potency for activating AMPK in a variety of tissues in vivo, compared to other AMPK activators, such as AICAR and metformin (Huang et al., 2008; Shackelford et al., 2013). Both PLX4720 and phenformin were given to mice by oral gavage in a twice-daily schedule, with phenformin given one day prior to the start of a 2-day PLX4720 treatment (Figure 7B). We observed that treatment of PLX4720 induced a moderate but significant increase in epidermal thickness in mouse dorsal skin, compared to the vehicle-treated control, as shown by the quantitative analysis of epidermal layers (Figure 7C). Importantly, co-treatment with phenformin significantly reduced the epidermal hyperplasia induced by PLX4720, while phenformin alone did not show any apparent effect on epidermal layer thickness (Figure 7C). Immunohistochemical analysis on the treated dorsal skin samples demonstrated that PLX4720 treatment led to an increase of pERK staining and proliferation index as estimated based on epidermal Ki67 staining, both of which were attenuated by pretreatment with phenformin (Figures 7D and 7E). Similar effects on the epidermal thickness and keratinocyte proliferative response were also observed in mice receiving PLX4720 through oral gavage and phenformin through topical treatment, or PLX4720 through topical treatment and the AMPK activator A-769662 through oral gavage (Figure S5). Together, these findings indicate that activation of AMPK attenuates the hyper-proliferative response induced by BRAF inhibitors, and suggest that AMPK activators are potential therapeutic strategy for preventing BRAF inhibitor-induced cutaneous SCC.
Figure 7. Activation of AMPK prevents PLX4720-induced hyper-activation of ERK and proliferative response in keratinocytes.
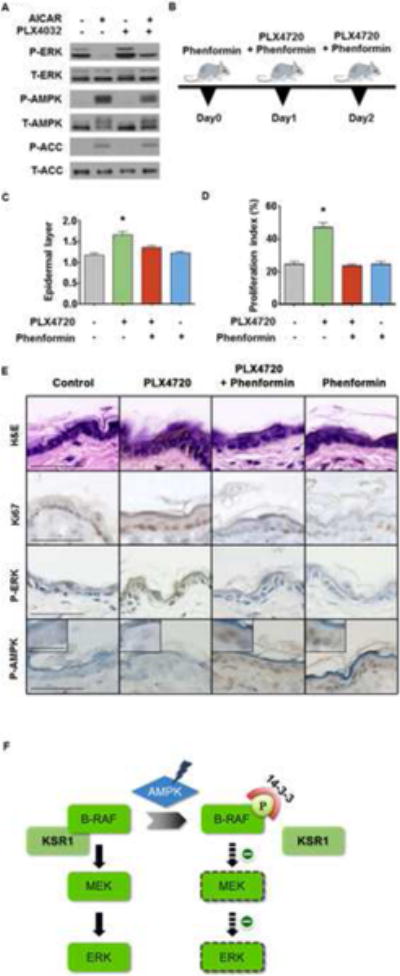
(A) Activation of AMPK by AICAR attenuates PLX4032-induced hyperactivation of ERK. CCD1106 keratinocytes were treated with 2 mM AICAR for 1 hr before adding 10 μM PLX4032 for an additional hour as indicated.
(B) Schematic diagram of drug treatment. FVB/n mice were given PLX4720, 100 mg/kg (bid) via oral gavage for two days. In an additional group, mice were treated twice daily with 100mg phenformin/Kg (bid) via oral gavage, one day before PLX4720 treatment and during the 2-day PLX4720 treatment.
(C, D) Phenformin prevents the PLX4720-induced epidermal hyperplasia (C) and increase of proliferation index (D) in mouse skin. Five mice were included in each group. Epidermal thickness and proliferation index were measured as described in methods. Paired t test was used for comparisons between groups. Data are represented as mean +/- SEM
(E) Treatment with phenformin leads to activation of AMPK in mouse skin and prevents PLX4720-induced activation of ERK and increase of Ki67 staining. Representative images of mouse skin samples subjected for various immunohistochemical analyses are shown. Bar indicates 50 μm.
(F) A schematic model for regulation of BRAF-MEK-ERK signaling pathway by AMPK.
See also Figure S6.
Discussion
The regulation of BRAF kinase activity is complex and involves multiple mechanisms such as membrane association, protein phosphorylation, intra-molecular interaction, and dimerization with RAF family members and scaffold proteins. Phosphorylation of BRAF Ser729 has been identified through 32P-labelled phospho-peptide mapping studies (Ritt et al., 2010). Mutation of Ser 729 to Ala abolishes its binding to 14-3-3 adaptor proteins, which was shown to be critical for the regulation of BRAF activity (Ritt et al., 2010) (MacNicol et al., 2000) (Brummer et al., 2006). However, the kinase responsible for the phosphorylation of BRAF Ser729 has been elusive. In this report, we have identified and characterized AMPK as a kinase for this site. Phosphorylation of BRAF at Ser729 by AMPK promotes its association with 14-3-3. Moreover, this phosphorylation disrupts the BRAF-KSR1 association, leading to attenuation of downstream MEK-ERK signaling (Figure 7F). KSR proteins are scaffolds for the RAF-MEK-ERK signaling cascade and facilitate the phosphorylation and activation of MEK by RAF. While KSR is believed to bind to MEK constitutively, it's binding to RAF is dependent on the stimulation of the RAS/RAF pathway (Udell et al., 2011). A recent structural study has suggested that the RAF-KSR dimerization induces a conformational change in MEK and enhances its phosphorylation and activation by RAF, further supporting a critical role of RAF-KSR heterodimers in regulating this pathway (Brennan et al., 2011). Our data uncover an important mechanism by which the association between RAF and the KSR scaffold protein is negatively regulated by AMPK through phosphorylation of BRAF at Ser729.
In addition to its association with KSR, BRAF also dimerizes with CRAF in response to mitogen stimulation and activation of Ras. Recent structural studies have suggested that the dimerization of RAF proteins is mediated by their kinase domains and may be critical for allosteric activation of the kinase activity (Rajakulendran et al., 2009). Both 14-3-3 binding and negative feedback phosphorylation of B-RAF by ERK (Rajakulendran et al., 2009; Ritt et al., 2010; Rushworth et al., 2006) have previously been proposed to regulate the BRAF-CRAF dimerization. Interestingly, we found that activation of AMPK by AICAR promotes the binding of BRAF to 14-3-3 and also disrupts the association between BRAF and CRAF, which based on previous observations, may result in the observed AICAR induced down-regulation of the BRAF-MEK-ERK signaling. Paradoxically, similar to a previous report (Ritt et al., 2010), we found that the S729A phosphorylation-deficient mutant of BRAF also failed to bind to CRAF. These results suggest that the hydroxy moiety of Ser729, in addition to being a substrate for AMPK and thereby mediating 14-3-3 binding, may also be critical for stabilizing the BRAF-CRAF heterodimer when not bound to 14-3-3. Consistent with this model, high affinity binding of 14-3-3 to BRAF required that both Ser729 and Ser365 (another 14-3-3 binding site) be available for phosphorylation (Figure 5G). Mutation of Ser365 to Ala eliminated the ability of AMPK activators to block the BRAF-CRAF interaction (Figure 5G). Interestingly, Braf-null MEFs reconstituted with BRAF S729A mutant showed similar basal ERK activity to WT BRAF-expressing cells, suggesting that, in these cells dimerization of BRAF with CRAF is not critical for ERK activation and that the BRAF-KSR1 interaction is more important.
The Ser729 of BRAF and its flanking sequences are well conserved among all RAF kinase family members (Fig. 2B). In fact, CRAF has been reported to be phosphorylated by AMPK in vitro at the analogous site, Ser621 (Sprenkle et al., 1997). However, subsequent studies suggested that CRAF Ser621 is auto-phosphorylated in vivo, rather phosphorylated by AMPK (Noble et al., 2008). Our data derived from Ampk-null MEFs support the second observation that activation of AMPK by AICAR does not induce phosphorylation of CRAF Ser621 or promote its association with 14-3-3. Considering the apparent differential effect of AMPK on BRAF versus CRAF, it is tempting to speculate that the effect of AMPK activation on ERK signaling may well depend on the abundance of BRAF versus CRAF in a particular cell. BRAF is known to differ significantly from CRAF and ARAF in the regulation of kinase activation. BRAF has a higher basal and constitutive activation of its kinase activity, compared to CRAF and ARAF (Roskoski, 2010). Whether any of these differences contribute to the differential effects of AMPK on BRAF remains to be examined.
BRAF is a major oncogenic driver in human cancer. Most cancer associated mutations in BRAF occur within the kinase domain, of which a V600E mutation accounts for ~90% of all BRAF mutations in cancer. The BRAF V600E mutant exhibits constitutively active kinase activity and apparently bypasses the negative regulation of wild-type BRAF proteins. Significantly, in the context of this common BRAF V600E mutant, additional mutation of Ser729 to Ala did not affect its transformation activity in NIH3T3 cells and the S729A mutant still transformed fibroblast (Brummer et al., 2006) (Ritt et al., 2010). We and others previously showed that, in contrast to cells with WT BRAF, cells with V600E mutant BRAF are impaired in their ability to activate AMPK in response to energy stress (Zheng et al., 2009; Esteve-Puig et al., 2009). Consistent with this observation, AICAR does not stimulate phosphorylation of Ser729 of the V600E mutant BRAF and does not stimulate association of this mutant protein with 14-3-3, and fails to dissociate BRAF V600E mutant from KSR (Figure S4).
The LKB1-AMPK and RAF-MEK-ERK signaling pathways exhibit an intimate but yet complex relationship. We and others previously reported that impairment of AMPK activation in BRAF V600E mutant melanoma cells is due to phosphorylation and inhibition of the AMPK upstream activating kinase, LKB1 by ERK and RSK (Esteve-Puig et al., 2009; Zheng et al., 2009). C-TAK1 (Cdc25C-associated protein kinase 1, also known as MARK3, MAP/microtubule affinity-regulating kinase 3), an AMPK-like kinase activated by LKB1, has been shown to be a negative regulator of KSR1 (Müller et al., 2001). C-TAK1 phosphorylates KSR1 at Ser392 and keeps it in its inactive state in the cytoplasm through 14-3-3 binding (Müller et al., 2001). More recently, KSR2 was reported to interact with AMPK, and loss of KSR2 in mice led to impaired AMPK signaling, which was proposed to explain metabolic phenotypes observed in KSR2 knock-out mice, such as increased triglyceride storage and impaired fatty acid oxidation (Costanzo-Garvey et al., 2009). In this study, we uncover a way of cross-talk between the LKB1-AMPK and RAF-MEK-ERK signaling pathways, by which AMPK attenuates MEK-ERK signaling through phosphorylation of wild-type BRAF. It remains to be seen whether this regulatory mechanism completely or partially explains previous observations of attenuated ERK signaling in response to various agents that activate AMPK (Kim et al., 2001; Green et al., 2011). It's worth-noting that long-term treatment with AMPK activators such as AICAR and metformin was recently suggested to enhance ERK activation in A375 melanoma cells, through promoting degradation of DUSP6, a dual-specificity phosphatase that negatively regulates ERK (Martin et al., 2012). Further experiments are needed to test whether induction of this feedback loop is a general phenomenon or is specific to a subset of cells types. Due to the complex regulation of RAF-MEK-ERK signaling, it is conceivable that the cross-talk between the BRAF-MEK-ERK and LKB1-AMPK pathways is highly influenced by the composition of various genetic mutations in cancer cells. Further knowledge of this complex network should lead to insight about therapeutic strategies for preventing resistance to drugs that target the RAF-MEK-ERK signaling pathway.
BRAF selective inhibitors, such as Vemurafenib and Dabrafenib, have shown great improvement in both progress-free survival and overall survival for melanoma patients with BRAF V600E mutations, compared to conventional chemotherapy (Ribas and Flaherty, 2011). However, about 15-30% patients treated with these inhibitors developed well-differentiated cSCCs and keratoacanthomas. More recently, mutations in KRAS or HRAS have been found in ~ 20 - 60% of these BRAF inhibitor-associated cSCCs (Oberholzer et al., 2012; Su et al., 2012). Although the cSCCs described so far are relatively easily identified and removed by surgery, there is an alarming possibility that BRAF inhibitors may accelerate other internal Ras-mutant premalignant lesions that are much more difficult to detect. Recent studies identified progression of a Ras-mutant leukemia during treatment with Vemurafenib BRAF inhibitor (Callahan et al., 2012).
The molecular mechanism underlying the development of BRAF inhibitor-associated cSCCs is under active investigation. The prevailing hypothesis is that, in striking contrast to inhibition of ERK signaling in BRAF-mutant cancer cells, BRAF inhibitors paradoxically activate ERK signaling in wild-type cells or RAS-mutant cells, leading to rapid growth and progression of premalignant lesions. In those cells without BRAF mutations, BRAF inhibitors have been shown to promote BRAF-CRAF heterodimer or CRAF-CRAF homodimer formation, in which one BRAF inhibitor bound RAF protein transactivates the adjacent CRAF subunit of the dimer, leading to downstream activation of MEK-ERK (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010). At sufficiently high doses of the drug, both subunits are inhibited and MEK-ERK signaling is attenuated, however such high doses have toxic side effects and are difficult to maintain. More recently, KSR proteins have also been implicated in mediating the hyperactivation of MEK-ERK pathway by BRAF inhibitors, although the details remain controversial. In one study, KSR1 was suggested to compete with CRAF for inhibitor-bound BRAF, leading to attenuation of ERK activation (McKay et al., 2009); whereas in another study, KSR1 was found to be required for the inhibitor-induced hyperactivation through promoting KSR1-CRAF dimerization (Hu et al., 2011).
Based on the inhibitory effect of AMPK on BRAF-MEK-ERK signaling, we examined whether AMPK activators would attenuate BRAF inhibitor-induced ERK hyperactivation in keratinocytes. We found that indeed PLX4720 BRAF inhibitor promotes activation of ERK in keratinocytes and this activation is prevented by addition of the AMPK activator, AICAR (Figure 7A). More importantly, we observed that the AMPK activators, phenformin and A-769662, attenuate PLX4720-indcued epidermal hyperplasia in mouse skin (Figure 7). Although short-term treatment (3 days) of PLX4720 in mice led to hyperplasia, we did not observe formation of cSCC with long-term chronic treatment of PLX4720 using a two-stage skin carcinogenesis model (Data not shown), which is consistent with a recent report (Su et al., 2012). Hence, the effect of AMPK activators on the cSCC formation and progression needs to be examined once appropriate mouse tumor models are available. Nonetheless, our study suggests a potential role of AMPK activators in preventing formation of SCC in patients treated with BRAF inhibitors.
Experimental Procedures
In vitro kinase assay
For AMPK kinase assays using immunoprecipitated BRAF proteins, immunoprecipitated FLAG-BRAF proteins from HEK293 cells were incubated with recombinant AMPK α1/β1/γ1 proteins (Millipore) in the presence of γ-32P-ATP (Perkin Elmer) at 37°C for 30 min. For AMPK kinase assay using peptides, various concentrations of BRAF S729 and SAMS peptides were used in the kinase reactions at 30 °C for 20 min. To terminate the reactions, 20 μl of reaction mixture were spotted onto phosphor-cellulose P81 paper. Air-dried P81 paper was sequentially washed in 1% phosphoric acid, before subjected for liquid scintillation counting to measure the radioactivity incorporated. For BRAF kinase assay, endogenous BRAF proteins were immunoprecipitated from C140 cells and the activities of BRAF kinase were measured using the BRAF Kinase Cascade Assay Kit (Millipore), according to manufacturer's instructions.
Animal Studies
Eight-week old female FVB/n mice (Charles River Laboratories) were given PLX4720, 100 mg/kg (bid), formulated in DMSO:1%Carboxy Methyl Cellulose (1:9), via oral gavage for two days. In an additional group, animals were treated twice daily with 100 mg phenformin/Kg of body weight via oral gavage, one day before PLX4720 treatment as well as during the 2-day PLX4720 treatment period. Back skin was harvested 2h after the last treatment. All animals were kept under controlled environment of temperature and humidity and a 12 h light/dark cycle. Animal studies were conducted under approved institutional protocols.
Tissue analysis
To determine epidermal thickness, the number of cell layers in the epidermis was blindly counted every 20 basal cells in neutral buffered formalin-fixed, hematoxylin and eosin stained skin sections. Cell proliferation was measured using Anti-Ki67 (VECTOR laboratories) immunohistochemical staining and expressed as the percentage of positive cells among total basal cells per section. Phospho-AMPK immunohistochemical staining was done in NBF-fixed, paraffin-embedded skin sections after heat-induced antigen retrieval using citrate buffer. Phospho-ERK staining was done using 70% ethanol-fixed, paraffin embedded skin sections. In all cases, staining was performed using HRP-conjugated antibodies and DAB (VECTOR laboratories) as chromogen.
Supplementary Material
Highlights.
AMPK is the long-sought kinase for BRAF Ser729.
Phosphorylation of BRAF by AMPK disrupts its association with KSR.
Phosphorylation of BRAF Ser729 modulates cell cycle progression and proliferation.
Phenformin prevents BRAF inhibitor-induced epidermal hyperplasia in mouse skin.
Acknowledgments
We thank Jaewoo Choi, Lee Hedden, Yannawan Wongchai and Xuemei Yang for technical assistance, Rong Du at the Skin Disease Research Center of Columbia University Medical Center for providing human primary keratinocytes, and members of the Zheng Lab for helpful discussions. We also would like to thank Ken Swanson for critical comments on the manuscript. This work is supported by National Institutes of Health grants R00-CA133245 and R01-CA166717, a pilot project grant from the Alexander and Margaret Stewart Trust, a V foundation Scholar award and the Elizabeth and Oliver Stanton young investigator award from the Melanoma Research Alliance to B.Z, and NIH CA102694 grant to L.C.C. The mass spectrometry experiments were partially supported by NIH grants 2P01CA120964 (J.M.A.) and 5P30CA00651648 (J.M.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villén J, Wang B, Kim SR, Sakamoto K, et al. Chemical Genetic Screen for AMPKα2 Substrates Uncovers a Network of Proteins Involved in Mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan DF, Dar AC, Hertz NT, Chao WCH, Burlingame AL, Shokat KM, Barford D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–369. doi: 10.1038/nature09860. [DOI] [PubMed] [Google Scholar]
- Brummer T, Martin P, Herzog S, Misawa Y, Daly RJ, Reth M. Functional analysis of the regulatory requirements of B-Raf and the B-RafV600E oncoprotein. Oncogene. 2006;25:6262–6276. doi: 10.1038/sj.onc.1209640. [DOI] [PubMed] [Google Scholar]
- Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, Wolchok JD, Solit DB, Rosen N, Abdel-Wahab O, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–2321. doi: 10.1056/NEJMoa1208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG, et al. KSR2 Is an Essential Regulator of AMP Kinase, Energy Expenditure, and Insulin Sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAF: trials and tribulations. Nat Med. 2011;17:286–288. doi: 10.1038/nm0311-286. [DOI] [PubMed] [Google Scholar]
- Esteve-Puig R, Canals F, Colomé N, Merlino G, Recio JÁ. Uncoupling of the LKB1-AMPKα Energy Sensor Pathway by Growth Factors and Oncogenic BRAFV600E. PLoS ONE. 2009;4:e4771. doi: 10.1371/journal.pone.0004771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Green CJ, Macrae K, Fogarty S, Hardie DG, Sakamoto K, Hundal HS. Counter-modulation of fatty acid-induced pro-inflammatory nuclear factor κB signalling in rat skeletal muscle cells by AMP-activated protein kinase. Biochem J. 2011;435:463–474. doi: 10.1042/BJ20101517. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Kaempgen E, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, Shaw AS. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc Natl Acad Sci USA. 2011;108:6067–6072. doi: 10.1073/pnas.1102554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5'-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochemical and Biophysical Research Communications. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kim J, Yoon MY, Choi SL, Kang I, Kim SS, Kim YS, Choi YK, Ha J. Effects of stimulation of AMP-activated protein kinase on insulin-like growth factor 1- and epidermal growth factor-dependent extracellular signal-regulated kinase pathway. J Biol Chem. 2001;276:19102–19110. doi: 10.1074/jbc.M011579200. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- MacNicol MC, Muslin AJ, MacNicol AM. Disruption of the 14-3-3 binding site within the B-Raf kinase domain uncouples catalytic activity from PC12 cell differentiation. J Biol Chem. 2000;275:3803–3809. doi: 10.1074/jbc.275.6.3803. [DOI] [PubMed] [Google Scholar]
- Martin MJ, Hayward R, Viros A, Marais R. Metformin Accelerates the Growth of BRAFV600E-Driven Melanoma by Upregulating VEGF-A. Cancer Discovery. 2012;2:344–355. doi: 10.1158/2159-8290.CD-11-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci USA. 2009;106:11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8:983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- Noble C, Mercer K, Hussain J, Carragher L, Giblett S, Hayward R, Patterson C, Marais R, Pritchard CA. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol Cell. 2008;31:862–872. doi: 10.1016/j.molcel.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, Roden C, Chalk CJ, Ardlie K, Palescandolo E, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. Journal of Clinical Oncology. 2012;30:316–321. doi: 10.1200/JCO.2011.36.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JK, Zaganjor E, Cobb MH. Signal control through Raf: in sickness and in health. Cell Research. 2012;22:14–22. doi: 10.1038/cr.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Lorenzo R, Zheng B. Targeted inhibition of BRAF kinase: opportunities and challenges for therapeutics in melanoma. Biosci Rep. 2012;32:25–33. doi: 10.1042/BSR20110068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8:426–433. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30:806–819. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Current Opinion in Oncology. 2011;23:177–182. doi: 10.1097/CCO.0b013e3283436e8c. [DOI] [PubMed] [Google Scholar]
- Roskoski R. RAF protein-serine/threonine kinases: Structure and regulation. Biochemical and Biophysical Research Communications. 2010;399:313–317. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- Rushworth LK, Hindley AD, O'Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate1. Journal of Molecular Biology. 2002;317:309–323. doi: 10.1006/jmbi.2001.5316. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Sprenkle AB, Davies SP, Carling D, Hardie DG, Sturgill TW. Identification of Raf-1 Ser621 kinase activity from NIH3T3 cells as AMP-activated protein kinase. FEBS Lett. 1997;403:254–258. doi: 10.1016/s0014-5793(97)00062-8. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiological Reviews. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udell CM, Rajakulendran T, Sicheri F, Therrien M. Mechanistic principles of RAF kinase signaling. Cell Mol Life Sci. 2011;68:553–565. doi: 10.1007/s00018-010-0520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.