

Abstract
The blood–brain barrier (BBB) is a dynamic system which maintains brain homeostasis and limits CNS penetration via interactions of transmembrane and intracellular proteins. Inflammatory pain (IP) is a condition underlying several diseases with known BBB perturbations, including stroke, Parkinson’s, multiple sclerosis and Alzheimer’s. Exploring the underlying pathology of chronic IP, we demonstrated alterations in BBB paracellular permeability with correlating changes in tight junction (TJ) proteins: occludin and claudin-5. The present study examines the IP-induced molecular changes leading to a loss in functional BBB integrity. IP was induced by injection of Complete Freund’s Adjuvant (CFA) into the plantar surface of the right hindpaw of female Sprague–Dawley rats. Inflammation and hyperalgesia were confirmed, and BBB paracellular permeability was assessed by in situ brain perfusion of [14C]sucrose (paracellular diffusion marker). The permeability of the BBB was significantly increased at 24 and 72 h post-CFA. Analysis of the TJ proteins, which control the paracellular pathway, demonstrated decreased claudin-5 expression at 24 h, and an increase at 48 and 72 h post-injection. Occludin expression was significantly decreased 72 h post-CFA. Expression of junction adhesion molecule-1 (JAM-1) increased 48 h and decreased by 72 h post-CFA. Confocal microscopy demonstrated continuous expression of both occludin and JAM-1, each co-localizing with ZO-1. The increased claudin-5 expression was not limited to the junction. These results provide evidence that chronic IP causes dramatic alterations in specific cytoarchitectural proteins and demonstrate alterations in molecular properties during CFA, resulting in significant changes in BBB paracellular permeability.
Keywords: Blood-brain barrier, Tight junction, Occludin, Claudin-5, Inflammatory pain, Complete Freund’s adjuvant
1. Introduction
More than 85 million people in the United States suffer from chronic pain, costing billions of dollars yearly in medical treatments, productivity losses, legal fees and compensation. Inflammation is recognized not only as a burden to the health of the U.S. population, but also as an underlying basis of many diseases. Inflammatory pain constitutes a major component in multiple sclerosis (MS), Alzheimer’s dementia, meningitis, systemic lupus erythematosis, arthritis (both rheumatoid and osteo), Crohn’s disease, irritable bowel syndrome, diabetic neuropathy and several types of cancer (Edwards, 2005; Matter et al., 2005; Sharma et al., 2004; Wieseler-Frank et al., 2005; Wolka et al., 2003). Therapeutic agents used to treat these conditions often need to cross an endothelial or epithelial barrier. In particular, many of these drugs have sites of action within the central nervous system (CNS). The CNS, however, is protected by the presence of an endothelial barrier – the blood–brain barrier (BBB) – and therapeutic agents have limited entry, and thus limited efficacy, for treatment of these disease states.
The BBB is a metabolic and physical barrier that is strictly regulated to maintain separation of the CNS from systemic circulation. Although previously thought to be a static barrier, the BBB is a dynamic structure capable of rapid modulation to maintain homoeostasis within the CNS (Abbott, 2005). Tight regulation of the BBB is required to allow nutrient and oxygen passage, while preventing access to circulating toxins, ions, amino acids and xenobiotics. The BBB endothelial cells are characterized by a lack of fenestrations, decreased pinocytosis and the presence of tight junctional proteins, multiple transport systems and enzymatic detoxification enzymes (Hawkins and Davis, 2005; Loscher and Potschka, 2005). Each of these characteristics serves to maintain the homeostatic environment of the CNS, as well as limit the entry of many therapeutic substances into the brain (Pardridge, 1997).
Tight junctions (TJs) of endothelial cells within the brain govern the paracellular route of entry into the brain. TJs span the apical paracellular cleft, tightly linking neighboring endothelial cells together, and their presence serves to limit paracellular mediated distribution (Gonzalez-Mariscal and Nava, 2005). The TJ is formed through complex interactions of numerous transmembrane, accessory and cytoskeletal proteins. The primary seal of the TJs is formed by the transmembrane proteins occludin, JAM-1 and the claudins, with the accessory protein zonula occludens-1 (ZO-1) interacting with their C-termini, linking them to the actin cytoskeleton (Farshori and Kachar, 1999; Mitic et al., 2000). The TJs are dynamic structures modulated by physiological and pathological conditions (Hawkins and Davis, 2005; Wolka et al., 2003). Changes in expression and intracellular localization of the TJ proteins are associated with alterations in paracellular transport (Balda et al., 2000; Coyne et al., 2003; Stamatovic et al., 2005).
The contribution of individual TJ proteins on the development or progression of neurological conditions isvaried. In some cases, TJ alterations and subsequent increased BBB permeability are an effect of the underlying pathology; alternatively, it is a causative and mediating event in disease development. For example, TJ disruptions and subsequent BBB perturbations are involved in the development of MS (Kirk et al., 2003; Neuwelt, 2004), while ischemic stroke and traumatic brain injury lead to BBB perturbations (Ilzecka, 1996; Morganti-Kossmann et al., 2001). There are many diseases, such as Alzheimer’s disease, where the direct correlation is not yet known, but are currently being investigated (Wardlaw et al., 2003).
We have previously reported a compromise in BBB integrity and paracellular permeability in three different inflammatory hyperalgesic (pain) models-formalin, carrageenan and complete Freund’s adjuvant (CFA). Significant increases in uptake of the normally impermeable marker [14C]sucrose, as well as [3H]codeine, were observed. In addition to increased codeine transport into the brain, increased antinociceptive efficacy was noted. These changes in barrier function correlated with alterations in TJ proteins (Brooks et al., 2005; Hau et al., 2004; Huber et al., 2001, 2002a, b).
Chronic inflammatory hyperalgesia induced by CFA for 72 h was previously demonstrated to alter the expression of several transmembrane TJ proteins (Brooks et al., 2005). We observed a decrease in occludin expression and increased expression of the claudin proteins following CFA injection. Moreover, an increase in paracellular permeability of [14C]sucrose was demonstrated via multi-time uptake studies. There were no changes in either mean blood pressures or initial volume of distribution noted. This indicates an increase in [14C]sucrose paracellular diffusion. With hyperalgesia established for 3 days, alterations in BBB characteristics were apparent.
In the present study, we expand upon these findings by examining the functional and molecular integrity of the BBB at earlier time points than the previous report. We sought to do this in order to understand the manner and extent of molecular cytoarchitectural changes which may lead to alterations in BBB functional integrity. In particular, we examine paracellular permeability across the BBB at time points leading up to the previously demonstrated change at 72 h. Also, we describe the expression and co-localization of integral TJ proteins. In addition to the previously studied ZO-1, occludin and claudin-5 (Brooks et al., 2005), we have assessed JAM-1, a key transmembrane TJ protein which is related to immune modulation of the BBB (Mandell and Parkos, 2005). Each of these TJ proteins is integral to membrane function, and alterations in any of them may lead to a perturbation of barrier integrity (Balda et al., 2000; Coyne et al., 2003; Stamatovic et al., 2005). Understanding the molecular mechanisms that lead to alterations in brain permeability may lead to new drug delivery strategies for neuropharmaceuticals.
2. Results
2.1. Edema formation and thermal hyperalgesia
In order to confirm CFA injection and the onset of inflammatory hyperalgesia, both hindpaw volume and thermal sensitivity were measured from 0 to 72 h. Measurements were taken for both saline and CFA treated rats, in both the injected and the contralateral hindpaw. As reported previously, volume was not changed by saline over the 72 h time course in either the injected or the contralateral hindpaws (Brookset al., 2005). Similarly, the contralateral hindpaw of the CFA injected rats did not change from 0–72 h. However, a significant change in volume was noted in the CFA injected hindpaws, with the average volume increasing from a baseline size of 1.4±0.02 to 2.4±0.1, 2.1±0.1 and 2.1±0.1 mL at 24, 48 and 72 h, respectively (p<0.05).
Similar to edema formation, there was no change in thermal hyperalgesia for either the injected or the contralateral hindpaw following saline injection. The contralateral hindpaw of the CFA treated animal was also not altered. A significant onset of hyperalgesia, however, was noted in the CFA injected hindpaws where the baseline paw removal latency of 9.9±0.3 s was decreased to 3.1±0.4, 4.6±1.3 and 5.1±0.3 s at 24, 48 and 72 h post-injection, respectively (p<0.05).
2.2. Packed RBC, leukocyte counts and differentiation
From 0–72 h post-injection, whole blood was collected from saline and CFA treated rats in order to monitor the development of an immune response. Hematocrit analysis demonstrated no change in packed red blood cells (RBC), and total white blood cell (WBC) counts were not increased by CFA (Table 1). CFA treated rats demonstrated a leukocyte population shift from lymphocytes to neutrophils over 0–72 h (Figs. 1A–B). As compared to saline control animals at the same time points, the neutrophil population increased from 13.7±0.8 to 39.4± 6.6, from 9.7±3.0 to 28.7±3.1 and from 8.5±0.8 to 18.6±3.9% at 24, 48 and 72 h, respectively (Fig. 1A; p<0.05). Concurrently, there was a decrease in the percent population of lymphocytes, as compared to time-matched saline controls, from 84.5±1.4 to 60.1±6.6, from 88.1±2.8 to 70.1±2.9 and from 9.1±0.8 to 79.3±3.7%, respectively, at 24, 48 and 72 h post-CFA (Fig. 1B; p<0.05).
Table 1.
Whole blood counts following CFA-induced inflammatory pain
| 24 h
|
48 h
|
72 h
|
||||
|---|---|---|---|---|---|---|
| Sal | CFA | Sal | CFA | Sal | CFA | |
| Hematocrit | 41.6±0.6 | 41.1±1.3 | 41.3±0.6 | 40.5±0.7 | 37.1±3.0 | 40.4±2.1 |
| WBC (×)103/mm3 | 5.96±1.1 | 7.86±0.4 | 7.75±0.9 | 6.43±1.3 | 7.19±0.0 | 6.18±0.8 |
Values are expressed as means±SE; n=2–6 per time point. Results indicate that neither packed red blood cells (RBC) nor WBC counts change as a function of time following CFA treatment.
Fig. 1.
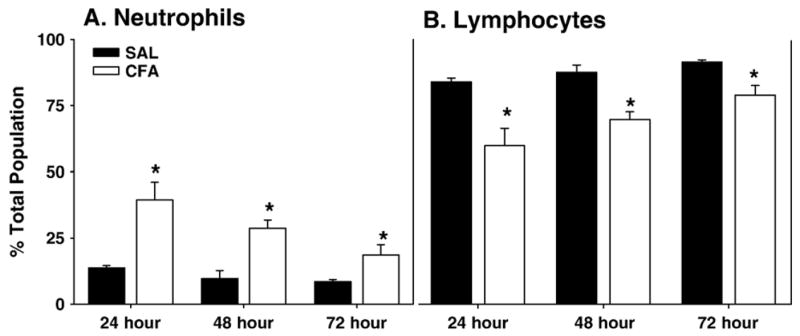
WBC differentiation following CFA-induced inflammatory pain. Percent of overall population is expressed for the differentiated white blood cells (WBC). (A) Neutrophils and (B) lymphocytes following 24–72 h of saline (filled bars) or CFA (open bars) treatment. CFA caused a significant (*p<0.05) increase in neutrophils and a concomitant decrease in lymphocytes from 24–72 h. Data are expressed as mean±standard error, n=4–8 per group.
2.3. In situ brain perfusions
Functional integrity of the BBB within the cerebrum was assessed by 20 min brain perfusions with [14C]sucrose (a marker of paracellular transport and BBB integrity). Visualization of the whole brain immediately following the perfusion indicated no leakage of Evans Blue Albumin, a marker for parenchymal leakage, indicating no gross or focal damage to the barrier by the surgical procedure. A significant increase (22%) in paracellular permeability of [14C]sucrose in the cerebrum was noted following 24 h of CFA treatment (Fig. 2, p<0.05). This increase was not evident at 48 h post-CFA treatment. However, a large and significant increase in paracellular permeability was noted at 72 h post-CFA injection when %Rbr increased by 81% in CFA treated rats (Fig. 2, p<0.001).
Fig. 2.
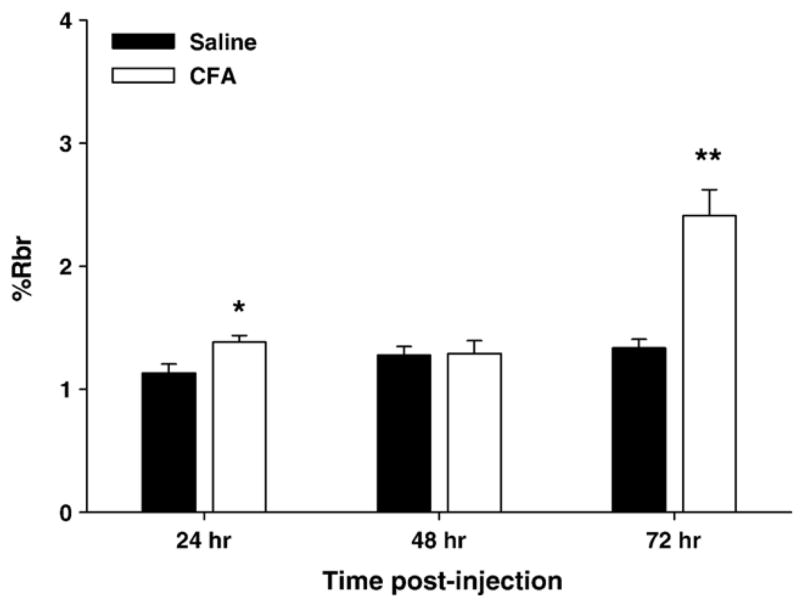
CFA-induced changes in [14C]sucrose brain uptake. [14C]sucrose was used as a marker of BBB paracellular transport in the cerebrum of saline (filled bars) and CFA (open bars) treated animals at 24, 48 and 72 h. Mean %Rbr ±standard error is represented for each group; n=4–8. CFA significantly altered the cerebral paracellular permeability at 24 and 72 h, but not at 48 h. *p<0.05; **p<0.005.
2.4. TJ protein expression and localization
Following the noted changes in [14C]sucrose paracellular permeability across the BBB, studies of the TJ proteins ZO-1, occludin, JAM-1 and claudin-5 were performed from isolated microvessels. ZO-1 was used as the standard protein, representative of the junction (Fig. 3), for confocal co-localization studies with the transmembrane TJ proteins occludin, JAM and claudin-5, as no changes were noted with ZO-1 protein expression, or localization in the cell (Figs. 4, 5) from 0 to 72 h following CFA treatment. Western blots of all naïve and saline controls (over time) demonstrated no change in protein expression (data not shown). Confocal micrographs confirm no effect on expression or localization of these specific TJ proteins with saline injection, and as compared to naïve animals (Fig. 5).
Fig. 3.
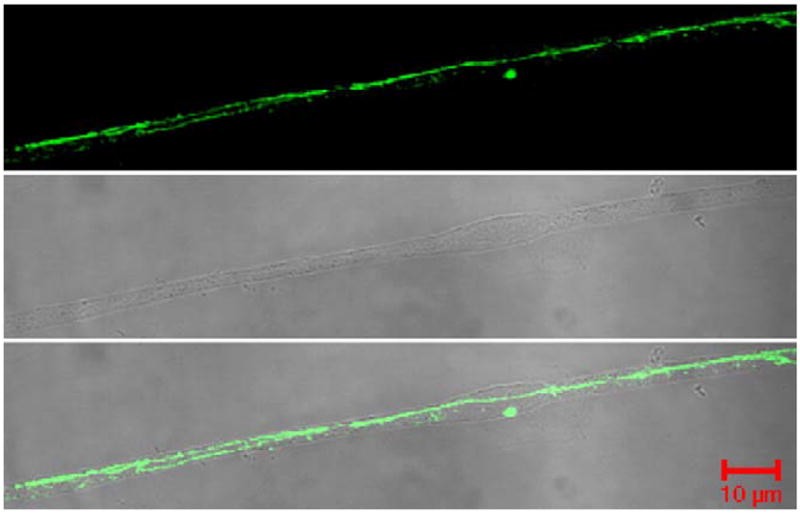
ZO-1 localization within the microvessel. Confocal micrographs reflect ZO-1 protein localization as continuous and junctional within the TJ. Phase contrast micrographs show the microvessel purity, and the merged micrograph demonstrates ZO-1 expression only within the microvessel. Micrographs were taken with an LSM 510 confocal laser scanning microscope at 63× with an oil immersion lens.
Fig. 4.
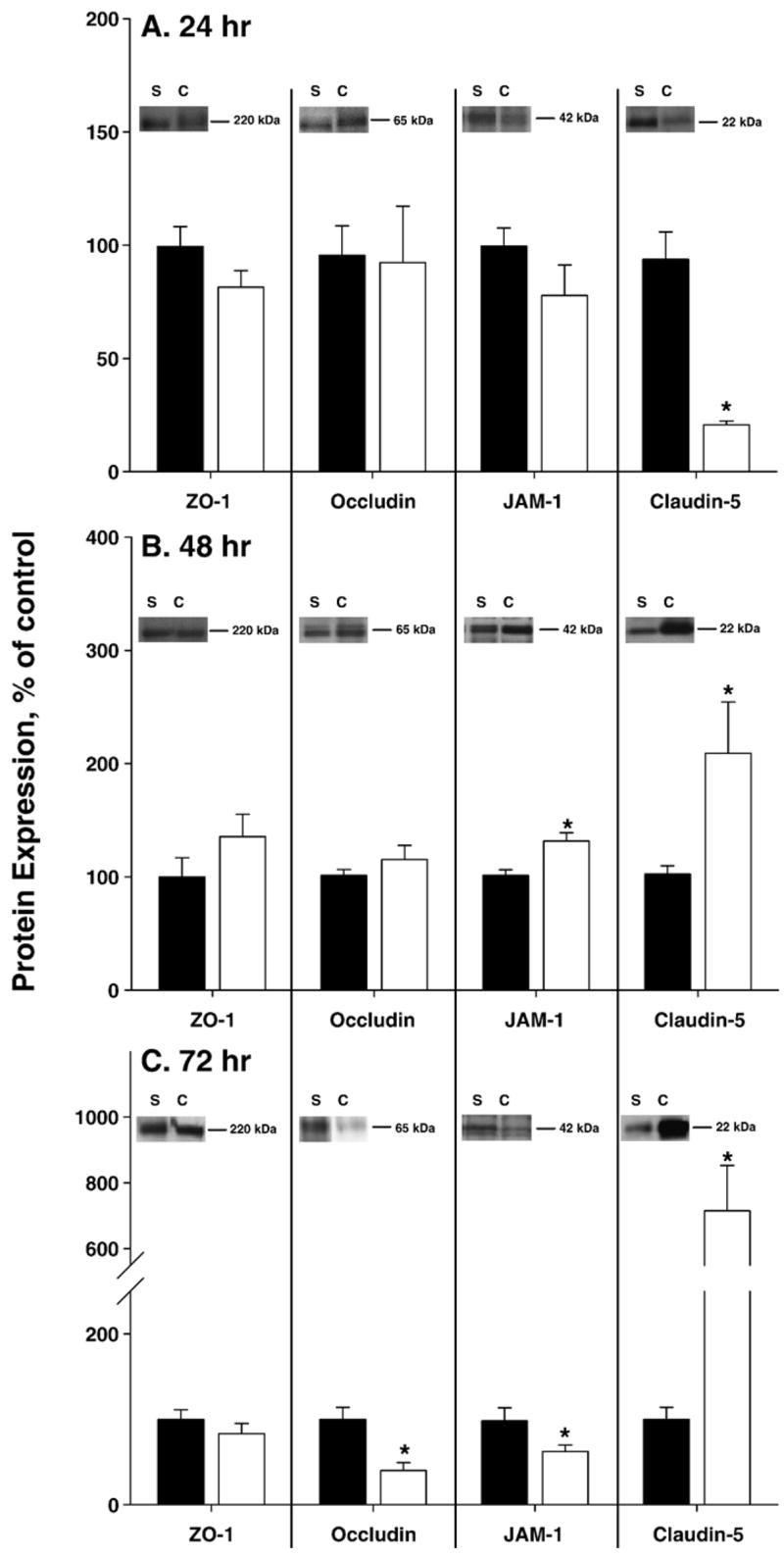
CFA induced changes in expression of the TJ proteins ZO-1, occludin, JAM-1 and claudin-5. Expression of the TJ proteins was measured 24 (A), 48 (B) and 72 (C) h post-injection of saline (filled bars) or CFA (open bars). CFA did not cause any alterations in the expression of ZO-1. Occludin expression decreased by 60% at 72 h. JAM-1 expression was modulated over time, increasing at 48 h and decreasing at 72 h post-CFA. Claudin-5 expression also changed with time, decreasing at 24 h, and increasing at 48–72 h post-CFA treatment. Western blots, inset, are representative of independent experiments, run minimally in triplicate. Gels were stained with GelStain Blue as a loading control. Optical density of each protein band was measured and normalized to saline control expression on each membrane. Data are presented as mean % of saline control±standard error (B); n≥6 per group, *p<0.05.
Fig. 5.

Effect of CFA on the localization of the TJ proteins. Confocal micrographs demonstrate protein localization within, or outside of, the TJ. Saline does not alter the apparent expression or localization of the TJ proteins ZO-1, occludin, JAM-1 or claudin-5, as compared to naïve. Following CFA injection, localization of ZO-1 does not change with time and is used for co-staining and for junctional localization. The representative micrographs, taken at 100× with an oil immersion lens on an LSM 510 confocal laser scanning microscope, qualitatively confirm the changes in protein expression seen by western blot for occludin and JAM-1, and demonstrate junctional localization at all time points studied. Confocal micrographs also qualitatively confirm claudin-5 protein alterations. Claudin-5 moves out of the junction at 24 h post-CFA, as observed by co-localization with ZO-1. Claudin-5 expression is increased and co-localizes with ZO-1 at 48 and 72 h post-CFA, but not all protein is junctional: claudin-5 staining is separate from that of ZO-1. Confocal micrographs of cerebral capillaries (vessel diameter 4–10 μm) are representative of at least 3 separate and independent experiments; n≥6 per group, *p<0.05.
Expression of occludin did not change until 72 h post-CFA treatment. At that time, protein expression decreased by 60%, as compared to time-matched saline controls (p<0.05; Figs. 4A–C). Confocal microscopy of microvessels co-stained for ZO-1 and occludin indicates consistent co-localization, and the merged images indicate a continuous and junctional (i.e. co-localized with ZO-1) localization for occludin (Figs. 5, 6). At 72 h post-CFA treatment, the decrease in occludin protein expression is evident by microscopy, and leads to a significant decrease in the ratio of Occludin:ZO-1 (Fig. 6, p<0.001).
Fig. 6.

Co-localization of ZO-1 with transmembrane TJ proteins. Confocal micrographs demonstrate co-localization of ZO-1 with each transmembrane protein – occludin, JAM-1 and claudin-5 – in isolated microvessels from both saline and CFA (72 h) treated rats. The representative micrographs, taken at 100× with an oil immersion lens on an LSM 510 confocal laser scanning microscope, qualitatively confirm the changes in protein expression seen by western blot for transmembrane TJ proteins. Higher magnification micrographs correlate to portions of the inset microvessels, outlined in red. A 1:1 ratio of protein co-localization with ZO-1 appears yellow, and any alterations from there are evident as increased green or red fluorescent channel. MetaMorph software was used to examine the relative fluorescence intensities of each channel, and is expressed as ratios in terms of the proteins they represent. These ratios demonstrate decreased co-localization of ZO-1 with both occludin and JAM-1, and an increase with claudin-5. Confocal micrographs of cerebral capillaries (vessel diameter 4–10 μm) are representative of at least 3 separate and independent experiments; n≥6 per group, *p<0.01, **p<0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Changes in the expression of JAM-1 were measured through 72 h post-saline or CFA. No changes in protein expression were noted at 24 h (Figs. 4A–C). Significant changes were evident in protein expression at 48 and 72 h post-CFA, with an increase and then a decrease, respectively. At 48 h post-CFA, JAM expression increased 30%, but subsequently decreased by 36% at 72 h post-CFA injection (Figs. 4A–C; p<0.05). Confocal microscopy images qualitatively reflect the changes in protein expression. Moreover, merged images indicate a continuous and junctional localization of JAM-1 as it co-localizes with ZO-1 (Figs. 5, 6). This co-localization is decreased 72 h post-CFA treatment, likely due to the decrease in JAM-1 protein expression, leading to a significant decrease in the ratio of JAM-1:ZO-1 (Fig. 6, p<0.01).
Expression and localization of claudin-5 were also studied. In contrast to the previously discussed transmembrane proteins, a change in claudin-5 expression is evident 24 h post-CFA injection, where it is decreased by 73% (p<0.05). Forty-eight hours post-CFA injection, expression had increased by 106% (p<0.05), and by 72 h, claudin-5 had increased by 615% (Figs. 4A–C, p<0.05). Microvessels were co-stained with ZO-1 antibody. Images from saline treated animals demonstrate that claudin-5 localization is primarily continuous and junctional, as it co-localizes with ZO-1. However, unlike the other TJ proteins examined, claudin-5 also appears non-continuous, and thus punctate, throughout the cell. This is most evident in the merged claudin-5/ZO-1 images from the saline control, where staining for claudin-5 (in green) is noted both co-localized with ZO-1 in the junction (yellow) and away from it in a more scattered manner (green alone) (Figs. 5, 6).
Following 24 h of CFA treatment, claudin-5 staining appears more punctate and less junctional than in a saline control microvessel. Co-localization with ZO-1 is decreased, as evident in the merged images, with a more diffuse pattern of localization throughout the vessel. By 48 h post-CFA injection, the increase in protein expression is qualitatively evident, and co-localization with ZO-1 has increased, comparable to that of saline control rats. A diffuse pattern of localization is still evident. Upon 72 h of CFA treatment, increased expression of claudin-5 is marked. Merged images demonstrate increased co-localization of ZO-1 with claudin-5, with a continuous and junctional localization pattern, and an increase in the claudin-5:ZO-1 ratio within the junction. Additionally, claudin-5 which is not co-localized with ZO-1 appears non-junctional and punctate, dispersed throughout the microvessel (Figs. 5, 6).
3. Discussion
In the present study, we demonstrated that CFA causes the onset of inflammatory hyperalgesia, as characterized by increased paw volume, enhanced thermal hyperalgesia and a shift in the population of white blood cells from lymphocytes to neutrophils, with no other significant alterations in the amount of white blood cell or packed red blood cell volume. This inflammatory hyperalgesia/pain (over 72 h) led to a temporal disregulation of key transmembrane TJ proteins (i.e. occludin, JAM and claudin-5). This TJ disruption correlated with increased BBB paracellular permeability of [14C]sucrose within the cerebral hemispheres. These data are in agreement with our previous findings that inflammatory hyperalgesia leads to functional changes in the BBB (Brooks et al., 2005; Hau et al., 2004; Huber et al., 2001, 2002a, b, 2006), and extends those findings to describe the earlier (i.e. 0–72 h) molecular alterations in TJ expression and co-localization, and changes in paracellular BBB integrity. In addition, the data presented agree with previous reports describing the importance of the expression and localization of transmembrane TJ proteins in the maintenance of BBB integrity (Hawkins and Davis, 2005; Martin et al., 2006).
The TJ is the result of complex interactions between several transmembrane and cytoskeletal proteins. The claudins, occludin and JAM-1 are linked to the actin cytoskeleton via ZO-1 (Farshori and Kachar, 1999; Mitic et al., 2000). Alterations in expression or location of these proteins have been correlated to decreased barrier function. Decreased occludin expression, such as seen in both the present and previous studies 72 h post-CFA treatment, will disrupt BBB function (Bolton et al., 1998; Brooks et al., 2005; Brown and Davis, 2005; Hawkins and Davis, 2005; Huber et al., 2002a). Similarly, modulation of JAM-1 protein expression and/or localization is linked to a decrease in normal barrier function, and increased paracellular transport (Dobrogowska and Vorbrodt, 2004; Liang et al., 2002; Mandell and Parkos, 2005). In the present study, we show alterations in the protein expression and localization of occludin, JAM-1 and claudin-5 which correlate to an early (24 h) and a late (72 h) period of increased BBB paracellular permeability to [14C]sucrose.
Of all the proteins examined, the dramatic expression and localization alterations of claudin-5 were a most striking finding. Claudin-5 belongs to a family of proteins with over 20 members, characterized by 4 membrane-spanning domains, two extracellular loops and two cytoplasmic termini (Bazzoni and Dejana, 2004; Tsukita and Furuse, 1998, 2000). In the current study, disruption of paracellular permeability to [14C] sucrose correlated with both a decrease and a 6-fold increase in claudin-5 expression at 24 and 72 h post-CFA, respectively. Disruption of barrier function has been reported with both decreased and increased claudin-5 expression, indicating complex regulation of the protein within the TJ. Reduced expression of claudin-5 as seen in the present study has been associated with increased permeability in several previous studies (Kirk et al., 2003; Kitajima et al., 2006; Kniesel and Wolburg, 2000; Tsukita and Furuse, 1998). While transfection of claudin-5 into epithelial cells with a low electrical resistance enhances barrier function (Amasheh et al., 2005), overexpression of claudin-5 results in a loss of barrier function and increased permeability and paracellular volume of distribution (Coyne et al., 2003; Kojima et al., 2002; Wang et al., 2003). Knocking out the claudin-5 gene is lethal within 10 h of birth, and BBB TJs become more permeable to molecules <800 Da, but not larger (Nitta et al., 2003). This size-selective loss in barrier function correlates with the presented work demonstrating increased paracellular transport of the small molecular weight sucrose (342 Da), but no extravasation of the larger compound, Evans Blue Albumin (~70,000 Da).
Modulation of barrier function has also been observed without any changes in TJ protein expression. Rather, function can be affected by alterations in protein–protein interactions and protein localization within the junction (Martin et al., 2006). In the present study, claudin-5 co-localized with ZO-1 to the TJ in control animals. The early period of increased paracellular permeability of [14C]sucrose (24 h) correlated with not only reduced claudin-5 expression, but also with a decrease in its co-localization with ZO-1. Similar to these presented data, increased permeability has also been linked to a loss of claudin-5 expression and movement away from the junction in gastric epithelium (Fedwick et al., 2005). During the later period of disruption in paracellular transport, claudin-5 co-localized with ZO-1, but was also visible in a discontinuous and non-junctional pattern throughout the endothelial cell (Figs. 3, 6). Presence of claudins, as opposed to other integral TJ proteins, is critical to cell-to-cell adhesion of the TJ and occludes the intercellular space to form characteristic TJ “kisses” (Kubota et al., 1999). Taken together, these studies demonstrate the complex regulation of claudin proteins, and the intricate balance of protein expression and localization critical for maintaining proper BBB function and integrity.
While the claudins are critical for cell–cell adhesion, their function is dependent on the interaction with other TJ proteins (Bazzoni and Dejana, 2004; Hawkins and Davis, 2005; Huber et al., 2002a; Wolka et al., 2003). A disruption of TJ protein interactions may be responsible for the BBB integrity loss observed in this study, as opposed to changes in only a single protein. In the present study, we have demonstrated both an early (24 h) and a late (72 h) disruption in BBB paracellular transport, which correlate to distinct molecular alterations in the TJ, suggesting differential regulation of the BBB during inflammatory pain states. Such a biphasic trend in TJ protein alterations and the loss BBB functional integrity has also been seen with other models of inflammatory pain, and highlights the dynamic nature of the barrier (Huber et al., 2002a).
During the first phase of altered paracellular permeability, claudin-5 protein expression and co-localization with ZO-1 are decreased. As described above, a decrease in claudin-5 expression and junctional localization can lead to a loss of barrier function. By 48 h post-CFA, the paracellular permeability of [14C] sucrose had returned to control levels, and claudin-5 expression had rebounded from the early time point and increased 2-fold, coupled with increased JAM-1 expression. The increase in JAM-1 may be an adaptive response to the earlier claudin-5 decrease and subsequent loss of barrier function. It is also possible that the increase in JAM-1 expression is related to the CFA-induced increase in circulating neutrophils, as JAM-1 is involved in leukocyte diapedesis (Muller, 2003). As the CFA-induced inflammatory hyperalgesia continues, paracellular permeability is again increased, where at 72 h, we described an 81% increase in sucrose paracellular permeability across the BBB. The TJ has a number of observed molecular alterations during this later period, including decreased occludin and JAM-1 expression and a dramatic increase in claudin-5 expression.
Many studies have demonstrated a dynamic regulation of ZO-1, with a correlating effect on the TJ integrity (Martin et al., 2006; Polette et al., 2005; Salama et al., 2006; Shen et al., 2006). In the current study, however, we noted no effect of either saline or CFA-injection on ZO-1 expression or localization. Previous studies have reported ZO-1 localization within and along the interendothelial cleft, as measured by immunogold labeling (Vorbrodt and Dobrogowska, 2003; Vorbrodt et al., 2006). Therefore, ZO-1 was utilized as representative of cell-to-cell contact and the junctional space. Changes were noted in the degree of ZO-1 co-staining with various transmembrane proteins. In particular, 72 h post-CFA injection, there was a marked decrease in both occludin and JAM-1 co-staining with ZO-1 and an increase in co-localization with claudin-5, most evident in the significant changes of protein ratios in relation to ZO-1 (Fig. 6). ZO-1 is the key accessory protein of the TJ which links each of these transmembrane proteins at their C-termini to the actin cytoskeleton (Farshori and Kachar, 1999; Mitic et al., 2000). In addition, ZO-1 has an apparent role in the organization and formation of the TJ by recruiting and trafficking transmembrane proteins to the TJ microdomain (Li et al., 2005; Sheth et al., 1997; Umeda et al., 2004). We hypothesize that the increased co-localization of claudin-5 and ZO-1 decreases the interactions of both occludin and JAM-1 with this support protein. As a result of the inability to bind to ZO-1 and link to the actin cytoskeleton, occludin and JAM-1 may then be targeted for degradation, which may lead to our observation of decreased protein expression. Such a disruption in protein–protein interactions could then lead to the noted alteration in paracellular integrity and the increased distribution of small, hydrophobic molecules within the CNS.
Disruptions in TJ interactions and subsequent barrier function have also been demonstrated in response to various immune modulators which are upregulated as a result of CFA treatment. In particular, IL-1β, IL-6, TNF-α and MCP-1 were increased following CFA treatment (Watanabe et al., 2005), and each of these cytokines is associated with increased disruptions in TJ proteins, such as JAM-1, occludin and claudin-5, and barrier permeability (Ahishali et al., 2005; Stamatovic et al., 2003, 2005, 2006; Ye et al., 2006). These cytokines also promote the expression of adhesion receptors such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) (Dunon et al., 1996; Springer, 1995). Enhanced expression of ICAM-1 on BBB endothelial cells has been observed following λ-carrageenan-induced inflammatory hyperalgesia (Huber et al., 2006). These specialized receptors allow leukocytes to incorporate chemoattractant signals from the endothelial cells, promoting immobilization and infiltration across the barrier.
Our laboratory has described alterations in functional BBB integrity with several models of inflammatory pain (Brooks et al., 2005; Hau et al., 2004; Huber et al., 2001, 2002a, b, 2006). We have focused our studies on acute and chronic inflammatory pain, by examining the effects of λ-carrageenan (3%) and CFA, respectively, and through these studies we have discovered some common molecular mechanisms contributing to increased paracellular permeability at the BBB. Specifically, decreases in occludin and increases in claudin-5 (Brooks et al., 2005; Huber et al., 2002a) coincide with increased paracellular permeability. These parallel molecular alterations leading to changes in functional integrity suggest a common underlying mechanism. It remains to be determined whether the impetus for the functional and molecular alterations at the BBB caused by inflammatory pain is due to an inflammatory event, the nociceptive input or a combination of both. Further characterization of these signaling and molecular mechanisms may lead to new therapeutic strategies for the delivery of drugs into the CNS. Such a pharmacological approach is currently being studied using various bacterial toxins for the enhancement of oral drug bioavailability and targeted therapy (Ebihara et al., 2006; Guttman et al., 2006; Salama et al., 2006). Extension of our studies and identification of a new and specific mechanism at TJs of the BBB would be of considerable therapeutic value in the treatment of CNS diseases such as epilepsy, CNS cancers, HIV encephalitis and Alzheimer’s.
4. Experimental procedures
4.1. Radioisotopes, antibodies and chemicals
[14C]sucrose (specific activity of 492 mCi/mmol, 99.5% purity) was purchased through ICN Pharmaceuticals (Irvine, CA). Primary antibodies for ZO-1 (mouse or rabbit), occludin (rabbit), JAM-1 (rabbit) and claudin-5 (mouse) were purchased from Zymed (San Francisco, CA). Conjugated anti-mouse immunoglobin G-horseradish peroxidase and anti-rabbit immunoglobin G-horseradish peroxidase were obtained from Amersham Life Science Products (Springfield, IL). Complete Freund’s adjuvant (CFA) and all other chemicals, unless otherwise stated, were of the highest analytical quality available and purchased from Sigma (St. Louis, MO).
4.2. Animals and treatments
All animal protocols were approved by the University of Arizona Institutional Animal Care and Use Committee and abide by the guidelines of the NIH for the proper treatment of animals. Female Sprague–Dawley rats (Harlan. Indianapolis, IN), weighing 250–300 g were housed under standard 12 h light/12 h dark conditions and received food ad libitum. Following baseline analysis of paw volume, rats were injected subcutaneously with 100 μL of either saline or CFA (1:1, CFA: saline, CFA 50 μg) into the plantar surface of their right hindpaw. Animals were assessed for hindpaw edema and hyperalgesia over a 72 h time period, and then anesthetized with pentobarbital sodium (64.7 mg/kg, i.p.) for in situ brain perfusion or with isofluorane for cerebral microvessel isolation.
4.3. Edema formation and thermal hyperalgesia
Hindpaw edema was measured by the displacement of electrolyte solution in a plesmythmometer (model 7141; Ugo Basile, Comerio VA, Italy) pre-injection and 24, 48 and 72 h post-injection. Thermal hyperalgesia was measured by paw removal latency upon exposure to infrared (IR) heat. The IR source heated linearly from 23.5 °C at time zero to 37 °C at 15 s. The rats were habituated to individual boxes on an elevated glass table for 20 min prior to exposure of the right hindpaw’s plantar surface to the mobile IR source. Paw withdrawal latencies were defined as the time (s) taken for the rat to remove its hindpaw from the heat source.
4.4. White blood cell counts, differentials and hematocrit
Whole blood (1–2 mL) was collected from the ascending venous route into EDTA coated tubes (BD Vacutainer; Franklin Lakes, NJ) as a function of time post-injection and subsequently used for analysis of differentiated white blood cells (WBC) and hematocrit analysis. For total WBC counts, whole blood was mixed with diluting fluid (1% gentian violet in 2% acetic acid) in a 1:20 ratio in order to lyse the RBC. Following manual mixing for 5 min, WBCs were counted with a hemacytometer. For WBC differentials, sample whole blood was smeared onto a glass slide and stained (fixative, xanthene dye, thiazine dye (Hema; Biochemical Sciences, Swedesboro, NJ); 30 s each followed by d-H2O rinse) in coplin jars. Leukocyte differentials were counted with a light microscope (Nikon TE3000) using a 60× oil objective. Differentiated WBCs were expressed as percent of total counts per slide (100). For hematocrit analysis, whole blood was drawn into 75 mm Mylar wrapped capillary tubes (Drummond, Broomall, PA) by capillary action. Tube ends were sealed with Cristaseal™ (AlC International, Cologno Monzese, MI Italy) and placed into a Jouan (Model A13) hematocrit centrifuge (St. Herblain, France). Following centrifugation (10 min; 13000 rpm), percent red blood cells was read using a rotoreader.
4.5. In situ brain perfusion
Animals were anesthetized with pentobarbital sodium (64.7 mg/kg) and heparinized by i.p. injection (10,000 U/kg). At the neck, a ventral midline incision was made; the common carotid arteries were exposed and cannulated with silicone tubing while the jugular veins were cut to allow outflow. The perfusion medium consisting of a Ringer’s solution (117 mM NaCl, 4.7 mM KCl, 0.8 mM MgSO4, 24.8 mM NaHCO3, 1.2 mM KH2PO4, 2.5 mM CaCl2, 10 mM D-glucose, 39 g/L dextran, 10 g/L BSA, pH 7.4) containing Evans blue-labeled albumin, was warmed to 37 °C, oxygenated with 95% O2 and 5% CO2, and passed through a peristaltic pump and a bubble trap. Upon reaching 100 mm Hg perfusion pressure and a flow rate of 3.1 mL/min, [14C]sucrose (10 μCi/20 mL Ringer) was infused (0.5 mL/min/hemisphere). The animals were perfused for 20 min, then decapitated and their brains harvested. The choroid plexus and meninges were removed and the cerebral hemispheres were sectioned and homogenized. Samples of the radioactive perfusate from each carotid cannula were collected as a reference. Brain tissue and 100 μL perfusate were prepared for liquid scintillation counting by incubation in 1 mL TS-2 tissue solubilizer for 2 days, and the amount of radioactivity in each sample radiation was determined following the addition of 100 μL 30% acetic acid and 2.5 mL Optiphase SuperMix (Perkin Elmer, Boston, MA) liquid scintillation cocktail. Results are reported as the ratio of radioactivity in the brain to that in the perfusate (Rbr):
4.6. Microvessel isolation
Under isofluorane anesthesia, rats were decapitated and their brains were removed. Meninges and choroid plexus were excised from the cerebral hemispheres, which were then homogenized in 4 mL of microvessel isolation buffer (103 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 15 mM HEPES, 2.5 mM NaHCO3, 10 mM D-glucose, 1 mM sodium pyruvate, dextran [MW 64,000, 10 g/L]; pH 7.4) with a protease inhibitor cocktail (0.2 mM phenylmethylsulfonyl fluoride, 1 mM benzamide, 1 mM NaVO4, 10 mM NaF, 10 mM sodium pyrophosphate and 10 μg/mL of aprotonin and leupeptin). Eight milliliters of 26% dextran was added, and the mixture was vortexed then centrifuged at 5600×g for 10 min. The supernatant was aspirated, the pellets were resuspended in 8 mL of microisolation buffer and were passed through a 70-μm filter (Falcon, Becton-Dickinson; Franklin, NJ). The filtered homogenates were centrifuged at 3000×g for 10 min at 4 °C. This isolation results in a highly enriched preparation of microvessels. Protein was extracted from the pellets with 6 M urea lysis buffer (6 M urea, 0.1% Triton X-100, 10 mM Tris, pH 8.0, 1 mM dithiothreitol, 5 mM MgCl2, 5 mM EGTA, 150 mM NaCl) with the protease inhibitor cocktail. Protein concentrations were determined by bicinchoninic acid protein assay (Pierce, Rockford, IL).
4.7. Tight junctional protein expression analysis
Protein isolated from microvessels was analyzed for expression of the TJ proteins ZO-1, occludin, JAM and claudin-5. The isolation procedure described above enriches for microvessels, sufficient to examine these proteins, which have minimal to no expression in other brain cell types. Microvessel protein samples (40 μg) were resolved on a 4–12% Tris–glycine gel (Novex, San Diego, ZA) for 1 h at 250 V and transferred to a nitrocellulose membrane for 45 min at 240 mA. Gel-Code Blue (Pierce, Rockford, IL) was utilized to stain the gels to ensure proper loading of protein. The nitrocellulose membranes were incubated with blocking buffer (20 mM Tris Base, 137 mM NaCl, 2 M HCl, 0.1% Tween 20; pH 7.6) with 5% non-fat milk overnight at 4 °C. Blots were incubated at 4 °C, overnight, with primary antibody (1:250–2000 dilution) washed in blocking buffer with 5% non-fat milk at room temperature for 1 h, and incubated with secondary antibody (1:1000–2000 dilution) for 45 min at room temperature. Blots were developed using enhanced chemiluminescence (ECL+, Amersham Life Science Products) and analyzed using Scion Image (Scion, Frederick, MD).
4.8. Confocal microscopy
Microvessels isolated as described above were smeared onto microscope slides and heat-fixed at 95 °C for 10 min. Following fixation (3.7% formaldehyde in PBS, 10 min) and permeabilization (0.1% Triton X-100 in PBS, 5 min), the vessels were blocked for 30 min in PBS with 1% BSA. Slides were incubated with primary antibody diluted in PBS with 1% BSA for 30 min (1:100 dilutions), rinsed and re-blocked for 30 min with 1% BSA in PBS and incubated with Alexafluor™ 488-conjugated anti-rabbit or anti-mouse IgG (R&D Systems, Minnesapolis, MN) diluted in 1% BSA in PBS for 30 min (1:500 dilutions). Vessels from saline and CFA treated animals were stained in parallel for each protein, in concert with vessels from a naïve rat. Slides were sealed with Vectashield and examined using an LSM 510 confocal laser scanning microscope controlled by LSM 5 software (Zeiss, Thornwood, NY USA). Alexafluor™ 488 and 546 secondary antibodies were excited with a 200 mW Argon multi-line laser (LASOS Lastertechnik GmbH, Jena, Germany). Fluorescence emission intensity was detected using a 10 nm LP505 longpass filter. Images were scanned under a 100× oil-immersion objective. Each day instrument settings were optimized to slides prepared from naïve rats as an internal control.
4.9. Statistical analysis
Data are presented as the mean±standard error. Analysis of data was performed by a one-way or two-way ANOVA, as applicable, followed by a post hoc Student–Newman–Keuls test.
References
- Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25(1):5–23. doi: 10.1007/s10571-004-1374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahishali B, Kaya M, Kalayci R, Uzun H, Bilgic B, Arican N, Elmas I, Aydin S, Kucuk M. Effects of lipopolysaccharide on the blood–brain barrier permeability in prolonged nitric oxide blockade-induced hypertensive rats. Int J Neurosci. 2005;115(2):151–168. doi: 10.1080/00207450590519030. [DOI] [PubMed] [Google Scholar]
- Amasheh S, Schmidt T, Mahn M, Florian P, Mankertz J, Tavalali S, Gitter AH, Schulzke JD, Fromm M. Contribution of claudin-5 to barrier properties in tight junctions of epithelial cells. Cell Tissue Res. 2005;321(1):89–96. doi: 10.1007/s00441-005-1101-0. [DOI] [PubMed] [Google Scholar]
- Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78(1):85–96. [PubMed] [Google Scholar]
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood–brain barrier breakdown in vivo. Neuroscience. 1998;86(4):1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood–brain barrier permeability and tight junction protein alterations. Am J Physiol: Heart Circ Physiol. 2005;289(2):H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC, Davis TP. Hypoxia/aglycemia alters expression of occludin and actin in brain endothelial cells. Biochem Biophys Res Commun. 2005;327(4):1114–1123. doi: 10.1016/j.bbrc.2004.12.123. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol: Lung Cell Mol Physiol. 2003;285(5):L1166–L1178. doi: 10.1152/ajplung.00182.2003. [DOI] [PubMed] [Google Scholar]
- Dobrogowska DH, Vorbrodt AW. Immunogold localization of tight junctional proteins in normal and osmotically-affected rat blood–brain barrier. J Mol Histol. 2004;35(5):529–539. doi: 10.1007/10.1007/s10735-004-1318-3. [DOI] [PubMed] [Google Scholar]
- Dunon D, Piali L, Imhof BA. To stick or not to stick: the new leukocyte homing paradigm. Curr Opin Cell Biol. 1996;8(5):714–723. doi: 10.1016/s0955-0674(96)80114-1. [DOI] [PubMed] [Google Scholar]
- Ebihara C, Kondoh M, Hasuike N, Harada M, Mizuguchi H, Horiguchi Y, Fujii M, Watanabe Y. Preparation of a claudin-targeting molecule using a C-terminal fragment of Clostridium perfringens enterotoxin. J Pharmacol Exp Ther. 2006;316(1):255–260. doi: 10.1124/jpet.105.093351. [DOI] [PubMed] [Google Scholar]
- Edwards T. Inflammation, pain, and chronic disease: an integrative approach to treatment and prevention. Altern Ther Health Med. 2005;11(6):20–27. quiz 28, 75. [PubMed] [Google Scholar]
- Farshori P, Kachar B. Redistribution and phosphorylation of occludin during opening and resealing of tight junctions in cultured epithelial cells. J Membr Biol. 1999;170(2):147–156. doi: 10.1007/s002329900544. [DOI] [PubMed] [Google Scholar]
- Fedwick JP, Lapointe TK, Meddings JB, Sherman PM, Buret AG. Helicobacter pylori activates myosin light-chain kinase to disrupt claudin-4 and claudin-5 and increase epithelial permeability. Infect Immun. 2005;73(12):7844–7852. doi: 10.1128/IAI.73.12.7844-7852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Nava P. Tight junctions, from tight intercellular seals to sophisticated protein complexes involved in drug delivery, pathogens interaction and cell proliferation. Adv Drug Deliv Rev. 2005;57(6):811–814. doi: 10.1016/j.addr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell Microbiol. 2006;8(4):634–645. doi: 10.1111/j.1462-5822.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- Hau VS, Huber JD, Campos CR, Davis RT, Davis TP. Effect of lambda-carrageenan-induced inflammatory pain on brain uptake of codeine and antinociception. Brain Res. 2004;1018(2):257–264. doi: 10.1016/j.brainres.2004.05.081. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood–brain barrier permeability and tight junctional protein expression. Am J Physiol: Heart Circ Physiol. 2001;280(3):H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- Huber JD, Hau VS, Borg L, Campos CR, Egleton RD, Davis TP. Blood–brain barrier tight junctions are altered during a 72-h exposure to lambda-carrageenan-induced inflammatory pain. Am J Physiol: Heart Circ Physiol. 2002a;283(4):1531–1537. doi: 10.1152/ajpheart.00027.2002. [DOI] [PubMed] [Google Scholar]
- Huber JD, Hau VS, Mark KS, Brown RC, Campos CR, Davis TP. Viability of microvascular endothelial cells to direct exposure of formalin, lambda-carrageenan, and complete Freund’s adjuvant. Eur J Pharmacol. 2002b;450(3):297–304. doi: 10.1016/s0014-2999(02)02150-7. [DOI] [PubMed] [Google Scholar]
- Huber JD, Campos CR, Mark KS, Davis TP. Alterations in blood–brain barrier ICAM-1 expression and brain microglial activation after lambda-carrageenan-induced inflammatory pain. Am J Physiol: Heart Circ Physiol. 2006;290(2):H732–H740. doi: 10.1152/ajpheart.00747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilzecka J. The structure and function of blood–brain barrier in ischaemic brain stroke process. Ann Univ Mariae Curie-Sklodowska, Med. 1996;51:123–127. [PubMed] [Google Scholar]
- Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J Pathol. 2003;201(2):319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- Kitajima Y, Endo T, Nagasawa K, Manase K, Honnma H, Baba T, Hayashi T, Chiba H, Sawada N, Saito T. Hyperstimulation and a gonadotropin-releasing hormone agonist modulate ovarian vascular permeability by altering expression of the tight junction protein claudin-5. Endocrinology. 2006;147(2):694–699. doi: 10.1210/en.2005-0700. [DOI] [PubMed] [Google Scholar]
- Kniesel U, Wolburg H. Tight junctions of the blood–brain barrier. Cell Mol Neurobiol. 2000;20(1):57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Rahner C, Peng S, Rizzolo LJ. Claudin 5 is transiently expressed during the development of the retinal pigment epithelium. J Membr Biol. 2002;186(2):81–88. doi: 10.1007/s00232-001-0137-7. [DOI] [PubMed] [Google Scholar]
- Kubota K, Furuse M, Sasaki H, Sonoda N, Fujita K, Nagafuchi A, Tsukita S. Ca(2+)-independent cell-adhesion activity of claudins, a family of integral membrane proteins localized at tight junctions. Curr Biol. 1999;9(18):1035–1038. doi: 10.1016/s0960-9822(99)80452-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Fanning AS, Anderson JM, Lavie A. Structure of the conserved cytoplasmic C-terminal domain of occludin: identification of the ZO-1 binding surface. J Mol Biol. 2005;352(1):151–164. doi: 10.1016/j.jmb.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Liang TW, Chiu HH, Gurney A, Sidle A, Tumas DB, Schow P, Foster J, Klassen T, Dennis K, DeMarco RA, Pham T, Frantz G, Fong S. Vascular endothelial-junctional adhesion molecule (VE-JAM)/JAM 2 interacts with T, NK, and dendritic cells through JAM 3. J Immunol. 2002;168(4):1618–1626. doi: 10.4049/jimmunol.168.4.1618. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev, Neurosci. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, Parkos CA. The JAM family of proteins. Adv Drug Delivery Rev. 2005;57(6):857–867. doi: 10.1016/j.addr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Martin TA, Das T, Mansel RE, Jiang WG. Synergistic regulation of endothelial tight junctions by antioxidant (Se) and polyunsaturated lipid (GLA) via Claudin-5 modulation. J Cell Biochem. 2006;98(5):1308–1319. doi: 10.1002/jcb.20860. [DOI] [PubMed] [Google Scholar]
- Matter K, Aijaz S, Tsapara A, Balda MS. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr Opin Cell Biol. 2005;17(5):453–458. doi: 10.1016/j.ceb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol: Gastrointest Liver Physiol. 2000;279(2):250–254. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16(3):165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24(6):327–334. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA. Mechanisms of disease: the blood–brain barrier. Neurosurgery. 2004;54(1):131–140. doi: 10.1227/01.neu.0000097715.11966.8e. discussion 141–2. [DOI] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. Drug delivery to the brain. J Cereb Blood Flow Metab. 1997;17(7):713–731. doi: 10.1097/00004647-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Polette M, Gilles C, Nawrocki-Raby B, Lohi J, Hunziker W, Foidart JM, Birembaut P. Membrane-type 1 matrix metalloproteinase expression is regulated by zonula occludens-1 in human breast cancer cells. Cancer Res. 2005;65(17):7691–7698. doi: 10.1158/0008-5472.CAN-04-4230. [DOI] [PubMed] [Google Scholar]
- Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006 doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Sharma PK, Hota D, Pandhi P. Biologics in rheumatoid arthritis. J Assoc Phys India. 2004;52:231–236. [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119(Pt 10):2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Sheth B, Fesenko I, Collins JE, Moran B, Wild AE, Anderson JM, Fleming TP. Tight junction assembly during mouse blastocyst formation is regulated by late expression of ZO-1 alpha+isoform. Development. 1997;124(10):2027–2037. doi: 10.1242/dev.124.10.2027. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116(Pt 22):4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV. Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J Cereb Blood Flow Metab. 2005;25(5):593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase C{alpha}-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281(13):8379–8388. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells. 1998;3(9):569–573. doi: 10.1046/j.1365-2443.1998.00212.x. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J Cell Biol. 2000;149(1):13–16. doi: 10.1083/jcb.149.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda K, Matsui T, Nakayama M, Furuse K, Sasaki H, Furuse M, Tsukita S. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J Biol Chem. 2004;279(43):44785–44794. doi: 10.1074/jbc.M406563200. [DOI] [PubMed] [Google Scholar]
- Vorbrodt A, Dobrogowska D. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Rev. 2003;42:221–242. doi: 10.1016/s0165-0173(03)00177-2. [DOI] [PubMed] [Google Scholar]
- Vorbrodt A, Dobrogowska D, Tarnawski M, Meeker HC, Carp RI. Immunogold study of altered expression of some interendothelial junctional molecules in the brain blood microvessels of diabetic scrapie-infected mice. J Mol Hist. 2006;37(1–2):27–35. doi: 10.1007/s10735-006-9026-9. [DOI] [PubMed] [Google Scholar]
- Wang F, Daugherty B, Keise LL, Wei Z, Foley JP, Savani RC, Koval M. Heterogeneity of claudin expression by alveolar epithelial cells. Am J Respir Cell Mol Biol. 2003;29(1):62–70. doi: 10.1165/rcmb.2002-0180OC. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Sandercock PA, Dennis MS, Starr J. Is breakdown of the blood–brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke. 2003;34(3):806–812. doi: 10.1161/01.STR.0000058480.77236.B3. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Guo W, Zou S, Sugiyo S, Dubner R, Ren K. Antibody array analysis of peripheral and blood cytokine levels in rats after masseter inflammation. Neurosci Lett. 2005;382(1–2):128–133. doi: 10.1016/j.neulet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J, Maier SF, Watkins LR. Central proinflammatory cytokines and pain enhancement. Neurosignals. 2005;14(4):166–174. doi: 10.1159/000087655. [DOI] [PubMed] [Google Scholar]
- Wolka AM, Huber JD, Davis TP. Pain and the blood–brain barrier: obstacles to drug delivery. Adv Drug Deliv Rev. 2003;55(8):987–1006. doi: 10.1016/s0169-409x(03)00100-5. [DOI] [PubMed] [Google Scholar]
- Ye D, Ma I, Mas TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol: Gastrointest Liver Physiol. 2006;290(3):G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]