

Abstract
Nur77 and Nor1 are highly conserved orphan nuclear receptors. We have recently reported that nur77−/−nor1−/− mice rapidly develop acute myeloid leukemia (AML) and that Nur77 and Nor1 transcripts were universally down-regulated in human AML blasts. These findings indicate that Nur77 and Nor1 function as leukemia suppressors. We further demonstrated silencing of Nur77 and Nor1 in leukemia stem cells (LSCs). We here report that inhibition of histone deacetylase (HDAC) using the specific class I HDAC inhibitor SNDX-275, restored the expression of Nur77/Nor1 and induced expression of AP1 transcription factors c-Jun and JunB, and of death receptor TRAIL, in AML cells and in CD34+38− AML leukemic stem cells (LSC). Importantly, SNDX-275 induced extensive apoptosis in AML cells, which could be suppressed by silencing nur77 and nor1. In addition, pro-apoptotic proteins Bim and Noxa were transcriptionally up-regulated by SNDX-275 in AML cells and in LSC. Our present work is the first report of a novel mechanism of HDAC inhibitor–induced apoptosis in AML that involves restoration of the silenced nuclear receptors Nur77 and Nor1, activation of AP1 transcription factors, a death receptor and pro-apoptotic proteins.
Keywords: SNDX-275, Nur77/Nor1, Apoptosis, AML
Introduction
Acute myeloid leukemia (AML) arises from the clonal expansion of aberrant myeloid progenitor cells; their increased proliferation and failure to differentiate results in an accumulation of myeloblasts. Only one third of younger adult patients diagnosed with AML can be cured, while older patients experience even lower survival rates.(1) Although many preclinical and clinical studies have sought to elucidate the biological and molecular basis of AML, the mechanism of AML pathogenesis remains undefined in the majority of cases. Recently, we reported that mice deficient in Nur77 and Nor1 develop AML-like disease and that reduced gene dosage of Nur77 and Nor1 leads to mixed myelodysplastic/myeloproliferative neoplasms (MDS/MPN) with progression to AML in rare cases (2, 3) Orphan nuclear receptors Nur77 (also termed NR4A1, TR3, or NGFI-B) and Nor1 (also termed NR4A3 or MINOR) are highly conserved members of the nuclear receptor family. As transcription factors, they are involved in various physiological and cellular functions, including apoptosis, mitosis, inflammation, and differentiation.(4–7) Induction and activation of Nur77 and/or Nor1 plays a central role in negative selection of T lymphocytes, macrophage activation-induced cell death, and apoptosis induced by antineoplastic agents in various cancer cells.(5, 8–11) Consistent with the parallel functions of these transcription factors, mice deficient in either nur77 or nor1 display relatively subtle abnormalities (12, 13) while mice deficient in both genes develop rapidly lethal and transplantable AML.(2) Importantly, the expression of Nur77 and Nor1 transcripts was profoundly decreased in leukemic blasts from all AML patients studied, compared to the levels in normal bone marrow cells (NBM), regardless of cytogenetics.(2) This finding suggests that silencing of Nur77 and Nor1 is a critical step in the pathogenesis of AML. Nur77 and Nor1 share 90% homology in the DNA-binding domain and they can both bind as monomers to a consensus NGFI-B response element (NBRE) sequence, or as homodimers to Nur-responsive element (NurRE),(14, 15) hence sharing a set of common target genes. Nur77 has been shown to regulate the induction of Fas-L, TRAIL, and pro-opiomelanocortin in lymphocytes or CNS cells;(16, 17) however, the common targets of Nur77 and Nor1 are not well characterized. Molecular analysis of nur77−/−/nor1−/− mice revealed that the absence of both nur77 and nor1 was associated with down-regulation of the activator protein 1 (AP-1) transcription factors c-Jun and JunB and down-regulation of the extrinsic apoptosis inducers TRAIL and FasL in myeloid leukemia cells.(2)
Epigenetic modifications have been shown to play critical roles in regulating gene expression. (18) Human tumor cells, including leukemia cells, exhibit a global loss of monoacetylation of histone H4, and aberrant histone acetyltransferase (HAT)/(HDAC) activity has been shown to be associated with cancer development (19–22) and hematological malignancies. (23–27) HDAC inhibitors display selective antitumor activity by inducing apoptosis, growth arrest, and differentiation and induce the expression of cell-cycle inhibitors p21, p19, and p57 and of the pro-apoptotic gene TRAIL in leukemia cells.(28–30) Notably, two HDAC inhibitors, vorinostat (SAHA) and romidepsin (Istodax) have been approved by FDA for clinical treatment of cutaneous T-cell lymphoma (CTCL), and HDAC inhibitors valproic acid, SAHA and MGCD0103 have shown clinical efficacy in ongoing AML trials.(28, 31–33) We hypothesized that histone acetylation plays a role in gene silencing of Nur77/Nor1 and HDAC inhibitor SNDX-275 was used in our study. SNDX-275 (Entinostat), a synthetic benzamide derivative, selectively inhibits the activities of class I HDACs, (HDAC1, -2, and -3), (34) has shown impressive efficacy in vitro and in vivo against a variety of tumors and is currently in phase I/II clinical trials.(34)
In this study, we demonstrate that Nur77 and Nor1 are profoundly silenced not only in bulk leukemia cells but also in leukemia stem cells (LSCs). This silencing was largely reversed by the HDAC inhibitor SNDX-275 alone or in combination with another Nur77/Nor1 inducer ionomycin. The restoration of Nur77/Nor1 by SNDX-275 was. accompanied by upregulation of c-Jun, JunB, TRAIL, Bim and Noxa in AML cells and LSCs. This resulted in extensive apoptosis of bulk AML and of leukemia stem cells.
Materials and Methods
Chemicals and cell cultures
SNDX-275 (Entinostat) was kindly provided by Dr. Peter Ordentlich (Syndax Pharmaceuticals, Inc., Waltham, MA). Trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), depsipeptide (FK228) and ionomycine were purchased from Sigma-Aldrich (St. Louis, MO), AtonPharma (Lawrenceville, NY), and Fujisawa Pharmaceutical Co. Ltd (Osaka, Japan), respectively. AML cell lines HL-60, MOLM13, OCI-AML3, and OCI-AML2 were purchased from ATCC (Manassas, VA). Bone marrow or peripheral blood samples were obtained consecutively for in vitro studies from patients diagnosed with AML during routine diagnostic workup under informed consent in accordance with regulations and protocols approved by the Institutional Review Board Committee of The University of Texas M. D. Anderson Cancer Center. Primary AML samples were harvested and no selection criteria were applied. Patient information is listed in Table 1. Mononuclear cells were separated with lymphocyte separation media (Mediatech, Manassas, VA) by density-gradient centrifugation resulting in ≥90% pure blast populations. Both cell lines and AML blast cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 mmol/L L-glutamine, and 50 μg/mL penicillin/streptomycin.
Table 1.
| Patients | Blasts % | Source | Cytogenetics | Sample status |
|---|---|---|---|---|
| 1 | 80 | PB | 46,XX[19] | New |
| 2 | 70 | PB | 46,XX,inv(12)(p11.2p13)[18] | New |
| 3 | 94 | PB | 46,XX,t(6;9)(p23;q34)[20] | New |
| 4 | 82 | PB | 46,XY[20] | Relapsed |
| 5 | 60 | PB | 46,XY[20] | New |
| 6 | 79 | PB | 41,XX, −5,der(7)t(7;12)(p11.1;p11.2),i(8)(q10),inv(9) (p11q12), −12, −16, −17, −18, −19,del(20)(q11.2q13.3),+der(20) t(12;20)(q12;q13.3), −21,add(21)(p11.2),+mar[9] | New |
| 7 | 98 | PB | 44,sl,+der(1;19)(q10;p10), −18[1] | Relapsed |
| 8 | 90 | BM | 46,XY[20] | New |
| 9 | 80 | BM | 46,XY[20] | New |
| 10 | 96 | PB | 46,XY,inv(16)(p13q22)[2];48,idem,t(9;22)(q34;q11.2),+ 13,+22[16];47,XY,inv(16)(p13q22),+13[1];48,XY,inv(16) (p13q22),+13,+22[1] | New |
| 11 | 79 | PB | 46,XX[19] | New |
| 12 | 75 | PB | 46,XY[20] | Relapsed |
| 13 | 73 | BM | 46,XY,(5)(q13q33) [20] | New |
| 14 | 92 | PB | 43−48,XY,+X, −5, −7,inv(9)(p11q12),add(13)(p13),add(17) (q25), −20, −21,+0-1r,+1-4mar[ 19] | New |
| 15 | 88 | PB | 46,XX,t(7;16;9)(q22;q22;q22), (12)(p12p13),t(12;22) (p13;q11.2), ins(17;12)(p11.2;p13p12)[15] | Refractory |
| [ ]: number of metaphase |
Abbreviations: BM, bone marrow; PM, peripheral blood.
Flow cytometry
The viable cell number (AnnV−, million/ml) and the % apoptosis (AnnV+ %). in drug treated samples was determined by using annexin V-FITC (BD Biosciences) as reported (35) and were used to calculate % surviving cells and % specific apoptosis. % surviving cells= viable cell number in treated sample × 100/viable cell number in control sample. % specific apoptosis = [% annexin V+ in treated sample) − (% annexin V+ in control sample) × 100] ÷ [100 − (% annexin V+ in control sample)] (36). The number of surviving leukemia stem cells from primary patient samples was determined using a method described previously.(35) % surviving cells= viable CD34+/38− cell number in treated sample×100/viable CD34+/38− cell number in control sample. CD34+/38−, CD34+/38+, and CD33+/34− cell populations were isolated from primary AML samples as previously described. (35)
Gene expression assay
Total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. 500 ng of cDNA was used for each real-time PCR reaction. All Taqman assays for measuring gene expression were purchased from Applied Biosystems (Foster City, CA), and the real-time PCR was conducted and analyzed with the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The abundance of each transcript relative to that of Abl1 was calculated as follows: Copies per 100 Abl1 = 100 × 2 exp [−ΔCt], where ΔCt is the mean Ct of the transcript of interest less the mean Ct of the transcript for Abl1.
Western blotting
Lysates of 1 × 106 cells were loaded onto a 10% or 12% SDS-PAGE gel (Bio-Rad, Hercules, CA) and proteins were transferred to Hybond-P membranes (Amersham Pharmacia Biotech, Piscataway, NJ), followed by immunoblotting. Signals were detected using a PhosphorImager (Storm 860, version 4.0; Molecular Dynamics). The following antibodies were used: mouse monoclonal histone3 (Upstate, Billerica, MA), rabbit polyclonal histone4 (Upstate, Billerica, MA), rabbit polyclonal acetyl-histone3 (Upstate, Billerica, MA), rabbit polyclonal acetyl-histone4 (Upstate, Billerica, MA), rabbit polyclonal c-Jun (Santa Cruz, Santa Cruz, CA), rabbit polyclonal caspase 8 (Cell Signaling, Danvers, MA), rabbit polyclonal Bim (Calbiocam, Danvers, MA), mouse monoclonal Noxa (Abcam Inc. Cambridge, MA), mouse monoclonal beta-actin (Cell Signaling, Danvers, MA), goat anti-mouse and goat anti-rabbit-horse radish peroxidase conjugate secondary antibodies(Bio-Rad).
Silencing of Nur77 and Nor1 with siRNA
Silencing of nur77 nor1, noxa and bim gene expression in HL-60 or primary AML samples was achieved by the small interfering RNA (siRNA) technique. All SiRNAs are ON-TARGETplus SMARTpools purchased from Dharmacon and were used individually or as 1:1 mixture. Transfection of leukemic cells was carried out by electroporation using the Amaxa Nucleofection system (Lonza, Cologne) following the manufacturer’s instructions
Chromatin immunoprecipitation (CHIP)
ChIP assays were conducted using the enzymatic chip Kit (Cell Signaling 9002) according to the manufacturer’s instructions. Primers amplifying human Nur77 and Nor1 promoters were: Nur77-1 forward GGA TTT GGG GGC TGT CGT, reverse TCC TCA CAG CTC TGG ATG; Nur77-2 forward TCC CTT TTT ATG CCT TCG, reverse ATC AGC ACC CAG TGG GCA; Nor1 forward TAA GAA ACC CAC GCC GTA; reverse GAG GTG CAG CTG CAC TTT. An aliquot of chromatin that was not incubated with an antibody was used as the input control sample (Input).
Statistics
Unless otherwise indicated, results are expressed as mean ± SD of 3 independent experiments. For immunoblot analyses, a representative immunoblot from 3 independent experiments is shown. P values were determined by one-way ANOVA followed by F statistics. A P value less than 0.05 was considered significant.
Results
HDAC inhibitor SNDX-275 restores Nur77 and Nor1 expression
First, we determined the universal histone acetylation level could be changed by SNDX-275. By immunoblotting, both Ac-H3 and Ac-H4 were strongly increased at 24 hrs at non-toxic concentrations of SNDX-275, whereas total H3 and H4 levels were not affected (Fig. 1). H4 acetylation was also observed in primary AML samples. At concentrations activating histone acetylation, SNDX-275 strongly induced gene expression of Nur77 and Nor1 in a dose-dependent manner in AML cell lines HL-60, MOLM13, OCI-AML3 and OCI-AML2 (Fig. 2Ai, iii). Importantly, SNDX-275 likewise restored Nur77 and Nor1 expression in primary AML blasts, although the increase was variable between different samples (Fig. 2Aii, iv). In addition, three other HDAC inhibitors TSA, SAHA and FK228 displayed effective gene induction by SNDX-275, and all of them induced robust restoration of both Nur77 and Nor1 at sub-cytotoxic concentrations (Fig. 2B). CHIP assays were performed to assess the acetylation levels of H4 on promoters of Nur77 and Nor1. Results demonstrated that SNDX-275 enhanced the amount of Ac-H4 on the promoter of Nur77 and Nor1, suggesting that SNDX-275 mediated the hyperacetylation of H4 which represents an active transcription status. (Fig. 2C) These results indicate that histone deacetylation plays a role in the epigenetic regulation of gene silencing of Nur77 and Nor1 and that this silencing can be reversed by HDAC inhibitors.
Figure 1. HDAC inhibitor SNDX-275 induces histone acetylation in AML cells.
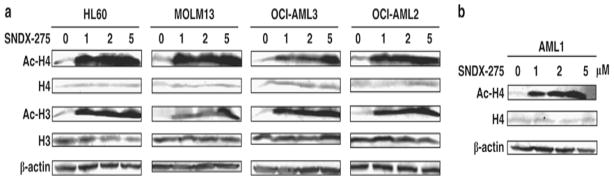
AML cell lines HL-60, MOLM13, OCI-AML3, and OCI-AML2 (A) and primary AML cells (B) were treated with vehicle (0) or indicated concentrations of SNDX-275 for 24 hr and subjected to Western blotting utilizing antibodies against acetyle-histone 4 (Ac-H4), histone 4 (H4), acetyle-histone 3 (Ac-H3), and histone 3 (H3). Beta-actin was used as loading control. Due to limited sample size, effects on Ac-H4/totalH4 only were assessed in primary AML blasts.
Figure 2. HDAC inhibitor SNDX-275 restores Nur77 and Nor1 expression in AML cells.

(A) AML cell lines HL-60, MOLM13, OCI-AML3, OCI-AML2, (i and iii) and primary AML blasts (n=5) (ii and iv) were treated with vehicle (0) or indicated concentrations of SNDX-275 for 24 hr. Nur77 (i and ii) and Nor1 (iii and iv) gene expression was determined by real-time RT-PCR analysis. (B) Gene expression of HL-60 cells treated with, vehicle or TSA (400nM), SAHA (500nM) or FK228 (2 nM), respectively, for 24 hr. (C) CHIP assay. HL-60 cells were treated with 1 μM SNDX-275 for 24 hours. Chromatin immunoprecipitations were performed for AcH4 and the immunoprecipitated chromatin and sonicated chromatin inputs were PCR amplified for the Nur77 and Nor1 promoters. An aliquot of chromatin that was not incubated with an antibody was used as the input control sample (Input). Chromatin incubated with rabbit serum was used as negative control (IgG).
SNDX-275 up-regulates the expression of c-Jun, JunB and TRAIL
We further investigated whether SNDX-275 treatment results in the induction of the hypothetical target genes of Nur77 and Nor1. Gene expression analysis performed in the same samples shown in Fig 2 revealed that SNDX-275 strongly enhanced c-Jun expression at both, RNA and protein levels (Fig. 3A, B), and enhanced JunB transcripts. SNDX-275 also up-regulated the expression of the death receptor family member TRAIL in AML cell lines and in primary AML samples (Fig. 3C). The increase in TRAIL was accompanied by caspase 8 cleavage (Fig. 3D). The association of upregulation of c-Jun, JunB and TRAIL with Nur77/Nor1 restoration implicated the potential regulation of those genes by Nur77 and Nor1.
Figure 3. SNDX-275 induces expression of C-Jun, JunB and TRAIL.

Sample treatment and gene expression analysis are the same as described in Fig. 2 (A) Gene expression of C-Jun (i and ii) and JunB (iii and iv) (B) Western blotting analysis of c-Jun. (C) Gene expression of TRAIL. (D) Western blotting analysis of Caspase 8 cleavage at 24 hr.
SNDX-275 induces Nur77 and Nor1-dependent apoptosis
As an antineoplastic agent, SNDX-275 induced extensive apoptosis in AML cell lines and primary samples in a time- and dose-dependent manner (Fig. 4 and Supplemental Figure 1). To determine the role of Nur77/Nor1 in SNDX-275-induced apoptosis, gene silencing with siRNAs was conducted on HL-60 (with the best electroporation efficiency and tolerance among AML cell lines) and primary AML samples. siRNAs successfully reduced the level of Nur77/Nor1 (Fig. 5A). Double silencing of nur77 and nor1 significantly reduced apoptosis induction by SNDX-275 (Supplemental Fig. 2B and Fig. 5Cii), and attenuated the growth-inhibitory effects of SNDX-275 (Supplemental Fig. 2A and Fig. 5Ci) while silencing of single gene was insufficient to suppress apoptosis. In primary AML samples, SNDX-275-induced reduction in cell numbers and apoptosis were significantly attenuated by the double silencing (Fig. 5D–F, panels iii–iv iii–iv, Supplemental Figure 3A–C). Simultaneously, samples with nur77/nor1 siRNA silencing were used for gene expression analysis of Nur77/Nor1 hypothetical target genes. Double silencing of nur77 and nor1, led to a significant but only partial decrease of SNDX-275-induced c-Jun and JunB upregulation (Fig. 5B). At variance with the results in HL-60 cells, silencing of nur77 and nor1 diminished SNDX-275-induced upregulation of c-Jun and JunB only in the AML11 sample, but not in other two samples (Supplemental Fig. 3D, E, F). The SNDX-275-induced TRAIL upregulation was not significantly affected by silencing nur77 and nor1 in HL60 and primary samples (Supplemental Fig. 2Bi, Supplemental Fig. 3D, E, F). This result suggested that these genes may not be the direct targets of Nur77/Nor1.
Figure 4. SNDX-275 induces apoptosis in AML cells.
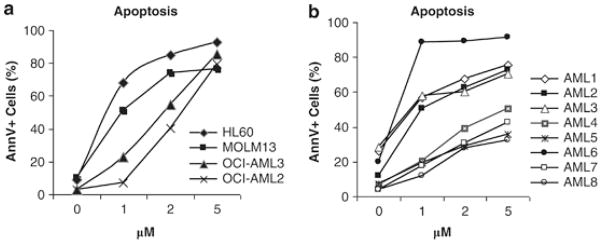
(A) AML cell lines HL-60, MOLM13, OCI-AML3, and OCI-AML2 and (B) eight primary AML samples were treated with vehicle (0) or indicated concentrations of SNDX-275. Apoptosis was determined as the percentage of annexin V-positive cells at 48 hr.
Figure 5. Silencing nur77 and nor1 suppresses SNDX-275-induced apoptosis.

HL-60 cells (A, B) or primary AML samples (AML10, AML11 and AML12, D–F) were transfected with scrambled siRNA (Scr), siRNA targeting nur77 (SiNur77) or nor1 (SiNor1), or mixture of SiNur77 and SiNor1 (SiMix). Twenty-four hours later, cells were treated with vehicle (0) or 1 μM SNDX-275 for another 24 hours. (A) (i) Nur77 (ii) Nor1 (B) (i) c-Jun and(ii) JunB gene expression in HL-60 cells treated for 24 hr. (C) % of surviving cells (i), and specific apoptosis (ii) was calculated as described in Material and Methods. In (D), (E), and (F), from left to right, the gene expression of Nur77 (i) and Nor1(ii), % surviving cells (iii) and specific apoptosis (iv) are shown for AML10, 11, 12, respectively. The raw data (viable cell numbers and % annexinV(+) cells) are shown in Supplemental Fig. 2Ai, ii for HL-60 and Supplemental Fig. 3A–C for AML1, AML2 and AML3.
We next sought to determine whether activation of Nur77/Nor1 through mechanisms other than HDAC blockade will promote apoptosis in AML cells. Calcium ionophore ionomycin and TPA are well recognized inducers of Nur77 and Nor1 whose pulse stimulation leads to apoptosis in lymphocytes and thymocytes. (37) In AML cells, 6-hr exposure to ionomycin and TPA (data not shown) induced brisk upregulation of Nur77 and Nor1 at protein levels (Fig. 6A) followed by apoptosis at 48 hours (Fig. 6B). Since the induction of Nur77 and Nor1 by ionomycin involves the release of CabinI/HDACs complex from gene promoters (38), we tested the effect of pretreatment of SNDX-275 on ionomycin-induced Nur77/Nor1 expression and apoptosis. Combined use of SNDX-275 dramatically furthered the ionomycin-induced Nu77/Nor1 protein in AML cells and induced a significantly higher degree of apoptosis than ionomycine alone. (Fig. 6A, B) Most importantly, apoptosis induced by SNDX-275 and ionomycin was attenuated by silencing Nur77/Nor1. (Fig. 6C, D) This observation supported the notion of a functional role of Nur77/nor1 in apoptosis induction of AML cells and suggested additive activation mechanism in Nur77/Nor1 expression Altogether, these findings suggest that apoptosis induced by SNDX-275 is mediated at least in part by the restoration of Nur77 and Nor1 function, however additional mechanisms are likely operational in AML cells.
Figure 6. Silencing of nur77 and nor1 suppresses apoptosis induced by combination of SNDX-275 and ionomycine.

AML cell lines HL-60 and OCI-AML3, and one primary AML sample were pretreated with SNDX-275 (0.5μM, 1μM and 1μM, respectively) for 16 hrs followed by treatment with ionomycine (1μg/ml). Protein lysates were collected at 6 hours after ionomycin treatment for detection of Nur77/Nor1 protein (A), and apoptosis induction was analyzed at 48 hours (B). HL-60 transfected with scrambled siRNA (Scr), siRNA or mixture of SiNur77 and SiNor1 (SiMix) were pretreated with 0.5μM SNDX- 275 followed by 1ug/ml ionomycin. Protein lysates were collected at 6 hours after ionomycin treatment for detection of Nur77/Nor1 protein (C), and apoptosis induction was analyzed at 48 hours (D).
SNDX-275 induces pro-apoptotic proteins Bim and Noxa
SNDX-275 induces apoptosis in tumor cells by a variety of different mechanisms, eg inducing Bim and Noxa in chronic lymphocytic leukemia.(39) Since SNDX-275-induced apoptosis was in part independent on Nur77/Nor1 restoration, we sought to characterize the contribution of the mitochondrial apoptotic pathway in SNDX-275-induced apoptosis, in particular through the pro-apoptotic Bcl2 family members Bim and Noxa. Real-time RT-PCR analysis and Western blotting showed that both Bim and Noxa were elevated by the compound in AML cell lines and primary samples, in association with caspase 9 cleavage (Fig. 7A, B, C). Besides, these elevations are not dependent on Nur77/Nor1 restoration (Supplemental Fig. 2B, Supplemental Fig. 3D–F). We confirmed the role of Noxa and Bim in SNDX-mediated apoptosis with siRNA gene silencing. SiNoxa led to reduction of SNDX-275-induced gene upregulation and this resulted in significantly diminished SNDX-275-induced apoptosis in HL-60 cells (Fig.7D) and two primary AML samples (Fig.7E, F). SiBim also significantly protected HL-60 cells and primary AML cells from SNDX-275-induced apoptosis (Supplemental Fig. 4). Our observation suggests that SNDX-275-induced upregulation of pro-apoptotic BH3-only Bcl-2 members in AML likely contribute to its pro-apoptotic activity.
Figure 7. SNDX-275 induces the expression of pro-apoptotic proteins Bim and Noxa in AML cells.

(A) Bim (i and ii) and Noxa (iii and iv) gene expression were detected with real-time RT-PCR in AML cell lines (I and iii) and primary AML samples (ii and iv). (B) Bim and Noxa protein expression measured by Western bloting. (C) Caspase 9 cleavage at 24 hr. HL-60 cells and 2 primary AML samples were used to determine the effect of Noxa in SNDX-275-induced apoptosis by SiRNA knockdown. In (D), (E), (F), Noxa gene expression detected by RT-PCR and immunoblotting (i); viable cell numbers (ii) and % apoptotic cells (iii) of HL-60, AML10 and AML11 transfected with siRNA targeting noxa(SiNoxa) is shown.
SNDX-275 induces apoptosis in AML stem cells (CD34+/38−)
AML is a disease of hematopoietic stem cells (HSCs) and progenitors cells and relapse after chemotherapy is thought to originate from an outgrowth of leukemia stem cells, a population of malignant CD34+CD38− progenitor cells. In our previous study, we observed the universal down-regulation of Nur77 and Nor1 gene expression in primary AML blasts.(2) Here, we measured the expression profiles of Nur77 and Nor1 in putative AML stem cells defined as CD34+/38−. Our results demonstrated the remarkable down-regulation of both Nur77 and Nor1 transcripts in leukemia stem cells (CD34+/38−), as well as in myeloid progenitors (CD34+/38+) and in committed progenitors (CD33+/34−) (Fig. 8A). We next evaluated the effects of SNDX-275 on leukemia stem cells. Our results demonstrated that SNDX-275 remarkably reduced the viable cell numbers and induced apoptosis in the LSC fraction of primary AML samples (Fig. 8B). Gene expression analysis revealed that in the CD34+/38− population, SNDX-275 restored Nur77 and Nor1 and induced c-Jun, TRAIL, Bim and Noxa (Fig. 8C).
Figure 8. SNDX-275 induces apoptosis in AML stem cells (CD34+/38−).

(A) Gene expression of Nur77 and Nor1 in subpopulations of primary AML samples. Six primary AML samples were used to isolate CD34+38−, CD34+38+, and CD33+34− populations by FACS sorting. As control, CD34+ cells were isolated from two normal bone marrow samples. (B) Viable cell number (left) and % apoptosis (right) in CD34+38− LSC of primary AML samples treated with 1μM SNDX-275 for 24 hr. (C) Gene expression of CD34+38− subpopulation sorted from AML14 treated with 1μM SNDX-275 for 24 hr. Results are presented as fold difference in normalized RNA expression of SNDX-275- over vehicle-treated cells. “‘Supplementary information is available at Leukemia’s website.”
Discussion
We here extend our previous observation of silencing of Nur77 and Nor1 in acute myeloid leukemias to leukemic stem/progenitor cells and demonstrate that blockade of histone deacetylation by HDAC inhibitor SNDX-275 efficiently restores Nur77/Nor1 expression in AML cells and LSCs and induces apoptosis. The induction of Nur77 and Nor1 involves the hyperacetylation of their promoters. To our knowledge, our current report is the first to describe the restoration of Nur77 and Nor1 by HDAC inhibition in AML. These findings implicate histone deacetylation as an important mechanism of Nur77 and Nor1 silencing in AML, and suggest that Nur77 and Nor1 play an important role in survival and proliferation of leukemic cells.
Restoration of Nur77 and Nor1 and apoptosis induction by HDAC inhibition has been reported in cutaneous T-cell lymphomas (CTCL) (40) involving a mechanism mediated by HDAC7, a class IIA HDAC which represses Nur77 expression in thymocytes/lymphocyte. (41) (42) We attempted identification of HDAC7 in several myeloid leukemic cell lines (HL-60, MOLM13 and OCI-AML3) but its expression was below detectable at both mRNA or protein levels (data not shown), implying tissue specificity of this mechanism. Recently, it was reported that Nur77 expression is also regulated at the post-transcriptional level by acetylation and HDAC1 mediated its deactivation leading to ubiquitinilation.(43) With its specificity on class I HDACs (including HDAC1, 2, 3), SNDX-275 might restore gene expression at both transcriptional and post-transcriptional levels. In the context of our study, SNDX-275 regulated Nur77 and Nor1 gene expression mainly at the transcriptional level, since treatment with SNDX-275 only did not induce pronouncedly induction of proteins. However, the cause-effect relationship between gene induction and apoptosis is unequivocal (Fig. 5 & 6) Recently, bifunctional RNA has been identified in many species from bacteria to mammals: RNA contains both coding capacity and regulatory activity. (44) SRA (Steroid Receptor RNA), with a long ORF and no initiating methionines, has been shown to co-regulate with the progesterone and several other steroid receptors (45); p53 mRNA was shown to interact with Mdm2 protein to prevent Mdm2 from promoting p53 protein ubiquitination and degradation and ultimately to stimulate p53 synthesis. (46, 47) While such effects are speculative in the context of our work, they are nevertheless a distinct possibility and could explain functionality without direct protein synthesis. In addition, histone deacetylation may not be the only mechanism responsible for Nur77 and Nor1 silencing, which was implied by the ionomycin combination experiment and HDAC inhibiton as single treatment may not be sufficient to restore silenced gene expression completely. Still, reactivation is a vital step in overcoming gene silencing.
We have demonstrated that restoration of Nur77 and Nor1 was associated with c-Jun and JunB induction and that silencing Nur77 and Nor1 significantly prevented c-Jun and JunB upregulation in HL-60. This effect, however, were not consistently seen in primary AML samples. This discrepancy may be explained by alternative mechanisms regulating expression of these transcription factors in primary samples. Furthermore, multiple AP-1 like binding sites were identified in the Nur77 promoter (48). However, silencing of c-Jun with siRNA did not affect upregulation of Nur77/Nor-1 by SNDX-275 (data not shown). Further studies to delineate the hierarchical relations of Jun transcriptional factors and Nur77/Nor1 are clearly warranted. Similarly, Our findings indicate that at least in human AML cells TRAIL may not a direct target of Nur77/Nor1
The induction of pro-apoptotic BH3-only proteins Noxa and Bim contributes to SNDX-275-induced apoptosis in AML, indicating involvement of the mitochondrial apoptosis pathway in addition to TRAIL-mediated activation of extrinsic apoptotic signaling. In contrast to a published observation(49) we did not observe modulation of Bcl-2 protein expression by SNDX-275 (data not shown). NR4As have also been implicated in the regulation of apoptosis via mechanisms independent of their primary transcription regulatory function, via translocation to mitochondria and interaction with the antiapoptotic protein, BCL2, leading to NR4A dependent conversion of BCL2 into a proapoptotic mediator.(8, 50) The relative contribution of genomic versus non-genomic mechanisms of apoptosis induction in leukemic cells remains to be established
Targeting LSCs remains the key for eradicating persistent leukemia. The consistent down-regulation of leukemia suppressors Nur77 and Nor1 among sub-populations reinforces that the silencing of these genes is a common event in AML cells, irrespective of the differentiation status, and that it is a critical step in leukemogenesis. In our present report, we demonstrate that SNDX-275 restores the expression of Nur77 and Nor1 in LSCs and induces cell death in these cells. The restoration of Nur77 and Nor1, their relative genes, and the induction of pro-apoptotic genes in LSCs may explain how SNDX-275 kills LSCs. Our results strongly suggest the potential ability of SNDX-275 to affect LSCs. This would endow SNDX-275 the capacity to kill not only bulk leukemic cells but also leukemia-initiating cells.
In conclusion, our findings suggest that HDAC-mediated gene silencing of the nuclear receptors Nur77 and Nor1 is a novel mechanism critical for survival of AML blasts and stem cells. Re-expression of these nuclear receptors may be considered as a valuable biomarker for the ability of HDAC inhibitors to induce apoptosis of LSC. We propose that therapeutic approaches directed toward the reexpression/activation of Nur77 and Nor1 could be of significant benefit in the treatment of human AML, irrespective of karyotype. To this aim, further understanding of the mechanism of Nur77/Nor1 silencing in AML and the exploration of alternative mechanisms other than histone deacetylation would provide clues for drug combination in leukemia therapy.
Acknowledgments
Supported by grants from the National Institutes of Health (P01 CA055164, P30 CA016672) (to MA), a Baylor College of Medicine (BCM) and the University of Texas M. D. Anderson Cancer Center collaboration grant from the BCM Dan L. Duncan Cancer Center (to OC and MK), RO1CA111411 (to OC) and Syndax Pharmaceuticals, Inc. The authors thank Karen C. Dwyer, Wendy D. Schober, and Jared K. Burks from the CCSG Flow Cytometry and Cellular Imaging Core Facility for their excellent technical support.
Footnotes
Conflict of Interest Disclosure
This work was in part supported by Syndax Pharmaceuticals, Inc.
References
- 1.Stone RM, O’Donnell MR, Sekeres MA. Acute myeloid leukemia. Hematology. 2004;1:98–117. doi: 10.1182/asheducation-2004.1.98. [DOI] [PubMed] [Google Scholar]
- 2.Mullican SE, Zhang S, Konopleva M, Ruvolo V, Andreeff M, Milbrandt J, et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13(6):730–735. doi: 10.1038/nm1579. [DOI] [PubMed] [Google Scholar]
- 3.Ramirez-Herrick AM, Mullican SE, Sheehan AM, Conneely OM. Reduced NR4A gene dosage leads to mixed myelodysplastic/myeloproliferative neoplasms in mice. Blood. 2010 Mar 3;117(9):2681–2690. doi: 10.1182/blood-2010-02-267906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolluri SK, Bruey-Sedano N, Cao X, Lin B, Lin F, Han Y-H, et al. Mitogenic Effect of Orphan Receptor TR3 and Its Regulation by MEKK1 in Lung Cancer Cells. Mol Cell Biol. 2003 Dec 1;23(23):8651–8667. doi: 10.1128/MCB.23.23.8651-8667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem. 2005 Aug 12;280(32):29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- 6.Pei L, Castrillo A, Tontonoz P. Regulation of Macrophage Inflammatory Gene Expression by the Orphan Nuclear Receptor Nur77. Mol Endocrinol. 2006 Apr 1;20(4):786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 7.Chao LC, Bensinger SJ, Villanueva CJ, Wroblewski K, Tontonoz P. Inhibition of adipocyte differentiation by Nur77, Nurr1 and Nor1. Mol Endocrinol. 2008 Oct 22; doi: 10.1210/me.2008-0161. 2008:me.2008–0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson J, Winoto A. During negative selection, Nur77 family proteins translocate to mitochondria where they associate with Bcl-2 and expose its proapoptotic BH3 domain. The Journal of Experimental Medicine. 2008 May 12;205(5):1029–1036. doi: 10.1084/jem.20080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sibayama-Imazu T, Fujisawa Y, Masuda Y, Aiuchi T, Nakajo S, Itabe H, et al. Induction of apoptosis in PA-1 ovarian cancer cells by vitamin K2 is associated with an increase in the level of TR3/Nur77 and its accumulation in mitochondria and nuclei. Journal of Cancer Research and Clinical Oncology. 2008;134(7):803–812. doi: 10.1007/s00432-007-0349-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y-L, Jian M-H, Lin C-C, Kang J-C, Chen S-P, Lin P-C, et al. The Induction of Orphan Nuclear Receptor Nur77 Expression by n-Butylenephthalide as Pharmaceuticals on Hepatocellular Carcinoma Cell Therapy. Mol Pharmacol. 2008 Oct 1;74(4):1046–1058. doi: 10.1124/mol.107.044800. [DOI] [PubMed] [Google Scholar]
- 11.Ye X-f, Wu Q, Liu S, Lin X-f, Zhang B, Wu J-f, et al. Distinct role and functional mode of TR3 and RAR[alpha] in mediating ATRA-induced signalling pathway in breast and gastric cancer cells. The International Journal of Biochemistry & Cell Biology. 2004;36(1):98–113. doi: 10.1016/s1357-2725(03)00143-2. [DOI] [PubMed] [Google Scholar]
- 12.Lee S, Wesselschmidt R, Linette G, Kanagawa O, Russell J, Milbrandt J. Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77) Science. 1995;269(5223):532–535. doi: 10.1126/science.7624775. [DOI] [PubMed] [Google Scholar]
- 13.Ponnio T, Burton Q, Pereira FA, Wu DK, Conneely OM. The nuclear receptor Nor-1 is essential for proliferation of the semicircular canals of the mouse inner ear. Mol Cell Biol. 2002 Feb 1;22(3):935–945. doi: 10.1128/MCB.22.3.935-945.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winoto A, Littman DR. Nuclear Hormone Receptors in T Lymphocytes. Cell. 2002;109(2, Supplement 1):S57–S66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- 15.Li Q-X, Ke N, Sundaram R, Wong-Staal F. NR4A1, 2, 3 - an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol. 2006;21:533–540. doi: 10.14670/HH-21.533. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, DeYoung A, Kasler HG, Kabra NH, Kuang AA, Diehl G, et al. Receptor-mediated apoptosis in T lymphocytes. Cold Spring Harb Symp Quant Biol. 1999;64:363–371. doi: 10.1101/sqb.1999.64.363. [DOI] [PubMed] [Google Scholar]
- 17.Philips A, Lesage S, Gingras R, Maira MH, Gauthier Y, Hugo P, et al. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol Cell Biol. 1997 Oct 1;17(10):5946–5951. doi: 10.1128/mcb.17.10.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 19.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 20.Giles RH, Peters DJM, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends in Genetics. 1998;14(5):178–183. doi: 10.1016/s0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- 21.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 22.Sakai K, Nagahara H, Abe K, Obata H. Loss of heterozygosity on chromosome 16 in hepatocellular carcinoma. J Gastroenterol Hepatol. 1992;7:288–292. doi: 10.1111/j.1440-1746.1992.tb00982.x. [DOI] [PubMed] [Google Scholar]
- 23.Borrow J, Stanton VP, Andresen JM, Becher R, Behm FG, Chaganti RSK, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14(1):33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda A, Shankar DB, Watanabe M, Tamanoi F, Moore TB, Sakamoto KM. Molecular targets and the treatment of myeloid leukemia. Molecular Genetics and Metabolism. 2006;88(3):216–224. doi: 10.1016/j.ymgme.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Rowley JD, Reshmi S, Sobulo O, Musvee T, Anastasi J, Raimondi S, et al. All patients with the t(11; 16)(q23; p13. 3) that involves MLL and CBP have treatment-related hematologic disorders. Blood. 1997 Jul 15;90(2):535–541. [PubMed] [Google Scholar]
- 26.Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. PNAS. 1998 Sep 1;95(18):10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Saunthararajah Y, Redner RL, Liu JM. Inhibitors of Histone Deacetylase Relieve ETO-mediated Repression and Induce Differentiation of AML1-ETO Leukemia Cells. Cancer Research. 1999 Jun 1;59(12):2766–2769. [PubMed] [Google Scholar]
- 28.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 29.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11(1):71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 30.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008 Aug 15;112(4):981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006 Nov 15;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008 Feb 1;111(3):1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 34.Hess-Stumpp H, Bracker TU, Henderson D, Politz O. MS-275, a potent orally available inhibitor of histone deacetylases--The development of an anticancer agent. The International Journal of Biochemistry & Cell Biology. 2007;39(7–8):1388–1405. doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Kojima K, Konopleva M, Tsao T, Nakakuma H, Andreeff M. Concomitant inhibition of Mdm2-p53 interaction and Aurora kinases activates the p53-dependent postmitotic checkpoints and synergistically induces p53-mediated mitochondrial apoptosis along with reduced endore duplication in acute myelogenous leukemia. Blood. 2008 Oct 1;112(7):2886–2895. doi: 10.1182/blood-2008-01-128611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youn H-D, Sun L, Prywes R, Liu JO. Apoptosis of T Cells Mediated by Ca2+-Induced Release of the Transcription Factor MEF2. Science. 1999 Oct 22;286(5440):790–793. doi: 10.1126/science.286.5440.790. [DOI] [PubMed] [Google Scholar]
- 38.Youn H-D, Liu JO. Cabin1 Represses MEF2-Dependent Nur77 Expression and T Cell Apoptosis by Controlling Association of Histone Deacetylases and Acetylases with MEF2. Immunity. 2000;13(1):85–94. doi: 10.1016/s1074-7613(00)00010-8. [DOI] [PubMed] [Google Scholar]
- 39.Inoue S, Riley J, Gant TW, Dyer MJS, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;21(8):1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Fiskus W, Eaton K, Fernandez P, Wang Y, Rao R, et al. Cotreatment with BCL-2 antagonist sensitizes cutaneous T-cell lymphoma to lethal action of HDAC7-Nur77-based mechanism. Blood. 2009 Apr 23;113(17):4038–4048. doi: 10.1182/blood-2008-08-176024. [DOI] [PubMed] [Google Scholar]
- 41.Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, et al. HDAC7, a Thymus-Specific Class II Histone Deacetylase, Regulates Nur77 Transcription and TCR-Mediated Apoptosis. Immunity. 2003;18(5):687–698. doi: 10.1016/s1074-7613(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 42.Scott FL, Fuchs GJ, Boyd SE, Denault J-B, Hawkins CJ, Dequiedt F, et al. Caspase-8 Cleaves Histone Deacetylase 7 and Abolishes Its Transcription Repressor Function. J Biol Chem. 2008 Jul 11;283(28):19499–19510. doi: 10.1074/jbc.M800331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang S-A, Na H, Kang H-J, Kim S-H, Lee M-H, Lee M-O. Regulation of Nur77 protein turnover through acetylation and deacetylation induced by p300 and HDAC1. Biochemical Pharmacology. 2010;80(6):867–873. doi: 10.1016/j.bcp.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 44.Ulveling D, Francastel C, Hubé F. When one is better than two: RNA with dual functions. Biochimie. 2011;93(4):633–644. doi: 10.1016/j.biochi.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Colley SM, Leedman PJ. Steroid Receptor RNA Activator – A nuclear receptor coregulator with multiple partners: Insights and challenges. Biochimie. 2011;93(11):1966–1972. doi: 10.1016/j.biochi.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 46.Gajjar M, Candeias Marco M, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, et al. The p53 mRNA-Mdm2 Interaction Controls Mdm2 Nuclear Trafficking and Is Required for p53 Activation following DNA Damage. Cancer Cell. 2012;21(1):25–35. doi: 10.1016/j.ccr.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 47.Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, et al. p53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10(9):1098–1105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 48.Williams GT, Lau LF. Activation of the inducible orphan receptor gene nur77 by serum growth factors: dissociation of immediate-early and delayed-early responses. Mol Cell Biol. 1993 Oct 1;13(10):6124–6136. doi: 10.1128/mcb.13.10.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romanski A, Bacic B, Bug G, Pfeifer H, Gul H, Remiszewski S, et al. Use of a novel histone deacetylase inhibitor to induce apoptosis in cell lines of acute lymphoblastic leukemia. Haematologica. 2004 Apr 1;89(4):419–426. [PubMed] [Google Scholar]
- 50.Lin B, Kolluri SK, Lin F, Liu W, Han Y-H, Cao X, et al. Conversion of Bcl-2 from Protector to Killer by Interaction with Nuclear Orphan Receptor Nur77/TR3. Cell. 2004;116(4):527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]