

SUMMARY
Differences in global levels of histone acetylation occur in normal and cancer cells, although the reason why cells regulate these levels has been unclear. Here we demonstrate a role for histone acetylation in regulating intracellular pH (pHi). As pHi decreases, histones are globally deacetylated by histone deacetylases (HDACs), and the released acetate anions are coexported with protons out of the cell by monocarboxylate transporters (MCTs), preventing further reductions in pHi. Conversely, global histone acetylation increases as pHi rises, such as when resting cells are induced to proliferate. Inhibition of HDACs or MCTs decreases acetate export and lowers pHi, particularly compromising pHi maintenance in acidic environments. Global deacetylation at low pH is reflected at a genomic level by decreased abundance and extensive redistribution of acetylation throughout the genome. Thus, acetylation of chromatin functions as a rheostat to regulate pHi with important implications for mechanism of action and therapeutic use of HDAC inhibitors.
INTRODUCTION
Targeted acetylation of lysine residues of histone proteins at distinct genomic loci is linked to regulation of essentially all DNA-templated processes, including transcription, replication, repair, recombination, and the formation of specialized chromatin structures such as heterochromatin (Kouzarides, 2007). For example, alterations in histone acetylation at select gene promoters—via recruitment of histone acetyltransferases (HATs) and histone deacetylases (HDACs) by sequence-specific DNA-binding transcription factors—regulate the transcriptional activity of the targeted genes (Ferrari et al., 2012). Histone acetylation regulates such DNA-templated processes by influencing the local chromatin structure and by regulating the binding or exclusion of bromo-domain-containing proteins to and from the chromatin (Shogren-Knaak et al., 2006; Taverna et al., 2007). The role of histone acetylation has largely been interpreted in this local, site-specific context (Margueron et al., 2005; Zhou et al., 2011). However, histone acetylation levels also differ at a cellular or global level (Horwitz et al., 2008; Vogelauer et al., 2000). Examination of acetylation by methods that assess total histone content—such as western blotting (WB) or immunohistochemistry (IHC)—has revealed heterogeneity in the levels of global histone acetylation in different tissues and cell types (Ferrari et al., 2012; Iwabata et al., 2005; Suzuki et al., 2009). IHC studies on a variety of primary cancer tissues have shown that an increased prevalence of cells with lower cellular levels of histone acetylation is associated with more aggressive cancers and poorer clinical outcome such as increased risk of tumor recurrence or decreased survival rates (Elsheikh et al., 2009; Fraga et al., 2005; Manuyakorn et al., 2010; Seligson et al., 2005, 2009). Such associations underscore the biological relevance of global differences in histone acetylation levels. However, very little is known about what function(s) the changes in global levels of histone acetylation serve for the cell. While a few studies have shown the necessity for a pool of acetyl coenzyme A (ac-CoA) to maintain global histone acetylation (Friis et al., 2009; Takahashi et al., 2006; Wellen et al., 2009), the biological factor(s) in response to which global histone acetylation levels change and what cellular processes are affected by this outcome have remained unknown (Friis and Schultz, 2009).
Cycles of histone acetylation and deacetylation occur continuously and rapidly throughout the genome, consuming ac-CoA and generating negatively charged acetate anions in the process. Since ac-CoA and acetate anions participate in many metabolic processes, we hypothesized that histone acetylation may be linked to certain metabolic or physiologic cues. We therefore systematically studied how global levels of histone acetylation change in response to alterations of various components of the standard tissue culture medium (Dulbecco’s modified Eagle’s medium, DMEM). Strikingly, we found that as intracellular pH (pHi) is decreased, histones become globally hypoacetylated in an HDAC-dependent manner. The resulting free acetate anions are transported with protons by the proton (H+)-coupled monocarboxylate transporters (MCTs) to the extracellular environment, thereby reducing the intracellular H+ load and resisting further reductions in pHi. As pHi increases, the flow of acetate and protons is favored toward the inside of the cell leading to global histone hyperacetylation. Our data reveal that chromatin, through the basic chemistry of histone acetylation and deacetylation, coupled with MCTs, function as a system for rheostatic regulation of pHi.
RESULTS
Glucose, Glutamine, or Pyruvate Is Required to Maintain Global Histone Acetylation
The metabolites in standard DMEM that are required to maintain a pool of ac-CoA for histone acetylation have not been systematically identified. Thus, we began by asking if any or all of the ac-CoA producing sources in DMEM are required to maintain steady-state levels of histones H3 and H4 acetylation. These sources potentially include glucose (G), glutamine (Q), pyruvate (P) and the 14 other amino acids (aa) present in DMEM. HeLa and MDA-MB-231 (231) cells were cultured for 16 hr in complete medium or in medium lacking all or one of the potential ac-CoA sources. Simultaneous removal of GQP and aa led to significant (~40%–99%) reduction in the acetylation of multiple lysine residues on histones H3 and H4 (Figures 1A and S1A, lane 2, available online). Elimination of G, Q, P, or aa individually had little or no effect on histone acetylation. These results suggest that the pool of ac-CoA that is used for histone acetylation derives from one or more of these carbon sources.
Figure 1. Minimal Levels of G, Q, or P Maintain Global Levels of Histone Acetylation.

(A) WBs of histone acetylation in HeLa cells cultured for 16 hr in DMEM salts and vitamins with the indicated ac-CoA sources. The bar graph shows average values ± SD from 15 independent experiments and p value derived from the Student’s t test.
(B) WBs of acetylation in normal IMR90 fibroblasts cultured as in (A).
(C) WBs of tubulin and histone H4 acetylation in HeLa cells under conditions of G, Q, and P deprivation.
(D) WBs of acetylation in HeLa cells cultured at the indicated concentrations of G or Q.
(E) WBs showing the effects of varying concentrations of Ca2+ and phosphate on histone acetylation in HeLa cells. Lane 1 in each panel (A–E) is the reference condition, the values of which are set to 1. See also Figure S1.
To determine which of these carbon sources are necessary to maintain histone acetylation, HeLa, 231, and IMR90 normal primary lung fibroblasts were cultured for 16 hr in medium with all ac-CoA sources, none, or a single ac-CoA source. Cells that - were cultured in only G, Q, or P maintained normal levels of histone acetylation in cancer cells and substantial amounts in IMR90 fibroblasts (Figures 1A, 1B, and S1A). However, histone acetylation was significantly reduced in the presence of all other aa. We conclude that G, Q, or P—but not the remaining aa present in DMEM—are sufficient to maintain normal levels of histone acetylation. It is interesting that, in the absence of GQP and aa, although histone acetylation was significantly decreased, we observed up to an 8-fold increase in tubulin acetylation (Figures 1C and S1B). Higher tubulin acetylation suggests that cellular pools of ac-CoA are not completely depleted when GQP are removed. Therefore, the loss of histone acetylation may be partly due to selective allocation of ac-CoA to other molecular processes under GQP starvation.
Minimal Amounts of G or Q Are Required to Maintain Normal Levels of Histone Acetylation
We next tested whether global levels of histone acetylation change with varying concentrations of G or Q as the sole ac-CoA source. As expected, at zero concentration of G or Q, histone acetylation was significantly reduced after 16 hr of culture (Figures 1D and S1C, lanes 6 and 12). It is surprising however, that global levels of histone acetylation remained largely unchanged over a 45-fold range of G concentration (0.1–4.5 g/l) and a 20-fold range of Q concentration (0.1–2 mM) (Figures 1D and S1C), which include concentrations insufficient to maintain cell growth and replication. These data indicate that global levels of histone acetylation do not change dose-dependently in response to variation in G and Q concentrations within the range tested.
Vitamins, Calcium, and Phosphate Do Not Affect the Global Levels of Histone Acetylation
We next asked whether global levels of acetylation change in response to the vitamins, cofactors, or salts present in DMEM (see Supplemental Experimental Procedures). Culturing HeLa cells for 16 hr in media with varying amounts of vitamins and cofactors in the presence or absence of carbon sources did not significantly affect the global levels of histone acetylation (Figure S1D). Calcium (Ca2+) can enhance the permeability of the mitochondrial membrane to ac-CoA in the presence of phosphate (Benjamin et al., 1983). However, reducing the amount of Ca2+ in the presence or absence of phosphate did not lead to significant changes in the global levels of histone acetylation in HeLa or 231 cells after 16 hr of culture (Figures 1E and Figure S1E). We conclude that global levels of histone acetylation do not change significantly in the absence of vitamins and cofactors or Ca2+ and phosphate.
Global Levels of Histone Acetylation Change in Response to pH
Finally, we asked whether the buffering component and the resulting pH of the medium could affect histone acetylation. Changes in pHe are known to induce corresponding changes in pHi (Fellenz and Gerweck, 1988; Mizuno et al., 2002). We measured pHi in our cell lines at different bicarbonate concentrations (i.e., different pHe levels) using the pH-sensitive dye BCECF-AM (see Experimental Procedures). Consistent with previous reports (Wong et al., 2002), we found that decreasing pHe led to decreasing pHi, with the range of pHi changes (6.3–7.3) being more restricted than that of pHe changes (5.9–7.6) (see top graphs in Figures 2A and S2A). Varying bicarbonate concentration from 45 mM to 10 mM resulted in pHe values that led to slight changes in pHi and histone acetylation (Figures 2A and S2A, lanes 1–3). However, decreasing bicarbonate from 10 mM down to 1 mM resulted in pHe values that resulted in pronounced effects on pHi and significant reductions in histone acetylation as assessed by WB (Figures 2A and S2A, lanes 3–5) and immunofluorescence (Figure 2B). Acetylation of the cytoplasmic pool of histones was affected by pH in a similar manner (Figures 2C and S2B). The effect of pHe on histone acetylation was not limited to cancer cells but was also observed in H1 human embryonic stem cells, IMR90 normal primary lung fibroblasts, and even in Saccharomyces cerevisiae, suggesting that changes in histone acetylation in response to varying pH is a fundamentally conserved process (Figure S2C). It is important to note that acetylation in yeast was reduced at levels of pHe (~pH 4.0) that have the most pronounced effects on pHi (Valli et al., 2005).
Figure 2. Global Levels of Histone Acetylation Decrease when pHi Decreases.

(A) Measurements of pHi (mean ± SD) as a function of pHe and effects on histone acetylation in HeLa cells cultured at the indicated pHe for 16 hr in complete DMEM as assessed by WB. The reference condition is set to 25 mM bicarbonate (lane 2), which approximates the normal physiological concentration. The bar graph shows average values ± SD from seven independent experiments and p value derived from the Student’s t test.
(B) Immunofluorescent analysis of histone acetylation of HeLa cells cultured as in (A).
(C) WB analysis of H4K5ac in HeLa cells in the indicated histone fractions and pH.
(D) WBs of histone acetylation in HeLa cells cultured in media buffered with HEPES. The bar graph shows average values ± SD from 17 independent experiments and p value derived from the Student’s t test.
(E) Acetyl-CoA measurements (mean ± SD) in HeLa cells treated for 4 hr at the indicated pHe.
(F) WBs of histone acetylation in HeLa cells that were treated with or without 5 mM sodium butyrate, 500 nM TSA, or 2 mM nicotinamide (NAM) for 6 hr in complete DMEM at pHe 7.4, followed by incubation at the indicated pHe for 4 hr.
(G) WBs of histone methylation in HeLa cells cultured at the indicated pH.
(H) WBs of tubulin and histone H4 acetylation in HeLa cells treated for 16 hr at the indicated pH values. See also Figure S2.
Histone acetylation was also significantly reduced at low pH when the culture medium was buffered with HEPES instead of bicarbonate (Figures 2D and S2D). This suggests the reduction in acetylation at low pH was in fact due to changes in pH and not to a reduction in bicarbonate concentration. The reduction in acetylation was also not due to changes in acetyl-CoA concentration (Figures 2E and S2E) but was dependent on the activity of class I and II HDACs. Treatment of cells with HDAC inhibitors Trichostatin A (TSA) and sodium butyrate, but not nicotinamide, prior to lowering the pHe blocked the effects of pH on acetylation (Figures 2F and S2F). In addition, deletion of the major HDACs, RPD3 and HDA1, in yeast also greatly diminished the pH-induced hypoacetylation, confirming the requirement for HDAC activity (Figure S2C, lanes 13 and 14). In all pH-related experiments, histone methylation levels at multiple sites were largely unaffected, suggesting that the effect of pH on histone acetylation is not due to general impairment of the cellular posttranslational machinery (Figure 2G). It is interesting that tubulin acetylation was only minimally affected as pHi decreased, indicating some degree of specificity for histone acetylation (Figures 2H and S2G). We conclude that global levels of histone acetylation change in response to pHi, with increased deacetylation of histones occurring at lower values of pHi.
The pH-Induced Changes in Histone Acetylation Levels Do Not Require Specific Carbon Sources or Salts
Lowering pHe in the presence of only G, Q, or P resulted in reduction of histone acetylation (Figures 3A and S3A), indicating that the effect of pH on histone acetylation is independent of the source of ac-CoA. Next, we asked if specific salts are required for hypoacetylation of histones at low pH. Cells were cultured at normal and low pHe in the presence and absence of sodium (Na+) or chloride (Cl−), using Q as the ac-CoA source. We found that global histone acetylation decreased in low pHi regardless of the presence of either ion (Figures 3B, 3C, S3B, S3C). Ca2+ and phosphate were also not required for pH-mediated changes of histone acetylation levels, regardless of whether the cells were deprived of these salts for several days prior to lowering the pH (Figures 3D and S3D) or deprived at the same time the pH was decreased (Figure S3E). We conclude that the alteration of histone acetylation levels in response to pH does not depend on a specific carbon source or extracellular Na+, Cl−, Ca2+ or phosphate.
Figure 3. Changes in Histone Acetylation Levels in Response to pH Do Not Require Specific Carbon Sources or Salts.
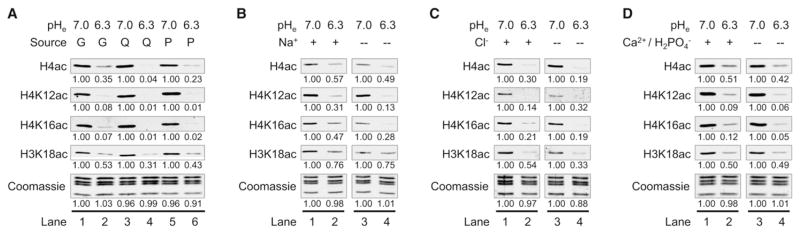
(A–D) WBs of histone acetylation in HeLa cells cultured for 16 hr in DMEM salts at pHe 7.0 or 6.3 either (A) including the indicated carbon source or including Q but lacking (B) Na+, (C) Cl−, and (D) Ca2+ and phosphate. In (D), cells were starved of Ca2+ and phosphate for 3 days prior to pHe treatment. See also Figure S3.
Acetylation of H4K16 Is Decreased and Drastically Redistributed at Low pH
To determine how the pH-induced changes in global levels of acetylation map to specific genomic loci, we treated HeLa cells for 4 hr at pHe 7.4 or pH 6.5 and then performed chromatin immunoprecipitation sequencing (ChIP-seq) analysis of H4K16ac. (For each pH treatment, both input and immunoprecipitated DNA were sequenced.) Consistent with the global decrease in H4K16ac levels, there were significant reductions in both the number of base pairs covered as well as the total number of genomic loci significantly associated with H4K16ac at low pH (Figure 4A). Clustering of the significant peaks of H4K16ac revealed drastic redistribution and decreased overall intensity of this acetylation site at low pH. Figure 4B shows the clusters of H4K16ac peaks (heat maps), the average value of each cluster (line graphs), and the genomic distribution of the peaks (pie charts) with associated gene ontology. Cluster 1 contained 2,751 peaks of H4K16ac in high pH, which were essentially fully deacetylated at low pH. These peaks are mostly near transcription start sites (TSS) and introns of genes whose gene ontology (GO) terms include in protein synthesis and cell cycle control. Cluster 2 contained 595 peaks that were present at both pH values but with much decreased intensity at low pH. More than 90% of cluster 2 peaks occur at TSS and introns of genes enriched in GO terms encoding ribonucleoproteins including mitochondrial and cellular ribosomal protein genes as well as, interestingly, the NuA4 HAT complex that acetylates histone H4 (Arnold et al., 2011). Cluster 3 contained 1,971 new peaks of H4K16ac at low pH, which are mostly in regions away from TSS and are associated with GO terms including spindle organization and replicative senescence.
Figure 4. Low pH Causes Global Decrease and Redistribution of H4K16ac throughout the Genome.

The distribution of H4K16ac in HeLa cells treated for 4 hr at pH 7.4 or 6.5 was determined by ChIP-seq.
(A) Number of DNA base pairs covered by significant (p < 10−4) peaks of H4K16ac and number of genomic regions (25 bp windows) associated with significant H4K16ac peaks.
(B) Heat maps showing the distribution of H4K16ac peaks at the indicated pH values as the −log p value of enrichment in ±1.5 kb region from the center of each peak. All peaks were clustered in three groups based on their combinatorial presence in each pH condition. The number of peaks in each cluster is indicated. The average value of enrichment as −log p value for each cluster is shown as a line chart below each heat map. The distribution of peaks in each cluster is shown as a pie chart. The gene ontology (GO) of genes associated with the peaks of H4K16ac in each cluster is shown. The GO terms include biological process, cellular component, molecular function, and mouse phenotypes of homologous gene knockout.
(C) Box plots showing the distribution of expression of genes associated with the H4K16ac peaks in each cluster under the indicated pH conditions. See also Table S1.
We also performed mRNA sequencing (mRNA-seq) under the same conditions to correlate the redistribution of H4K16ac to changes in gene expression. Compared to high pH, 740 genes were upregulated greater than 2-fold at low pH, including histone genes and genes involved in regulation of transcription, signaling, and metabolism (Table S1). A total of 888 genes were downregulated greater than 2-fold at low pH with functions in plasma membrane and extracellular matrix biology (Table S1). It was surprising that there were no significant changes in the average expression of genes in the three acetylation clusters (Figure 4C). We conclude that treatment at low pH results in elimination of most H4K16ac peaks and re-establishment of less than half as many peaks with lower intensity at new genomic locations predominantly away from TSS regions. This redistribution does not significantly correlate with gene expression after 4 hr of treatment at low pH.
Changes in Histone Acetylation in Response to Nutrient Availability and pH Occur via Different Mechanisms
We next performed kinetic experiments to compare the histone acetylation changes that occur in response to the acidification of the intracellular milieu to those resulting from changes in nutrient availability. First, we performed time course experiments to examine the kinetics of the decrease in acetylation upon each type of treatment. HeLa and 231 cells were cultured for 1, 2, 4, 8, and 16 hr at normal or low pH in the presence or absence of GQP (Figures 5A and S4A). Removal of GQP at normal pH resulted in a gradual decrease in histone acetylation over 16 hr (compare solid and dashed green lines). In contrast, acidification of the medium in the presence of GQP resulted in an abrupt decrease in acetylation at 1 hr, which then remained relatively constant over time (compare solid green and orange lines). In addition, removal of GQP in the low pH medium resulted in a sharp decrease in acetylation during the first hour, followed by a gradual decline over 16 hr (compare solid and dashed orange lines). These data indicate that the kinetics of hypoacetylation in response to acidic pHi and to GQP deprivation are markedly different.
Figure 5. Nutrient Availability and pH Alter Histone Acetylation Differently.

(A and B) WBs of histone acetylation in HeLa cells cultured in DMEM salts (A) for the indicated amount of time and pHe in the presence (+) and absence (−) of GQP and (B) at pHe 7.4 with GQP (control), pHe 7.4 without GQP (for GQP recovery), or pHe 6.5 with GQP (for pH recovery) for 16 hr followed by treatment for the indicated amount of time in medium at pHe 7.4 with GQP. Membrane images were cropped in order to place them below the graph at the corresponding time points. See also Figure S4.
Second, we performed time course experiments to investigate the kinetics of the recovery of histone acetylation in cells that were returned to steady-state culture conditions after being treated at low pH or being deprived GQP. HeLa and 231 cells were cultured for 16 hr at pHe 7.4 or 6.5. A cohort group of cells were cultured for 16 hr in medium with or without GQP at pHe 7.4. In each case, the cells were then returned to normal culture conditions for various amounts of time as shown in Figures 5B and S4B. The recovery of acetylation upon returning GQP was gradual and required ~24 hr to return to steady-state levels (compare purple and green lines). The recovery of acetylation upon return to a normal pHe was much faster (compare purple and orange lines). These data show that the kinetics of acetylation recovery are also different for cells treated at low pH versus those that have been GQP starved. Together with the differential effects on tubulin acetylation, we conclude that medium acidification and nutrient deprivation affect histone acetylation by distinct mechanisms.
Decreased pH Results in an HDAC-Dependent Excretion of Acetate and Protons by MCTs
Since the levels of acetylation were regulated in response to pH, we hypothesized that histone acetylation may impact cellular control of pHi. While deacetylation of lysines in and of itself has no net effect on pHi, export of free, negatively charged acetate anions, which are membrane impermeable (Walter and Gutknecht, 1984), through the H+-coupled MCTs would buffer against further acidification of pHi by decreasing the intracellular H+ load. As obligate H+ symporters, MCTs 1–4 bidirectionally cotransport acetate and other small organic anions—such as lactate—with a H+, thereby participating in pHi regulation (Boron et al., 1988; Halestrap and Meredith, 2004).
To determine if HDAC-derived acetate anion is excreted increasingly as pH decreases, we incubated cells with 3H-acetate in complete medium for 1 hr in order to label histone acetyl groups (Carmen et al., 1996). Following a 30 min chase, HeLa and 231 cells were treated in media of varying pH, which was assayed for released tritium (3H) at the indicated time points in Figure 6A. (Note that at each time point, the entire volume of medium was removed for liquid scintillation counting and fresh medium was added.) Incubation in decreasing pHe resulted in immediate, dose-dependent increases in the appearance of 3H for up to 30 min, which we attribute to acetate anion released from the cell (Figure 6A). The fast nature of the response is consistent with other regulators of pHi (Hulikova et al., 2011). To determine if global histone acetylation is affected in a similarly short time period, we performed kinetic studies analogous to those done in Figure 5 but on a shorter time scale. Loss of global acetylation at most sites examined was detected by WB at 30 min after lowering pHe (Figure S5A). It is possible that histones are deacetylated at earlier time points but at levels below the sensitivity of the antibodies used in this study. Indeed, we detected recovery of acetylation as early as 5 min after return to normal pHe (Figure S5B). We conclude that global deacetylation of histones in response to pH is associated with increased excretion of acetate anion from cells.
Figure 6. Efflux of Acetate through the MCTs at Low pH Is Dependent on HDAC Activity.

(A–C) Liquid scintillation counts (mean ± SD) of media from HeLa and 231 cells labeled with 3H-acetate and treated as indicated in 10% FBS-containing DMEM.
(A) Cells were exposed to media of different pHe for the indicated time. (B) Cells were treated with the MCT inhibitor CNCn (10 mM) or dimethyl sulfoxide during the chase and during subsequent treatment at the indicated pHe. (C) Cells were treated with 500 nM TSA or DMSO prior to labeling and during subsequent treatment at varying pHe.
(D) Acetate excretion rate (mean ± SD) in 231 cells incubated for 30 min in 10% FBS-containing DMEM without P at the indicated pHe.
(E) WBs of histone acetylation in 231 cells treated for 16 hr in complete DMEM at the indicated pHe followed by treatment at the same pHe with or without 500 nM TSA. See also Figure S5.
The appearance of 3H in the media at both normal and low pH was significantly decreased by treating cells for 30 min with the MCT1/MCT4 inhibitor α-cyano-4-hydroxycinnamate (CNCn) after the labeling step but before addition of medium at normal or low pH (Figure 6B). In addition, the class I HDAC inhibitor TSA inhibited the increase in the appearance of 3H in the media at low pHe (Figure 6C). These data suggest that histones are globally deacetylated at low pH in an HDAC-dependent manner with subsequent excretion of acetate anion and protons from the cell through the MCTs.
The potential of HDAC-dependent, H+-coupled acetate excretion to regulate pHi depends upon sufficient rates of acetate excretion. To determine the rate of acetate excretion from cells, we used an enzyme-based assay to measure the amount of acetate in culture media from 231 cells incubated in DMEM of varying pHe for 30 min. Assuming an average cell volume of 500 fl (Kim et al., 2007), we found that, as the pHe was decreased in a stepwise manner, the rate of acetate anion excretion increased from ~0.43 mM/min at pHe 7.4 to ~0.74 mM/min at pHe 6.7, reaching a rate of ~0.96 mM/min at pHe 6.1 (Figure 6D). This rate of acetate and H+ secretion is within the range of other known regulators of pHi such as NHE1, which, for example, secretes protons from acid-loaded (pHi of 6.8) HeLa and MDA-MB-468 cells at a rate ~0.5 mM/min and ~2.0 mM/min, respectively, at a pHe of 6.8 (Hulikova et al., 2011). Since MCT-dependent efflux of acetate anions and protons has a 1:1 stoichiometry, the acetate efflux rate could significantly contribute to pHi regulation by enabling efflux of protons at the same rate. It is important to note that levels of acetylation were increased in 231 cells that were treated with TSA after they had been cultured at low pHe (Figure 6E). This suggests that HAT activity is maintained at low pH, thereby ensuring a steady supply of acetate anions.
Inhibition of HDACs or MCTs Decreases pHi
Our data predict that HDAC inhibition should decrease pHi by preventing the release of acetate from histones, thereby reducing the ability of MCTs to couple H+ efflux to acetate anion efflux. Treatment with increasing concentrations of TSA in complete medium at normal pHe resulted in a dose-dependent decrease of pHi in both HeLa and 231 cells (Figures 7A and S6A, left panel). This effect was not due to passive acidification
Figure 7. HDACs, MCTs, and Cellular Proliferation Affect pHi and Global Histone Acetylation Levels.

(A and B) pHi of HeLa cells treated in standard DMEM overnight with (A) the indicated concentration of TSA (left panel); 250 nM TSA, 50 μM ITSA-1, or both (middle panel); 100 nM TSA followed by 6 hr of culture at the pHe indicated (right panel); or (B) the listed HDAC inhibitors at the indicated concentrations (NAM = 2 mM nicotinamide).
(C) pHi of HeLa cells in which MCT function was inhibited by treatment for 1 hr with 10 mM CNCn (left panel) or by siRNA-mediated knockdown of MCT1 for 96 hr (right panel) followed by treatment for 30 min at the pHe shown.
(D) Rates of lactate excretion in HeLa cells after 30 min of treatment in DMEM without G and P at the indicated pHe (left panel); pHi measurements under identical conditions with 10 mM CNCn or DMSO (right panel).
(E) T cells were stimulated to proliferate by CD3/CD28-coated dynabeads and interleukin-2 treatment for 48 hr. EdU incorporation was determined by flow cytometry, and cell number was counted with and without 48 hr stimulation.
(F) pHe and pHi values, as well as histone acetylation, of resting and activated T cells after 48 hr. Note that pHi changes independently of pHe. All data (A–F) are presented as mean ± SD. See also Figure S6.
of the intracellular environment by TSA—which is weakly acidic—as cotreatment with TSA and ITSA-1, an inhibitor of TSA (Koeller et al., 2003), blocked the TSA-induced acidification of pHi (Figures 7A, middle panel and S6A, right panel). Since histone deacetylation and acetate anion excretion is increased as pHi decreases, we reasoned that inhibition of HDACs may have more pronounced effects on pHi in more acidic conditions. Indeed, this was the case. Treatment of cells with 100 nM TSA had little effect on pHi at normal pH but disrupted the ability of cells to maintain their pHi at lower values of pHe (Figure 7A, right panel). The effect of HDAC inhibition on pHi was not limited to TSA as the HDAC inhibitors Panobinostat (LBH589) and sodium butyrate (which are both weak acids) and Apicidin (a cyclic peptide antibiotic which is not an acid) all caused dose-dependent decreases in pHi (Figure 7B). Nicotinamide, a sirtuin HDAC inhibitor, had essentially no effect on pHi at any pHe tested (Figures 7B and S6B).
Our data also predict that inhibition of MCTs should lead to decreases in pHi by diminishing the rate of H+-coupled acetate excretion. Indeed, treatment with CNCn in complete medium with decreasing pHe resulted in decreased pHi (Figures 7C, left panel and S6C, top left panel). It is important to note that the effects of CNCn on pHi of HeLa cells were most prominent at lower values of pHe, where global histone acetylation is decreased and rates of acetate anion excretion are increased. In addition, knockdown of MCT1 also resulted in reduced pHi compared to cells treated with nontargeting siRNAs (Figure 7C, right panel).
Since the effect of MCT inhibition on lowering pHi could potentially be explained by inhibition of lactate rather than acetate excretion, we examined the rates of lactate excretion at varying pHe. We found that, as pHe was decreased, the rate of lactate excretion was drastically reduced in both HeLa and 231 cells (Figures 7D and S6C, lower left panel) and became even slower than that of acetate excretion at lower values of pHe in G-deprived 231 cells (Figure S6C, lower right panel). It is important to note that MCT inhibition under G-deprived conditions still resulted in decreased pHi (Figures 7D, right panel and S6C, top right panel). It is interesting that MCT inhibition by CNCn did not affect pH-induced histone hypoacetylation, indicating that histone deacetylation is not dependent on acetate excretion (Figure S6D). Taken together, these results indicate that decreasing acetate release from histones by HDAC inhibition or efflux from the cell by MCT inhibition results in lower pHi.
Rapid Cell Proliferation Is Associated with Increased pHi and Histone Hyperacetylation
Although our pH experiments were all done at pHe values that are known to occur in certain cancer tissue (Zhang et al., 2010), we wished to investigate a system in which pHi changes independently of pHe. Considering that rapidly dividing cells have higher pHi than nondividing cells (Reshkin et al., 2000), our data predict
that changes in proliferation are associated with changes in histone acetylation. To test this prediction, we used an in vitro system that mimics the physiological stimulation of T cells to proliferate by antigen-presenting cells (Trickett and Kwan, 2003). Stimulation of proliferation of normal, primary peripheral blood T cells—as evidenced by the incorporation of 5-ethynyl-2′-deoxyuridine (EdU)—led to an increase in overall cell number after 48 hr (Figure 7E). In agreement with previous reports (Bental and Deutsch, 1994), the activation of T cells was associated with an increase in pHi independent of changes in pHe. As predicted, increased pHi was associated with an increase in global histone acetylation (Figure 7F). These data indicate that in physiological conditions in which pHi is affected independently of pHe, global histone acetylation levels change correspondingly.
DISCUSSION
Cells have developed multiple mechanisms to maintain and regulate pHi in response to changes in pHe or increased acid production within the cell (Casey et al., 2010; Parks et al., 2011). These mechanisms include Na+/H+ and Cl−/bicarbonate exchangers, H+ pump ATPases, and bidirectional transport of organic acids by H+-coupled MCTs (Cardone et al., 2005; Wahl et al., 2002). Notably, the MCTs contribute to pHi regulation particularly at low pHe values (Cardone et al., 2005) in tissues such as muscle during exercise (Messonnier et al., 2007) and in certain cancer cells (Wahl et al., 2002). Our data now reveal that the dynamic acetylation and deacetylation of histones together with flux of acetate anions and protons in and out of the cell through the MCTs provides an additional mechanism for cells to modulate their pHi. Deacetylation of histones releases acetic acid from chromatin. Owing to its pKa of 4.74, acetic acid immediately releases a proton that is readily accepted by the ε-amino group (pKa 10.5) of the deprotonated lysine within physiologically relevant pH range. The impermeability of the acetate anion to the plasma membrane necessitates a transporter for the movement of acetate in and out of the cell. This need is met in part by the proton-coupled MCTs, imparting chromatin with the capability to serve as a buffer of pHi. Whether the acetate molecules serve additional functions once excreted from the cell remains to be determined.
The mechanism(s) by which pH affects global histone acetylation levels remains to be determined, but our data suggest that HAT activity is not compromised and that the activity of HDACs is required. It is possible that, at low pHi levels, the balance of histone acetylation and deacetylation is shifted toward the latter, resulting in continual generation of free acetate anions for H+ transport by MCTs. This would result in the appearance of globally hypoacetylated histones but increased production of acetate anions due to continued activity of HATs (see Graphical Abstract). Different sites of histone acetylation may also respond differently to changes in pH. We consistently observed more reduction in global levels of histone H4 than H3K18ac, suggesting that some fraction of H3K18ac may be protected against or unresponsive to pH alterations.
At a molecular level, global hypoacetylation of H4K16 at low pH is associated with elimination of 82% of peaks that are normally present at high pH and with decreased intensity of the remaining 18% of the peaks. Conversely, novel peaks of H4K16ac are established at low pH, covering less than half as many regions of the genome as in a high pH condition. It is interesting that many more peaks of H4K16ac are associated with promoter regions at high pH compared to low pH. Despite the widespread decrease and redistribution of H4K16ac, there was essentially no correlation with the gene expression changes that occurred at low pH in the same time frame. It is possible that redistribution of H4K16ac precedes corresponding changes in gene expression that may occur at a later time point or that other sites of histone acetylation correlate better with transcriptional changes. Nonetheless, our data suggest that rapid changes in gene expression are decoupled from hypoacetylation and redistribution of H4K16ac in response to low pH.
Participation of histone acetylation in pH regulation is consistent with the long-standing phenomenon that acetyl groups of histones have a rapid turnover rate, on the order of a few minutes (Waterborg, 2002). Such rapid turnover would be required for cells to respond swiftly to pH fluctuations. Our data may also be of particular relevance to cancers with low levels of histone acetylation that display a poor clinical outcome (Manuyakorn et al., 2010; Seligson et al., 2009; Suzuki et al., 2009). Cancer tissues displaying low levels of histone acetylation may have enhanced acetate and proton efflux, thereby contributing to an alkaline pHi and an acidic pHe. Tumor microenvironments are commonly found to be acidic, and some with pH values below 6.5 have been reported (Zhang et al., 2010). In addition, tumors with an alkaline pHi and/or acidic microenvironment exhibit more aggressive phenotypes (Harguindey et al., 2005; Lora-Michiels et al., 2006; Moellering et al., 2008; Perona and Serrano, 1988; Reshkin et al., 2000; Rofstad et al., 2006). Our data also suggest that the use of HDAC inhibitors as therapeutic agents in cancer treatment should also be considered from the additional perspective that HDAC inhibition may disrupt the pHi-buffering capacity of cells. Indeed, HDAC inhibitors other than those used in this study also lower pHi (Chung et al., 2008), suggesting an alternative mechanism of action for this class of drugs. Altogether, our findings uncover chromatin as an essential component of an alternative pHi regulatory system involving histone acetylation and deacetylation and MCTs.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa, 231, and IMR90 cells were maintained in DMEM supplemented with G, Q, P, sodium bicarbonate, antibiotics, and 5% dialyzed fetal bovine serum (FBS). Experiments were performed in the absence of serum, unless otherwise noted. H1 cells were maintained in mTeSR1 medium (STEMCELL Technologies #05850) on Matrigel. T cells were purchased from AllCells (PB009-1F) and were maintained in RPMI-1640 (ATCC #30-2001) with 10% FBS (Hyclone #SV3001403) that was heat inactivated by treatment at 56°C for 1 hr.
WB Analysis
WBs were performed using the LI-COR Odyssey system. Blots were performed on acid-extracted histones from isolated nuclei or on whole-cell lysates in the case of tubulin, cytoplasmic histones, and MCT1. A representative blot of three or more independent biological replicates is shown. Statistical analysis was performed in cases where at least seven replicate data points were obtained.
Immunofluorescence
Cells were plated on chambered slides (Fisher #12-565-110N) in control medium and then treated for 16 hr at the indicated pH. Cells were fixed in methanol and stained with the indicated antibodies at 1:200 dilutions in PBS-Tween with 5% bovine serum albumin. Alexa Fluor 488 goat anti-rabbit secondary antibody was used at 1:1,000 dilution. Images were taken using an inverted fluorescent microscope and processed using Slidebook.
Measurements of ac-CoA
Cells grown on 10 cm dishes were treated for 4 hr in DMEM at pH 7.4 or 6.5. The plates containing cells were then directly submerged in liquid nitrogen and shipped on dry ice to the University of Michigan Metabolomics Resource Core for mass spectrometry measurements (Lorenz et al., 2011).
ChIP-Seq and mRNA-Seq
Cells were cultured for 4 hr at the indicated pH in complete DMEM. DNA and RNA were subjected to standard ChIP-seq and mRNA-seq as previously described (Ferrari et al., 2012). Briefly, sequenced reads were aligned to the human genome (Hg19), and only those that matched a unique location with up to two sequence mismatches were retained. To define peaks of enrichment, we segmented the human genome into 25 bp windows and compared the ChIP and normalized input DNA read counts in each window. Using the Poisson distribution, we calculated p values for the enrichment of ChIP reads in each window. A cutoff p value <10−4 was used to maintain a false discovery rate <1%. Significant peaks were defined as those with a p value <10−4 and with significant windows at the same p value in the two neighboring windows.
Total RNA was extracted from cells using QIAGEN RNeasy Mini kit and treated with Ambion’s Turbo DNase. Two micrograms of total RNA was used to start the library preparation according to the manufacturer’s instructions (Illumina TruSeq RNA Sample Preparation Kit). Libraries were sequenced using Illumina HiSeq system to obtain 50 bp-long reads. Alignment of mRNA-seq reads to Human genome (Hg19) was performed using default parameters of Tophat (Trapnell et al., 2009). SAMMate software (Xu et al., 2011) was used to determine the transcript RPKM (reads per kilobase of exon per million of reads).
Acetate and Lactate Excretion Assays
Cells grown in 10 cm dishes were incubated in 5 ml of medium at the indicated pHe for 30 min. Acetate was measured using the K-ACET kit, and lactate was measured using the K-LATE kit (Megazyme) according to manufacturer’s instructions. We confirmed the compatibility of all reagents used in experiments with this kit using the provided standards. Note that an unknown compound in HeLa cell culture medium interfered with the reactions of the K-ACET kit, precluding direct acetate measurement.
pHi Measurements
Adherent cells were grown on 35 mM poly-lysine-coated glass bottom dishes in the presence or absence of pharmacological inhibitors or siRNA. Cells were loaded with 5 nmol/ml of the pH-sensitive dye BCECF-AM in EBSS for 25 min. We then determined pHi by obtaining the ratio of emission at 535 nm wavelength for excitation wavelengths of 495 nm and 440 nm. The pHi of non-adherent T cells was obtained by flow cytometry. Cells were resuspended in Earle’s balanced salt solution (EBSS) and loaded with 5 nmol/ml of the pH-sensitive dye SNARF-1 for 30 min. The pHi was determined by obtaining the ratio of emission wavelength of 580 and 640 nm for excitation wavelength of 488 nm. Ratios were converted to pH using in situ calibration curves as described elsewhere (Nehrke, 2006).
T Cell Stimulation
T cells were stimulated to proliferate using recombinant human IL-2 (Invitrogen #CTP0021) and CD3/CD28-coated Dynabeads (Invitrogen #111.31D) according to manufacturer’s instructions. 5-ethynyl-2′-deoxyuridine (Invitrogen #C35002) incorporation was assessed by flow cytometry according to manufacturer’s instructions.
Additional details are available in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Klara Olofsdotter Otis and Kelsey Martin for the use of their fluorescent microscope; Yasutada Akiba and Jonathan Kaunitz for initial help with pHi measurement; and Steven Clarke, Sohail Tavazoie, and Saeed Tavazoie for discussions. M.A.M. was supported in part by a University of California, Los Angeles, Genetics Training Grant fellowship. This work was supported by grants from the California Institute for Regenerative Medicine and the American Cancer Society and by Beckman Young Investigator, Howard Hughes Medical Institute Early Career, and National Institutes of Health Director’s Innovator awards to S.K.K.
Footnotes
ACCESSION NUMBERS
ChIP-seq and mRNA-seq data are available for download at the NCBI Gene Expression Omnibus under accession number GSE40114.
Supplemental Information includes six figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2012.10.025.
References
- Arnold KM, Lee S, Denu JM. Processing mechanism and substrate selectivity of the core NuA4 histone acetyltransferase complex. Biochemistry. 2011;50:727–737. doi: 10.1021/bi101355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin AM, Murthy CR, Quastel JH. Calcium-dependent release of acetyl-coenzyme A from liver mitochondria. Can J Physiol Pharmacol. 1983;61:154–158. doi: 10.1139/y83-023. [DOI] [PubMed] [Google Scholar]
- Bental M, Deutsch C. 19F-NMR study of primary human T lymphocyte activation: effects of mitogen on intracellular pH. Am J Physiol. 1994;266:C541–C551. doi: 10.1152/ajpcell.1994.266.2.C541. [DOI] [PubMed] [Google Scholar]
- Boron WF, Siebens AW, Nakhoul NL. Role of monocarboxylate transport in the regulation of intracellular pH of renal proximal tubule cells. Ciba Found Symp. 1988;139:91–105. doi: 10.1002/9780470513699.ch6. [DOI] [PubMed] [Google Scholar]
- Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- Carmen AA, Rundlett SE, Grunstein M. HDA1 and HDA3 are components of a yeast histone deacetylase (HDA) complex. J Biol Chem. 1996;271:15837–15844. doi: 10.1074/jbc.271.26.15837. [DOI] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, Atadja P, Griffiths JR, Judson IR, Workman P, Leach MO, Beloueche-Babari M. Noninvasive magnetic resonance spectroscopic pharmaco-dynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia. 2008;10:303–313. doi: 10.1593/neo.07834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69:3802–3809. doi: 10.1158/0008-5472.CAN-08-3907. [DOI] [PubMed] [Google Scholar]
- Fellenz MP, Gerweck LE. Influence of extracellular pH on intra-cellular pH and cell energy status: relationship to hyperthermic sensitivity. Radiat Res. 1988;116:305–312. [PubMed] [Google Scholar]
- Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, Kurdistani SK. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012;22:1212–1221. doi: 10.1101/gr.132308.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Friis RM, Schultz MC. Untargeted tail acetylation of histones in chromatin: lessons from yeast. Biochem Cell Biol. 2009;87:107–116. doi: 10.1139/O08-097. [DOI] [PubMed] [Google Scholar]
- Friis RM, Wu BP, Reinke SN, Hockman DJ, Sykes BD, Schultz MC. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 2009;37:3969–3980. doi: 10.1093/nar/gkp270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Meredith D. The SLC16 gene family-from mono-carboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- Harguindey S, Orive G, Luis Pedraz J, Paradiso A, Reshkin SJ. The role of pH dynamics and the Na+/H+ antiporter in the etiopatho-genesis and treatment of cancer. Two faces of the same coin—one single nature. Biochim Biophys Acta. 2005;1756:1–24. doi: 10.1016/j.bbcan.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. Adenovirus small e1a alters global patterns of histone modification. Science. 2008;321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulikova A, Vaughan-Jones RD, Swietach P. Dual role of CO2/ HCO3(-) buffer in the regulation of intracellular pH of three-dimensional tumor growths. J Biol Chem. 2011;286:13815–13826. doi: 10.1074/jbc.M111.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabata H, Yoshida M, Komatsu Y. Proteomic analysis of organ-specific post-translational lysine-acetylation and -methylation in mice by use of anti-acetyllysine and -methyllysine mouse monoclonal antibodies. Proteomics. 2005;5:4653–4664. doi: 10.1002/pmic.200500042. [DOI] [PubMed] [Google Scholar]
- Kim U, Shu CW, Dane KY, Daugherty PS, Wang JY, Soh HT. Selection of mammalian cells based on their cell-cycle phase using di-electrophoresis. Proc Natl Acad Sci USA. 2007;104:20708–20712. doi: 10.1073/pnas.0708760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeller KM, Haggarty SJ, Perkins BD, Leykin I, Wong JC, Kao MC, Schreiber SL. Chemical genetic modifier screens: small molecule trichostatin suppressors as probes of intracellular histone and tubulin acetylation. Chem Biol. 2003;10:397–410. doi: 10.1016/s1074-5521(03)00093-0. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lora-Michiels M, Yu D, Sanders L, Poulson JM, Azuma C, Case B, Vujaskovic Z, Thrall DE, Charles HC, Dewhirst MW. Extracellular pH and P-31 magnetic resonance spectroscopic variables are related to outcome in canine soft tissue sarcomas treated with thermoradio-therapy. Clin Cancer Res. 2006;12:5733–5740. doi: 10.1158/1078-0432.CCR-05-2669. [DOI] [PubMed] [Google Scholar]
- Lorenz MA, Burant CF, Kennedy RT. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal Chem. 2011;83:3406–3414. doi: 10.1021/ac103313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, Hines OJ, Reber H, Seligson DB, Horvath S, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. 2010;28:1358–1365. doi: 10.1200/JCO.2009.24.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Messonnier L, Kristensen M, Juel C, Denis C. Importance of pH regulation and lactate/H+ transport capacity for work production during supramaximal exercise in humans. J Appl Physiol. 2007;102:1936–1944. doi: 10.1152/japplphysiol.00691.2006. [DOI] [PubMed] [Google Scholar]
- Mizuno S, Demura Y, Ameshima S, Okamura S, Miyamori I, Ishizaki T. Alkalosis stimulates endothelial nitric oxide synthase in cultured human pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L113–L119. doi: 10.1152/ajplung.00436.2001. [DOI] [PubMed] [Google Scholar]
- Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25:411–425. doi: 10.1007/s10585-008-9145-7. [DOI] [PubMed] [Google Scholar]
- Nehrke K. Intracellular pH measurements in vivo using green fluores-cent protein variants. Methods Mol Biol. 2006;351:223–239. doi: 10.1385/1-59745-151-7:223. [DOI] [PubMed] [Google Scholar]
- Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226:299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- Perona R, Serrano R. Increased pH and tumorigenicity of fibroblasts expressing a yeast proton pump. Nature. 1988;334:438–440. doi: 10.1038/334438a0. [DOI] [PubMed] [Google Scholar]
- Reshkin SJ, Bellizzi A, Caldeira S, Albarani V, Malanchi I, Poignee M, Alunni-Fabbroni M, Casavola V, Tommasino M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000;14:2185–2197. doi: 10.1096/fj.00-0029com. [DOI] [PubMed] [Google Scholar]
- Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. 2009;174:1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Chen YY, Scott GK, Devries S, Chin K, Benz CC, Waldman FM, Hwang ES. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin Cancer Res. 2009;15:3163–3171. doi: 10.1158/1078-0432.CCR-08-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickett A, Kwan YL. T cell stimulation and expansion using anti-CD3/CD28 beads. J Immunol Methods. 2003;275:251–255. doi: 10.1016/s0022-1759(03)00010-3. [DOI] [PubMed] [Google Scholar]
- Valli M, Sauer M, Branduardi P, Borth N, Porro D, Mattanovich D. Intracellular pH distribution in Saccharomyces cerevisiae cell populations, analyzed by flow cytometry. Appl Environ Microbiol. 2005;71:1515–1521. doi: 10.1128/AEM.71.3.1515-1521.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelauer M, Wu J, Suka N, Grunstein M. Global histone acetylation and deacetylation in yeast. Nature. 2000;408:495–498. doi: 10.1038/35044127. [DOI] [PubMed] [Google Scholar]
- Wahl ML, Owen JA, Burd R, Herlands RA, Nogami SS, Rodeck U, Berd D, Leeper DB, Owen CS. Regulation of intracellular pH in human melanoma: potential therapeutic implications. Mol Cancer Ther. 2002;1:617–628. [PubMed] [Google Scholar]
- Walter A, Gutknecht J. Monocarboxylic acid permeation through lipid bilayer membranes. J Membr Biol. 1984;77:255–264. doi: 10.1007/BF01870573. [DOI] [PubMed] [Google Scholar]
- Waterborg JH. Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem Cell Biol. 2002;80:363–378. doi: 10.1139/o02-080. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Kleemann HW, Tannock IF. Cytostatic potential ofnovel agents that inhibit the regulation of intracellular pH. Br J Cancer. 2002;87:238–245. doi: 10.1038/sj.bjc.6600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Deng N, Zhao Z, Judeh T, Flemington E, Zhu D. SAMMate: a GUI tool for processing short read alignments in SAM/BAM format. Source Code Biol Med. 2011;6:2. doi: 10.1186/1751-0473-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lin Y, Gillies RJ. Tumor pH and its measurement. J Nucl Med. 2010;51:1167–1170. doi: 10.2967/jnumed.109.068981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.