

Abstract
Seeding a protein solution with pre-formed fibrils can enhance dramatically the growth rate of amyloids. As the seeds do not need to be of the same protein, seeding may account for the observed correlations between amyloid diseases. In an effort to understand better the molecular mechanisms behind cross seeding we have studied in silico the effect of mutations on the seeding of amylin fibrils. Our investigations of the structural stability of decamers of wild type amylin peptides, of Y37L mutants, and of hetero-assemblies of wild-type and mutant amylin molecules, show that the experimental observed efficient cross seeding can be explained based on similarity in fibril structure of components. We find that amyloids with similar side chains packing at the β-sheet interface are structurally compatible, acting as a good template for the congruent incorporation of homologues peptides. In the Y37L mutants, lack of tyrosine-specific interactions causes significant higher flexibility of the C terminal than observed in the wild-type fibril. This effects elongation of the mutant fibril leading to the longer lag times during aggregation that are observed in experiments. Our study gives guidelines for the design of ligands that could stabilize amylin fibrils.
Keywords: cross-seeding, hetero-assembly, face–to-face hydrophobic contacts, π-π interactions
Introduction
Amyloid aggregates are implicated in at least 30 distinct diseases1,2. These aggregates result from failure of a specific peptide or protein to maintain its native (functional) conformation.3,1 Instead, they form amyloid filaments characterized by β-strands that are oriented perpendicularly to the fibril axis. The strands are connected through a dense hydrogen-bonding network and side-chain interactions between strands that drive their lateral association4 to supra-molecular β-sheets. The rate-limiting step in the growth process of these aggregates is the formation of an initial nucleus5, and in vitro, the seeding of the protein solution with preformed fibrils leads to dramatically faster fibril growth6. Human amyloids can be biochemically mixed; implying the possibility that one amyloidic peptide can cross-seed another one in vivo7. Such cross-seeding may account for the observed correlations between amyloid diseases. Hence, a detailed knowledge of the molecular mechanism of fibril seeding would provide a platform for the rational design and therapeutic intervention of disease states associated with mature amyloid fibrils and their precursors.
However, an accurate description of the aggregation process and seeding of fibrils is still missing, as these processes are difficult to explore in experiments8. For this reason, we rely in the present study on an alternative approach. Using molecular dynamics simulations, we study the molecular mechanisms of cross-seeding by probing the effects of mutations on the seeding of fibrils. Our underlying assumption is that cross-seeding depends on the similarity of the fibrils resulting from the different components. Hence, studying “cross-seeding” between wild type and mutants having similar fibrillar structures allows comparison of the various interactions that enable cross seeding. Key candidates are the stacking of aromatic residues along the outside of the fiber, hydrophobicity of key residues, and structural similarity between adjacent polypeptide strands9,10,11.
Our test system is amylin, a 37 residue hormone produced in the pancreas that is highly amyloidogenic and associated with type-2 diabetes mellitus through membrane permeabilization induced β-cell loss12,13. A fibril model of the full-length human amylin has been extrapolated from X-ray diffraction data of cross-β spine structures of two segments of human amylin (NNFGAIL and SSTNVG). 14 The topology of this model is similar to that reported by Luca et al14 and Bedrood et al15, and exhibits a β-strand-loop-β-strand motif, consisting of an N terminal β-stand residue 8–19, with the loop region located at residues 20–23 and C terminal β-stand comprising residues 24–3614. Experimental and computational studies have shown that the human amylin proto-fibril pair has molecular polymorphism 14, 16, 17,18,19,20,21,15 in which the two layer models could be packed either in antiparallel or parallel fashion. Depending of sample preparation, Middleton et al17 found parallel and anti-parallel mixed polymorphism, while Nielson et al16 found only anti-parallel fibrils from. Other simulations have shown the stability of the packing of proto-fibril pairs of amylin where one layer is shifted against the other by two residues20,21. This suggests that two-layer models such as in Figure 1C, packed in an antiparallel fashion, are good structural descriptions20,21 of such oligomers. Molecular dynamics simulations also indicate that X-ray models22,19 with more closely inter-digitated (interlocked β-strands that tighten the binding of two β-sheets) side chains are more compact and stable than the NMR models19. Amylin contains three aromatic residues, F15 is the only one that resides in the β-sheet core, while F23 is located in a bend and Y37 is exposed at the C-terminus. The fibril model of amylin has an in-register alignment of matching residues, generating a tight packing that maximizes favorable hydrophobic and van der Waals side-chain contacts along the long axis of the fibril, and may be stabilized in addition through π– π stacking of the F15, F23 and Y37 aromatic rings in the β-strand, loop and C terminal regions (Figure 1).
Figure 1.

The structure of human amylin fibril model10. (A) Sequence of human amylin, the aromatic amino acids (F15, F23 and Y37) are colored in red. (B) Single layer amylin decamer. Intra-molecular face-to-face side chain interaction that determines the stability of the U shaped in wild type, mutants and seeded oligomers, are emphasized by representing the corresponding side chains as balls and sticks. (C) Double layered amylin fibril model.
We will use molecular dynamics simulations to study the role of interactions involving the three aromatic residues F15, F23 and Y37 by comparing the wild type with mutants where a single aromatic residue is replaced by leucine (F15L, F23L and Y37L). Tu et al23 have already shown that fibril seeds from such mutants can still seed amyloid growth of wild-type amylin, suggesting that similarity in fiber structures is a key requirement for efficient mixed growth. Amyloid formation of the F15L mutant is almost twice as rapid than for the wild-type, while it is almost three times slower for the Y37L mutant, and two times slower for F23L mutant23. Molecular dynamics simulations of stability and conformational changes of such amyloid assemblies will enable us to measure directly changes in structural stability induced by specific alterations of the amino acid sequence of amyloid polypeptides. In-vivo conditions are mimicked and information that is currently unobtainable through experiment, such as real time structural data, will be obtained, making visual inspection possible. Especially, we aim to answer the following questions:
Is similarity in fibril structure the key requirement for effective cross seeding of single mutant seeds with wild type amylin?
Can differences in lag times observed in the self-assembly of single mutants amylin fibers be explained by the relative stability of the mutant structures during the simulation?
Does the loss of π-π interactions affect the stability of amylin mutant fibers? What is the effect of such mutation on the face–to-face hydrophobic contacts in the interior core?
Answering these questions may point the way to in-silico development of molecules that, by binding to the amylin fibril, shift the equilibrium of amylin toxic oligomer to the non-toxic fibers.24 Finally, we propose new mutation experiments that investigate the role of pairs of amino acids involved in face-to-face interactions between the β sheet regions.
Materials and Methods
For the wild type, we base the start configuration on the fibril models of amylin by the Eisenberg group14, and build out of these a single layer decamer (Figure 1B) and double layer decamer models of amylin consist of two pentamer layers (Figure 1C). This choice is motivated by recent work by Kahler et al 25 which showed that decamers of Aβ retained the fibrillar states and gain in stability with oligomer growth26 and are more stable than pentamers25. Since amylin has similar size and the same β-strand-loop-β-stand motif as Aβ, we assume that the same observations apply for amylin. We denote the so designed wild-type amylin decamer as WT. Mutant decamers are designed by replacing the aromatic residues, F15, F23, and Y37 with leucine, and denote them as F15L, F23L and Y37L, respectively (Table 1). These mutants are obtained from wild type coordinates by replacing the side chains of the targeted residues while retaining the original backbone conformations of the wild-type27. The structure of the designed mutants is minimized for 5000 steps using the steepest decent algorithm with the backbone of the protein restrained. The hetero-assemblies of wild type and mutants are a 1:1 mixture of wild-type (the first five strands) and mutants (the last five strands, consisting of F15L, F23L or Y37L in the above notation), and are denoted as WT-F15L, WT- F23L and WT-Y37L respectively.
Table 1.
Summary of amylin, mutants and mixed decamer models and simulation conditions. The simulations are performed at 310 K. The symbol * marks the cross-seeded aggregates where the first five strands are from the wild type amylin and the last five strands are from one of the mutants (i.e. F15L, F23L and Y37L, respectively). SL marks single layer decamers and DL double layer decamers, where each layer consists of five strands.
| System | # Atoms of peptide/# Atoms Water /Cl− | Simulation box dimensions (x, y, z[Å]) | Total simulation time, ns |
|---|---|---|---|
| WT (SL) | 5350/36665/20 | 106.8,106.8, 106.8 | 600ns (200×3) |
| F15L(SL) | 5340/36675/20 | 106.8,106.8, 106.8 | 600ns (200×3) |
| F23L(SL) | 5340/36396/20 | 106.6,106.6, 106.6 | 600ns (200×3) |
| Y37L(SL) | 5330/36695/20 | 106.8,106.8, 106.8 | 600ns (200×3) |
| WT- F15L*(SL) | 5345/36667/20 | 107.0,107.0, 107.0 | 600ns (200×3) |
| WT- F23L*(SL) | 5345/36677/20 | 106.9,106.9, 106.9 | 600ns (200×3) |
| WT- Y37L*(SL) | 5340/36681/20 | 106.9,106.9, 106.9 | 600ns (200×3) |
| WT(DL) | 5350/39644/20 | 108.1,108.1, 108.1 | 600ns (200×3) |
| F23L(DL) | 5340/39666/20 | 108.0,108.0, 108.0 | 600ns (200×3) |
| Y37L(DL) | 5330/39660/20 | 108.0,108.0, 108.0 | 600ns (200×3) |
Molecular dynamics simulations
Our molecular dynamics simulations rely on a combination of the AMBER ff99SB force field28 with explicit water (TIP3P)29,30, a common choice for exploring amyloid peptide aggregation31,32, as implemented in the GROMACS program version 4.5.5 33. Hydrogen atoms are added with the pdb2gmx module of the GROMACS suite. The start configurations for all proteins are put in the center of a cubic box, with at least 12 Å between the solute and the edge of the box. Periodic boundary conditions are employed, and electrostatic interactions are calculated with the PME algorithm34,35. We use a time step of 2 fs. Hydrogen atoms are constrained with the LINCS 36 algorithm while for water the Settle algorithm is used 37. The temperature of 310 K is kept constant by the Parrinello-Donadio-Bussi algorithm38(τ = 0.1 fs) which is similar to Berendsen coupling but adds a stochastic term that ensures a proper canonical ensemble38,39. In a similar way, the pressure is kept constant at 1 bar by the Parrinello-Rahman algorithm40 (τ = 1 fs). After energy-minimizing first the solvated start configuration using the steepest descent method, followed by conjugate gradient, the system is equilibrated in two steps of 500ps, first in an NVT ensemble and secondly in an NPT ensemble at 1 bar. After equilibrization, 200 ns of trajectories are analyzed for each system to monitor how the oligomer structures evolve with time. Data are saved at 4.0 ps intervals for further analysis. For each system (Table 1), we run three distinct simulations of 200 ns with different initial velocity distributions. This allows us to test that we reached equilibrium and guarantees three independent sets of measurements.
The molecular dynamics trajectories are analyzed with the tool set of the GROMACS package. Especially, we monitor conformational changes and stability of the oligomer models through the time evolution of root means square deviations of the Cα atoms (RMSD), root-mean-square fluctuation (RMSF), hydrophobic contacts distances and hydrogen bonds, measure with the g_hbond and g_dist modules in GROMACS. Hydrogen bonds are defined by a distance cut off between donor and acceptor of 0.36 nm and an angle cut off of 30°. Configurations are visualized using PyMOL41.
Result and discussion
The purpose of our simulations is to examine the stability of oligomers of amylin wild-type and single aromatic mutants in comparison with mixed decamers. The initial conformations and final structures obtained from the molecular dynamics simulations of the single layer aggregates11 are shown in Figure 2. We find by visual inspection that the β-strand-loop-β-strand topology of the decamers, the main structural feature of amyloidogenic fibrillar state, is maintained throughout the simulation (Figure 2). The structures from wild-type oligomer mixed with the mutants (the hetero-polymers WT-F15L, WT-F23L and WT-Y37L) exhibit the same shape and topology as the wild-type oligomer (WT) and mutants oligomer, pointing to structural similarity as major factor for the seeding observed in pervious experiments23.
Figure 2.
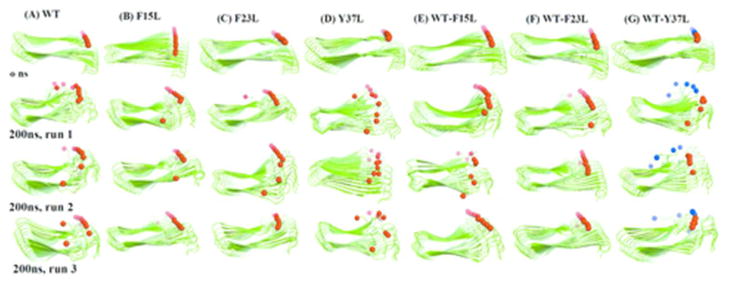
Snapshots of the amylin decamers of wild type, mutants and their seeded assembly before and after 200ns (water molecules are omitted for clarity; the snap shot are form three independent simulations). (A) Wild type (WT, red ball: Y37); (B) F15L (red ball: L37); (C) F23L (red ball: Y37); (D) Y37L (red ball: L37); (E) WT-F15L (red ball: Y37); (E) WT-F23L (red ball: L37); and (G) WT-Y37L (red ball: Y37 and blue ball: L37). The balls mark the residues 37 in the C-termini of each strand of the peptides.
In order to obtain a more detailed picture of the stability of the various amylin decamers we show in Figure 3A the Cα-RMSD as calculated from the molecular dynamic trajectory. The reference structure is the equilibrated start structure (i.e. the configuration at 0 ns). The faster this quantity grows throughout the simulation, the less stable and the more dynamic is a decamer. The average RMSD values, calculated over three independent trajectories of 200ns for each system, are within the range of 3.0 to 4.5 Å. These values are smaller than the RMSD value of more than 5.5 Å measured for the single layer amylin pentamer 42, and close to the 4.5 Å observed for double layer amylin decamer19. Thus the simulated oligomers appear to be stable, with no discernible difference in stability of the examined structures (Figure 3A).
Figure 3.

RMSD, Cα-Cα distances along strand 5-and 6 and hydrogen bonds of the studied oligomer systems. Average <RMSD> in Å (A); Average number of main chain hydrogen bonds (B); side chain hydrogen bonds (C); and Average <Cα-Cα distances>, in Å, between strands 5 and 6 (D).
We then examined the average inter-strand distance between stand 5 and strand 6 across the U shaped β-strand-loop-β-stand motif for the wild type, mutants and mixed oligomers (Figure 3D). This distance has been selected because in the mixed assemblies the two strands are located along the interface between wild type and mutant chain. This distance quantifies how close the two strands and therefore how strong and favorable the wild type and mutants are. On the other hand, an increase in the distance over that in the initial structures indicates detachment of the hetero-assembly system and unfavorable contacts between the peptides. The measured distances for all three cases, wild type, mutants and mixed oligomers are around ~5Å range, indicating that the intra-strand distance is close to the experimental inter strand distance of 4.8 Å for amyloid fibrils and oligomers43,14. Hence, in all three forms of oligomers the peptides interact strongly at this interface.
Protein aggregates are stabilizes through a network of side chain hydrophobic and hydrogen bonding interactions4,44. Main-chain (inter-backbone) hydrogen bonds link the β-strands within an amyloid fibril, while the side-chains hydrogen bonds modulate the intermolecular packing arrangement within and between the β-sheets of the fibril core45. For this reason, we have counted main-chain and side-chain hydrogen bonds during the simulations, and averaged the values over the three trajectories (Figure 3B and 3C). Again, we find little differences in the average side-chain and main chain hydrogen bond networks between wild type, mutants and mixed oligomers. On average, all decamer structures are stabilized by about 200 main chain and 50 side chain hydrogen bonds (Figure 3B and 3C).
An analysis of the root mean square fluctuation (RMSF), computed for the wild type, mutants and mixed decamers, shows that in all cases the residues in the turn region are more flexible than those in the β-strand regions. The exceptions are residues near the N-/C-termini that are exposed to the solvent (Figure 4). This is similar to pervious amylin simulations19. The RMSF describes where in the sequence of a protein structural stability is gained or lost, and therefore allows one to relate this change in stability to specific mutations. The smallest per residue fluctuation is observed in β1 region that includes residues L12ANFLV17 and residues F23GAIL27 of β2 region for all the systems studied indicating these regions are crucial for the stability of the oligomers. Phenylalanine to Leucine substitution at position 37 causes decreased stability at the C terminal (Figure 4), which may explain the experimentally observed longer lag time in the growth of this mutant. This can be seen by both visual inspection and by comparison of the RMSF values of each structure (Figure 4 and 2). This increased flexibility of the C terminal region may hinder the formation of contact between adjacent Y37 and between Y37 and F23 at the interface of the double layer oligomer during fibril elongation therefore contributing to the increased lag phase observed in experiments. The F15 is not involved in the sheet-to-sheet contact while F23 and Y37 are involved. The distance between the centers of mass of residues 37 of the C-terminal of the three different kinds of decamers change little during the 200 ns simulation from the initial distance (see Table 2). However, the distance between centers of mass of residues 37 of the C-terminal of amylin become within the range of 7–10 Å, therefore much larger than the value of about 4.8 Å of the initial conformation (i.e. at 0 ns of the simulation). Hence, mutation of Y37 to L results in the largest fluctuation in the C terminal of each monomer. This higher flexibility could slow down the elongation of the Y37L mutant and therefore contribute to the larger lag phase in its aggregation observed in a previous experimental study23. The simulation of decameric double layer wild-type amylin and its F23L and Y37L mutants (F23 and Y37 participate in sheet-to-sheet contacts between the β-sheets at the interface of the two U shaped oligomers, Figure 1C) shows no discernible difference in stability (Figure 5). The lack of susceptibility to destabilization of the initially preformed structure of the decamer double layer oligomers of both wild type and mutants suggests that the aromatic π-stacking interactions are not critical for aggregation stability as Leu is similar in size and hydrophobicity to F and Y but not capable of π-stacking. Note that our result does not exclude a role of the aromatic side-chain of F23 and Y37 for favoring aggregation nucleation, an effect which could contribute also to the experimentally observed differences in aggregation kinetics23. However, a combination of much longer simulation times and enhanced conformational techniques would be required to determine the equilibrium structures 46 of the wild type and mutants monomers and dimers, and to get an insight into the influence of π-stacking during the slow nucleation phase.
Figure 4.
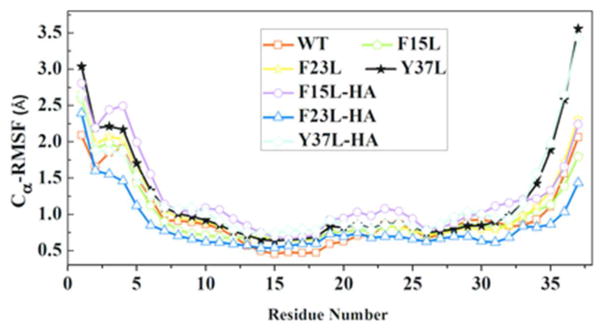
Average RMSF values for the 10 β-strands in single layer amylin, its mutants and cross-seeded aggregates. In the mixed aggregates the last five strands are from the wild type while the first five strands are from mutants. The results are calculated from three independent trajectories of 200 ns.
Table 2.
Center of mass distances between of residue 37 of the C-terminal of amylin, its mutants and the cross-seeded assemblies. The distances are measured between residue 37 of each strand and the adjacent successive one, excluding the less stable terminal chains 1 and 10.
| Y37/Y37, distance (Å) | WT oligomer | F15L oligomer | F23L oligomer | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | Run 1 | Run 2 | Run 3 | Run 1 | Run2 | Run 3 | |
| Chains-2_3 | 11.14(4.0) | 7.14(2.8) | 5.16(0.3) | 5.18(0.3) | 6.22(1.5) | 5.28(0.5) | 5.25(0.4) | 5.44(0.7) | 10.68(4.9) |
| Chains-3_4 | 6.02(2.5) | 5.13(0.3) | 5.06(0.2) | 5.05(0.2) | 5.14(0.3) | 5.03(0.3) | 5.14(0.3) | 5.14(0.3) | 5.14(0.3) |
| Chains-4_5 | 6.92(1.8) | 5.04(0.2) | 5.02(0.2) | 5.00(0.2) | 4.99(0.2) | 5.00(0.2) | 5.00(0.2) | 5.02(0.2) | 5.02(0.3) |
| Chains-5_6 | 5.14(0.4) | 5.02(0.3) | 5.00(0.3) | 4.95(0.2) | 4.95(0.2) | 4.99(0.2) | 4.95(0.2) | 4.97(0.2) | 4.99(0.2) |
| Chains-6_7 | 5.06(0.2) | 5.05(0.3) | 5.40(0.8) | 4.99(0.2) | 4.97(0.2) | 4.92(0.2) | 4.98(0.2) | 4.99(0.2) | 5.02(0.2) |
| Chains-7_8 | 5.01(0.1) | 5.19(0.4) | 8.70(4.0) | 5.06(0.2) | 5.03(0.2) | 4.93(0.2) | 4.98(0.2) | 5.55(1.4) | 5.05(0.2) |
| Chains-8_9 | 5.05(0.3) | 6.45(2.7) | 8.74(6.4) | 5.76(0.8) | 5.17(0.4) | 5.00(0.3) | 4.99(0.2) | 9.93(4.2) | 5.06(0.2) |
| Mean± SD | 6.33 ± 2.2 | 5.57± 0.8 | 6.15± 1.8 | 5.14 ±0.3 | 5.21 ± 0.4 | 5.02± 0.1 | 5.04 ±0.1 | 5.86 ±1.8 | 5.85 ±2.1 |
| L37/L37, distance (Å) | Y37L oligomer | Res37/Res37, distance (Å) | WT-F15L hetero assembly oligomer | ||||
|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run3 | Run 1 | Run 2 | Run 3 | ||
| Chains-2_3 | 10.35(1.1) | 13.01(2.4) | 10.67(3.2) | Chains-2_3 (WT) | 5.22(0.5) | 16.21(3.4) | 5.10(0.2) |
| Chains-3_4 | 6.08(0.5) | 7.73(0.8) | 6.54(1.1) | Chains-3_4 (WT) | 5.09(0.3) | 5.45(1.1) | 4.99(0.2) |
| Chains-4_5 | 7.25(0.7) | 6.58(1.0) | 7.79(1.6) | Chains-4_5 (WT) | 5.06(0.3) | 5.32(0.5) | 4.92(0.2) |
| Chains-5_6 | 6.51(0.9) | 5.58(0.3) | 8.98(3.2) | Chains-5_6(interface) | 5.47(0.3) | 5.58(0.5) | 4.88(0.2) |
| Chains-6_7 | 13.20(6.5) | 7.50(0.8) | 10.77(3.7) | Chains-6_7 (F15L) | 6.19(1.2) | 7.64(1.5) | 4.88(0.2) |
| Chains-7_8 | 17.89(6.5) | 5.24(0.3) | 12.17(6.2) | Chains-7_8 (F15L) | 6.30(1.7) | 11.62(3.0) | 4.98(0.2) |
| Chains-8_9 | 9.86(2.9) | 5.61(0.4) | 14.09(5.4) | Chains-8_9(F15L) | 5.35(0.4) | 10.66(2.0) | 5.09(0.2) |
| Mean± SD | 10.16± 4.2 | 7.3 ± 2.7 | 10.1 ± 2.6 | Mean ± SD | 5.52 ± 0.5 | 8.92 ± 4.1 | 4.98 ± 0.1 |
| Res37/Res37, distance (Å) | WT-F23L hetero assembly oligomer | Res37/Res37, distance (Å) | WT-Y37L hetero assembly oligomer | ||||
| Run 1 | Run 2 | Run 3 | Run 1 | Run 2 | Run 3 | ||
| Chains-2_3 (WT) | 5.16(0.3) | 5.36(0.6) | 5.11(0.3) | Chains-2_3 (WT) | 7.37(1.5) | 16.48(0.2) | 16.89(4.4) |
| Chains-3_4 (WT) | 5.02(0.2) | 5.04(0.2) | 5.00(0.2) | Chains-3_4 (WT) | 9.44(1.5) | 9.15(0.2) | 11.62(4.0) |
| Chains-4_5 (WT) | 4.93(0.2) | 4.94(0.2) | 4.97(0.2) | Chains-4_5 (WT) | 6.74(1.4) | 9.92(0.4) | 6.39(0.7) |
| Chains-5_6 (interface) | 4.94(0.2) | 4.90(0.2) | 4.91(0.2) | Chains-5_6(interface) | 7.92(0.2) | 9.51(0.2) | 7.57(0.9) |
| Chains-6_7 (F23L) | 5.00(0.2) | 4.98(0.2) | 4.97(0.2) | Chains-6_7 (Y37L) | 5.40(0.5) | 5.34(0.4) | 5.19(0.4) |
| Chains-7_8 (F23L) | 5.05(0.3) | 5.03(0.2) | 5.06(0.2) | Chains-7_8 (Y37L) | 5.55(0.5) | 5.19(0.3) | 5.02(0.2) |
| Chains-8_9(F23L) | 5.42(1.0) | 4.97(0.3) | 5.04(0.3) | Chains-8_9(Y37L) | 7.76(1.6) | 5.17(0.4) | 5.03(0.2) |
| Mean± SD | 5.07± 0.2 | 5.03 ±0.2 | 5.01± 0.1 | Mean ±SD | 7.17 ±1.4 | 8.68 ±4.1 | 8.24 ±4.5 |
Figure 5.

Cα root-mean-square deviations (RMSDs) with respect to the corresponding minimized start configurations of the average structures of (A) WT, (B) F23L and (C) Y37L. The average structures are calculated from the position of all heavy atoms of the protein over the 200 ns of each trajectory using the program g_covar of the Gromacs 4.5.5 package.
Face-to-face interactions between β-sheets are common in proteins and amyloids4. They involve hydrophobic surfaces with good shape complementarity held together through van der Waals and hydrophobic interactions4. For this reason, we have monitored also the face-to-face contacts between β-sheets (Figure 1B) for amylin wild type and its mutants, selecting residue pairs known to disrupt amylin amyloid formation47. These contacts are calculated also in the mixed aggregates. Results for all three cases are shown in Table 3. The side chains of L12, N14 and L16 (projecting from the lower face of the β1-region) in β1-region interact with the side chains of S28, I26 and V32 (projecting on the upper face of β2-region) in β2-region located on the opposite side of β1-region (Figure 1). The average face-to-face distances are measured for all ten strands of each system and are within 8.5 to 9.5 Å (Table 3) in agreement with experimental results 8–11Å48. Hence, in all oligomers, the β-strand-turn-β-strand motifs are stabilized by such face-to-face hydrophobic interactions. The face to face interactions between amino acid side chains (hydrogen bonds between aligned N14 with S28, or T30 residues; hydrophobic interactions between aligned L16, I26, L12, V32, or polar interaction between N14 and S28 residues) in β1 and β2 regions is important in keeping the U shaped structure. The cross-β-strand topology structure of the fibril, which is the main structural feature of amyloid fibril, is maintained throughout the simulation for the both hetero and homo-aggregates indicating stabilization by a network of hydrogen bond and hydrophobic interactions. The stability of the aggregates is a strong indicator of the role of structural similarity in explaining the efficient cross seeding23. Pinpointing the amino acids (the hydrophobic cores; L12ANFLV17 and F23GAIL27 and Y37 in our simulation) which can alter the stability of amylin could contribute to a rational design of aggregation inhibitors that trap the toxic species in the fibril form49,50. Our simulation indicates that the selected face-to-face contacts within the two β-sheet regions are preserved, indicating that such side chain contacts are important for retaining an overall U shaped structure. Interior contact distances are not significantly changed by the three substitutions. Future experiments and computer simulation involving single and double mutants of those amino acids mutations at residues 16, 26, 14, 28, 12 or 32 will elucidate the role of these residues in the stability of fibril structures.
Table 3.
Average inter-atomic distances between amino acids involved in a face-to-face interaction (L16-I26, N14-S28 and L12-V32), as calculated from the 200 ns trajectories. Results are listed for the single layer models of amylin wild type, mutants and cross-seeded decamers. The listed values are averages over three independent trajectories, s and are calculated using all ten strands.
| Average Cα -Cα distances | Runs | Models of amylin, mutants and hetero-assembly decameric oligomers | ||||||
|---|---|---|---|---|---|---|---|---|
| WT | F15L | F23L | Y37L | WT-F15L | WT-F23L | WT-Y37L | ||
| <L16-I26> | 1 | 9.21±0.6 | 9.14±0.3 | 9.03±0.3 | 9.51±1.0 | 9.12±0.4 | 9.09±0.3 | 9.09±0.4 |
| 2 | 9.43±0.8 | 9.34±0.8 | 9.08±0.4 | 9.10±0.5 | 9.49±1.2 | 9.38±0.9 | 9.56±1.1 | |
| 3 | 9.10±0.4 | 9.33±0.6 | 9.92±1.7 | 9.05±0.4 | 8.98±0.2 | 9.22±0.7 | 9.45±1.0 | |
| Mean±SD* | 9.25±0.2 | 9.27±0.1 | 9.34±0.5 | 9.22±0.2 | 9.20±0.3 | 9.23±0.1 | 9.37±0.2 | |
| <N14-S28> | 1 | 8.42±1.5 | 8.43±0.3 | 7.97±0.4 | 9.36±1.4 | 8.42±0.4 | 8.98±1.0 | 8.48±0.4 |
| 2 | 8.74±1.1 | 8.74±1.0 | 8.12±1.2 | 8.68±0.7 | 9.26±1.7 | 9.21±1.2 | 8.76±1.7 | |
| 3 | 8.44±1.2 | 8.89±0.9 | 9.44±1.7 | 8.33±0.4 | 7.67±0.4 | 9.19±0.9 | 9.18±1.7 | |
| Mean±SD* | 8.53±0.2 | 8.69±0.2 | 8.51±0.8 | 8.79±0.5 | 8.45±0.8 | 9.13±0.1 | 8.81±0.4 | |
| <L12-V32> | 1 | 9.12±0.4 | 9.53±0.2 | 8.87±0.4 | 8.89±0.3 | 8.75±0.3 | 8.80±0.4 | 9.23±0.1 |
| 2 | 8.98±0.3 | 8.76±0.4 | 9.27±0.4 | 8.90±0.5 | 9.11±0.3 | 8.75±0.3 | 9.18±0.5 | |
| 3 | 8.98±0.3 | 8.72±0.4 | 8.91±0.5 | 9.44±0.2 | 9.25±0.3 | 8.87±0.2 | 9.01±0.2 | |
| Mean±SD* | 9.02±0.1 | 9.00±0.5 | 9.02±0.2 | 9.08±0.3 | 9.04±0.3 | 8.81±0.1 | 9.14±0.1 | |
Mean values and standard deviation (SD) are calculated from the three values obtained by averaging over the 200 ns of each of the three independent runs of each model.
Using the DSSP51 software we have compared the changes in secondary structure encountered by wild types, mutants and their complex. Our analysis compares in each case the first and last 50 ns of the molecular dynamics trajectory. Our data listed in Table 4 indicate that the β-sheet content of the aggregates is maintained for wild type, mutants and their complex. This is another indication for the stability of the β-hair pin topology and provides additional evidence for the importance of fibril structural similarity in efficient seeding23.
Table 4.
Comparison of average percentage secondary structure contents of wild types, mutants and their complex from the first (0–50ns) and last (150–200 ns) 50 ns MD simulations.
| System | Secondary structure*, first 50 ns | Secondary structure, last 50 ns | ||||
|---|---|---|---|---|---|---|
| β-sheet | helix | turn | β-sheet | helix | turn | |
| WT (SL) | 60.24(1.21) | 0.04(0.02) | 39.73(1.20) | 56.11(0.50) | 0(0) | 43.89(0.50) |
| F15L (SL) | 62.09(2.27) | 0(0) | 37.91(2.28) | 59.44(1.80) | 0.04(0.07) | 40.53(1.82) |
| F23L (SL) | 61.80(1.94) | 0(0) | 38.20(1.94) | 59.97(2.99) | 0.02(0.05) | 42.34(1.17) |
| Y37L (SL) | 62.34(3.22) | 0(0) | 40.76(3.53) | 55.71(2.11) | 0.01(0.02) | 44.28(2.13) |
| WT- F15L*(SL) | 60.68(0.38) | 0(0) | 39.32(1.07) | 57.63(1.20) | 0.03(0.05) | 42.34(1.17) |
| WT- F23L*(SL) | 62.25(1.73) | 0(0) | 37.85(1.73) | 0.03(0.06) | 60.67(1.99) | 39.30(2.02) |
| WT- Y37L*(SL) | 59.03(1.04) | 0.09(0.15) | 40.88(0.96) | 56.11(0.50) | 0(0) | 43.89(0.50) |
| WT (DL) | 60.04(0.38) | 0(0) | 39.96(0.38) | 59.00(1.23) | 0(0) | 41.00(1.23) |
| F23L (DL) | 57.81(1.60) | 0.01(0.01) | 42.17(1.58) | 56.47(2.90) | 0.03(0.03) | 43.50(2.90) |
| Y37L (DL) | 61.80(2.54) | 0(0) | 38.20(2.54) | 57.87(4.43) | 42.12(4.42) | 0.01(0.01) |
β-sheet = β-strand + β-bridge, helix = α-helix +310-helix + π-helix, turn = turns + bend + coil. The results are the averages of three independent simulations and the standard deviation is given in parentheses.
Using single trajectory MM-PBSA52 we have also estimated the binding free energy for symmetrically segmented proto-filaments (single layer, between the first pentameric and the second pentameric units) and proto-filament pairs (double layer, between the upper pentameric and the lower pentameric β-hairpin units). This allows us to characterize favorable association between wild type and different mutants and related it to the experimentally observed efficient seeding between mutant and wild type amylin23. We have taken for this analysis an average over 2000 equally spaced (at an interval of 20 ps) snapshots from the 40ns production trajectory. Assuming equal entropic factors for the oligomers, their binding will be the stronger the larger their negative free energy, whereas a positive number or small negative number signifies a weaker binding44,53,54. The calculated binding free energies and energy components are shown in Table 5. The results show that the binding affinity of the hetero-assembly single layer complexes is comparable to the homogenous fibrils. However, the binding energy of the double layer assembly of the single aromatic amino acid mutants is less favorable than that of the wild type (Table 5) suggesting contribution of aromatic interactions to the thermodynamic stability. The polar energy term is unfavorable for all the aggregates while the nonpolar energy term contributes favorably to their association and stability. Note that the binding energy addresses only the stability of the preformed aggregates, it does not describe adequately the initial interaction between the chains (i.e the kinetic process). Hence, it is difficult to compare the values calculated by MMPBSA with experimental55 rates of aggregate formation and lag times.
Table 5.
Binding free energies and different Components of the binding free energy using the MM PBSA approximations for interaction within symmetrically segmented proto-filaments (single layer, between the first pentameric and the second pentameric units) and proto-filament pairs (double layer, between the upper and the lower β-hairpin units)
| Energy components | WT (SL) | F15L (SL) | F23L (SL) | Y37L (SL) | WT- F15L (SL) | WT- F23L (SL) | WT- Y37L (SL) | WT (DL) | F23L (DL) | Y37L (DL) |
|---|---|---|---|---|---|---|---|---|---|---|
| Δ Eelec | 1038.7±182.0 | 1055.1±94.9 | 1006.1±38.2 | 1047.9±146.2 | 1148.5±118.5.4 | 960.9±9.9 | 1057.5±162.8 | 453.7±20.0 | 570.2±12.0 | 482.8±25.4 |
| Δ Evdw | −188.9±6.1 | −187.3±2.6 | −187.5±11.1 | −187.0±4.5 | −190.8±5.0 | −186.7±6.2 | −190.9±0.1 | −206.5±17.7 | −98.8±12.0 | −211.8±3.7 |
| Δ EPB | −1004.7±179.0 | −1008.3±95.0 | −969.1±26.2 | −1007.7±145.0 | −1104.1±117.0 | −921.5±5.4 | −1020.7±159.1 | −395.5±24.9 | −531.4±18.6 | −397.4±14.8 |
| Δ ESA | 108.3±0.8 | 108.9±0.1 | 108.3±6.4 | 107.1±2.1 | 109.1±3.2 | −78.5±3.4 | 108.3±0.1 | 120.9±0.5 | 58.5±7.3 | 126.7±2.6 |
| Δ Epolar | 34.4±2.7 | 46.9±0.1 | 37.1±12.0 | 40.2±1.3 | 44.4±1.5 | 39.4±4.4 | 36.7±3.7 | 58.2±4.7 | 38.8±6.6 | 85.4±10.6 |
| Δ Enonpolar | −118.3±49.9 | −109.4±41.3 | −79.2±4.6 | −80.1±2.4 | −146.4±6.6 | −78.5±3.4 | −82.5±0.3 | −85.6±17.2 | −38.4±6.7 | −85.2±6.4 |
| Δ Gbinding | −46.3±2.6 | −31.6±2.6 | −42.1±7.4 | −39.9±1.1 | −37.3±3.3 | −39.1±7.8 | −45.8±3.4 | −27.5±12.5 | 0.4±0.2 | 0.2±4.2 |
The data are averages of two independent 40 ns simulation with the corresponding standard deviations. All values are in kcal/mol. The polar term is the sum of Coulomb interaction energy (Eelec) and polar contribution to the solvation free energy (EPB). The nonpolar term consists of takes the van der Waals interaction energies (EvdW) and the nonpolar contribution to the solvation free energy (ESA)
The commonly accepted mechanism for their toxicity is that amyloids oligomers interact with cell membranes compromising their structural integrity and lowering their permeability barrier56. This ability of amyloid oligomers to form membrane pores or channels is then responsible for their neurotoxicity57. Recent structural studies of the toxic oligomer models suggest that the hydrophobic surface facilitates insertion into the membrane while water pores formed from the hydrophilic groups interferes with cellular homeostatic by enabling water and ion transport 58. The hydrophobic surfaces (L12ANFLV17 and F23GAIL27) of the fibril like oligomer assembly of amylin may provide a means by which these structures can insert into lipid bilayer membranes, while the hydrophilic water channel observed in a recent simulation could be involved with toxicity of amylin oligomer. Studies have shown that amyloid aggregates in lipid bilayers adopt structures similar to that in an aqueous medium59,60,61. The experimentally determined structures of amyloid fibril 18,18 (including amylin) do not contain water molecules while various molecular dynamic simulations53,62 revealed embedded water molecules are integral part of the fibril models. Thus, we have examined the presence of the water trapped in amylin wild-type, mutants and cross-assembly pores. The results are shown in Figure 6 and Figure 7. The water molecules in amylin, its mutants and their hetero-assembly are found in the middle of the two β-stands near to a group of polar amino acids whose side chains points toward the interior of the oligomer cavity (N14, S28 and T30). The location of the hydration channel in our simulation is similar to that found in a previous study of amylin alone and in hetero-assembly with Aβ53.
Figure 6.

Average number of water molecules inside the hydrophilic cavities. Mean values and standard deviation (SD) are calculated for each model from the three values obtained by averaging each independent run over the full 200 ns.
Figure 7.

Water residing in the hydrophilic cavities. Snapshots taken after 10 ns for (a) Wild type (WT); (b) Y37L; (c) WT-Y37L; (d) F15L; (e) F23L; (f) WT-F15L; (g) WT-F23L. The red and white spheres represent water oxygen and hydrogen atoms, respectively.
Conclusion
We investigate stability and conformational changes of amyloid hetero-assemblies through molecular dynamics simulations as the structure and stability of hetero-polymeric fibrils are difficult to probe in experiments. Our all atom explicit solvent simulations give molecular level insight into the cross seeding between amyloids. We find no significant differences in the structure of the wild type, mutants and their hetero-assembly as all of them retain the original U-shaped fibril conformation over the 200ns time trajectories. Hence, amyloids with similar side chains packing at the β-sheet interface are structurally compatible, acting as a good template for the congruent incorporation of homologue peptides, which underlie efficient mixed growth. This points to structure similarity as key determinant for an efficient cross seeding between wild type and mutants. Replacement of aromatic amino acids with non-aromatic residues of similar size and hydrophobicity is not critical at position 15, which is not involved in any inter-sheet steric zipper interaction. On the other hand, the replacement of tyrosine with leucine at position 37 leads to a fibril structure with a greater flexibility resulting in a loss of steric zipper interaction. This loss of a steric zipper explains the slow growth of the aggregates of Y37L mutants compared to other mutants and wild type. Our results indicate that the residues L12ANFLV17 and F23GAIL27 which are located in the β-strand domain are more rigid and could be a pharmacophore for ligand binding,63,24 targeted to stabilize amylin fibril and thereby reducing its toxicity. We therefore propose to use these two segments of amylin for combined computational screening and experimental tests to find small molecules that effects the aggregation and toxicity of amylin. Jiang et al64 has recently found this approach to be successful in searching for compounds that reduced Aβ cytotoxicity.
Acknowledgments
This work is supported by the National Institutes of Health under Grant No. GM62838 and by the National Science Foundation under Grant No. EPS-0814361. This research used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Other parts of the simulations were done on the BOOMER cluster of the University of Oklahoma. Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation, the National Institutes of Health, or the Department of Energy.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Buxbaum JN, Linke RP. A Molecular History of the Amyloidoses. J Mol Biol. 2012;421:142–159. doi: 10.1016/j.jmb.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 3.Eisenberg D, Jucker M. The Amyloid State of Proteins in Human Diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng PN, Pham JD, Nowick JS. The Supramolecular Chemistry of beta-Sheets. J Am Chem Soc. 2013;135:5477–5492. doi: 10.1021/ja3088407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harper JD, Lansbury PT. Models of amyloid seeding in Alzheimier’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 6.Eisele YS. From Soluble A beta to Progressive A beta Aggregation: Could Prion-Like Templated Misfolding Play a Role? Brain Pathol. 2013;23:333–341. doi: 10.1111/bpa.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F. Amylin deposition in the brain: a second amyloid in Alzheimer’s disease? Ann Neurol. doi: 10.1002/ana.23956. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarrett JT, Lansbury PT. Seeding one-dimensional crystallization of amyloid - a pathogenic mechanism in Alzheimers-disease and Scrapie. Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 9.Profit AA, Felsen V, Chinwong J, Mojica ERE, Desamero RZB. Evidence of pi-stacking interactions in the self-assembly of hIAPP(22–29) Proteins. 2013;81:690–703. doi: 10.1002/prot.24229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doran TM, Kamens AJ, Byrnes NK, Nilsson BL. Role of amino acid hydrophobicity, aromaticity, and molecular volume on IAPP(20–29) amyloid self-assembly. Proteins. 2012;80:1053–1065. doi: 10.1002/prot.24007. [DOI] [PubMed] [Google Scholar]
- 11.Berhanu WM, Hansmann UHE. Side-chain hydrophobicity and the stability of Ab16–22 aggregates. Protein Sci. 2012;21:1837–1848. doi: 10.1002/pro.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weise K, Radovan D, Gohlke A, Opitz N, Winter R. Interaction of hIAPP with Model Raft Membranes and Pancreatic beta-Cells: Cytotoxicity of hIAPP Oligomers. ChemBioChem. 2010;11:1280–1290. doi: 10.1002/cbic.201000039. [DOI] [PubMed] [Google Scholar]
- 13.Sciacca MFM, Milardi D, Messina GML, Marletta G, Brender JR, Ramamoorthy A, La Rosa C. Cations as Switches of Amyloid-Mediated Membrane Disruption Mechanisms: Calcium and IAPP. Biophys J. 2013;104:173–184. doi: 10.1016/j.bpj.2012.11.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiltzius JJW, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, Eisenberg D. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedrood S, Li YY, Isas JM, Hegde BG, Baxa U, Haworth IS, Langen R. Fibril Structure of Human Islet Amyloid Polypeptide. J Biol Chem. 2012;287:5235–5241. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen JT, Bjerring M, Jeppesen MD, Pedersen RO, Pedersen JM, Hein KL, Vosegaard T, Skrydstrup T, Otzen DE, Nielsen NC. Unique Identification of Supramolecular Structures in Amyloid Fibrils by Solid-State NMR Spectroscopy. Angew Chem-Int Edit. 2009;48:2118–2121. doi: 10.1002/anie.200804198. [DOI] [PubMed] [Google Scholar]
- 17.Madine J, Jack E, Stockley PG, Radford SE, Serpell LC, Middleton DA. Structural Insights into the Polymorphism of Amyloid-Like Fibrils Formed by Region 20–29 of Amylin Revealed by Solid-State NMR and X-ray Fiber Diffraction. J Am Chem Soc. 2008;130:14990–15001. doi: 10.1021/ja802483d. [DOI] [PubMed] [Google Scholar]
- 18.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu WX, Su HB, Zhang JZH, Mu YG. Molecular Dynamics Simulation Study on the Molecular Structures of the Amylin Fibril Models. J Phys Chem B. 2012;116:13991–13999. doi: 10.1021/jp308708h. [DOI] [PubMed] [Google Scholar]
- 20.Zhao J, Yu XA, Liang GZ, Zheng J. Structural Polymorphism of Human Islet Amyloid Polypeptide (hIAPP) Oligomers Highlights the Importance of Interfacial Residue Interactions. Biomacromolecules. 2011;12:210–220. doi: 10.1021/bm101159p. [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Yu X, Liang GZ, Zheng J. Heterogeneous Triangular Structures of Human Islet Amyloid Polypeptide (Amylin) with Internal Hydrophobic Cavity and External Wrapping Morphology Reveal the Polymorphic Nature of Amyloid Fibrils. Biomacromolecules. 2011;12:1781–1794. doi: 10.1021/bm2001507. [DOI] [PubMed] [Google Scholar]
- 22.Berhanu WM, Masunov AE. Full length amylin oligomer aggregation: insights from molecular dynamics simulations and implications for design of aggregation inhibitors. J Biomol Struct Dyn. doi: 10.1080/07391102.2013.832635. in press. [DOI] [PubMed] [Google Scholar]
- 23.Tu LH, Raleigh DP. Role of Aromatic Interactions in Amyloid Formation by Islet Amyloid Polypeptide. Biochemistry. 2013;52:333–342. doi: 10.1021/bi3014278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang L, Liu C, Leibly D, Landau M, Zhao M, Hughes MP, DSE Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. Elife. 2013;2:e00857. doi: 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahler A, Sticht H, AHCH Conformational Stability of Fibrillar Amyloid-Beta Oligomers via Protofilament Pair Formation-A Systematic Computational Study. PLoS ONE. 2013;8:e70521. doi: 10.1371/journal.pone.0070521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Autieroa I, Saviano M, Langella E. In silico investigation and targeting of amyloid β oligomers of different size. Mol BioSyst. 2013;9:2118–2124. doi: 10.1039/c3mb70086k. [DOI] [PubMed] [Google Scholar]
- 27.Berhanu WM, Masunov AE. Alternative packing modes as basis for amyloid polymorphismin five fragments. Peptide Science. 2012;98:131–144. doi: 10.1002/bip.21731. [DOI] [PubMed] [Google Scholar]
- 28.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zachariae U, Schneider R, Briones R, Gattin Z, Demers JP, Giller K, Maier E, Zweckstetter M, Griesinger C, Becker S, Benz R, de Groot BL, Lange A. beta-Barrel Mobility Underlies Closure of the Voltage-Dependent Anion Channel. Structure. 2012;20:1540–1549. doi: 10.1016/j.str.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutzner C, Grubmuller H, de Groot BL, Zachariae U. Computational Electrophysiology: The Molecular Dynamics of Ion Channel Permeation and Selectivity in Atomistic Detail. Biophys J. 2011;101:809–817. doi: 10.1016/j.bpj.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berhanu Workalemahu M, Hansmann UHE. Structure and Dynamics of Amyloid-b Segmental Polymorphisms. PLoS ONE. 2012;7:e41479. doi: 10.1371/journal.pone.0041479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ndlovu H, Ashcroft AE, Radford SE, Harris SA. Effect of Sequence Variation on the Mechanical Response of Amyloid Fibrils Probed by Steered Molecular Dynamics Simulation. Biophys J. 2012;102:587–596. doi: 10.1016/j.bpj.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pronk SPS, Roland Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts Michael R, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darden T, York D, Pedersen L. Particle mesh ewald - an n. log(n) method for ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 35.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 36.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto S, Kollman PA. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 38.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 39.Bussi G, Zykova-Timan T, Parrinello M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J Chem Phys. 2009;130:074101. doi: 10.1063/1.3073889. [DOI] [PubMed] [Google Scholar]
- 40.Parrinello M, Rahman A. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 41.DeLano WL. PyMOL molecular graphics system Version 1.3.0.4. Schrödinger, LLC; 2002. [Google Scholar]
- 42.Liang GZ, Zhao J, Yu X, Zheng J. Comparative Molecular Dynamics Study of Human Islet Amyloid Polypeptide (IAPP) and Rat IAPP Oligomers. Biochemistry. 2013;52:1089–1100. doi: 10.1021/bi301525e. [DOI] [PubMed] [Google Scholar]
- 43.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berhanu Workalemahu M, Hansmann UHE. The stability of cylindrin β-barrel amyloid oligomer models-A molecular dynamics study. Proteins. 2013;81:1542–1555. doi: 10.1002/prot.24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang LC, Ladizhansky V, Muller SA, et al. Atomic structure and hierarchical assembly of a cross-beta amyloid fibril. Proc Natl Acad Sci U S A. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C, Shea JE. Structural Similarities and Differences between Amyloidogenic and Non-Amyloidogenic Islet Amyloid Polypeptide (IAPP) Sequences and Implications for the Dual Physiological and Pathological Activities of These Peptides. PLoS Comput Biol. 2013;9:e1003211–1003223. doi: 10.1371/journal.pcbi.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao P, Marek P, Noor H, Patsalo V, Tu LH, Wang H, Abedini A, Raleigh DP. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. Febs Letters. 2013;587:1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 49.Shahnawaz M, Soto C. Microcin Amyloid Fibrils A Are Reservoir of Toxic Oligomeric Species. J Biol Chem. 2012;287:11665–11676. doi: 10.1074/jbc.M111.282533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kabsch W, Sander C. Dictionary Of Protein Secondary Structure - Pattern-Recognition Of Hydrogen-Bonded And Geometrical Features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 52.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 53.Berhanu W, Fatih Y, Hansmann U. In Silico cross seeding of Aβ and amylin fibril-like oligomers. ACS neuroscience. doi: 10.1021/cn400141x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park J, Kahng B, Hwang W. Thermodynamic Selection of Steric Zipper Patterns in the Amyloid Cross-beta Spine. PLoS Comput Biol. 2009;5(9):e1000492. doi: 10.1371/journal.pcbi.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sorensen J, Palmer DS, Schiott B. Hot-Spot Mapping of the Interactions between Chymosin and Bovine kappa-Casein. J Agric Food Chem. 2013;61:7949–7959. doi: 10.1021/jf4021043. [DOI] [PubMed] [Google Scholar]
- 56.Butterfield SM, Lashuel HA. Amyloidogenic Protein Membrane Interactions: Mechanistic Insight from Model Systems. Angew Chem-Int Edit. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 57.Lasagna-Reeves CA, Glabe CG, Kayed R. Amyloid-beta Annular Protofibrils Evade Fibrillar Fate in Alzheimer Disease Brain. J Biol Chem. 2011;286:22122–22130. doi: 10.1074/jbc.M111.236257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pham JD, Chim N, Goulding CW, Nowick JS. Structures of Oligomers of a Peptide from β-Amyloid. J Am Chem Soc. 2013;135:12460–12467. doi: 10.1021/ja4068854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nanga RPR, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jang H, Connelly L, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. Mechanisms for the Insertion of Toxic, Fibril-like beta-Amyloid Oligomers into the Membrane. J Chem Theory Comput. 2013;9:822–833. doi: 10.1021/ct300916f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stroud JC, Liu C, Teng PK, Eisenberg D. Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc Natl Acad Sci U S A. 2012;109:7717–7722. doi: 10.1073/pnas.1203193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.GhattyVenkataKrishna PK, Mostofian B. Dynamics of water in the amphiphilic pore of amyloid β fibrils. Chem Phys Lett. 2013;582:1–5. [Google Scholar]
- 63.Skeby Katrine Kirkeby, Sørensen Jesper, Schiøtt aB. Identification of a Common Binding Mode for Imaging Agents to Amyloid Fibrils from Molecular Dynamics Simulations. J Am Chem Soc. 2013;135:15114–15128. doi: 10.1021/ja405530p. [DOI] [PubMed] [Google Scholar]
- 64.Jiang L, Liu C, Leibly D, Landau M, Zhao M, Hughes MP, DSE Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. Elife. 2013;16:e00857. doi: 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]