

Abstract
Massive scleractinian corals of the genus Porites are important reef builders in the Indo-Pacific, and they are more resistant to thermal stress than other stony corals, such as the genus Acropora. Because coral health and survival largely depend on the interaction between a coral host and its symbionts, it is important to understand the molecular interactions of an entire “coral holobiont”. We simultaneously sequenced transcriptomes of Porites australiensis and its symbionts using the Illumina Hiseq2000 platform. We obtained 14.3 Gbp of sequencing data and assembled it into 74,997 contigs (average: 1,263 bp, N50 size: 2,037 bp). We successfully distinguished contigs originating from the host (Porites) and the symbiont (Symbiodinium) by aligning nucleotide sequences with the decoded Acropora digitifera and Symbiodinium minutum genomes. In contrast to previous coral transcriptome studies, at least 35% of the sequences were found to have originated from the symbionts, indicating that it is possible to analyze both host and symbiont transcriptomes simultaneously. Conserved protein domain and KEGG analyses showed that the dataset contains broad gene repertoires of both Porites and Symbiodinium. Effective utilization of sequence reads revealed that the polymorphism rate in P. australiensis is 1.0% and identified the major symbiotic Symbiodinium as Type C15. Analyses of amino acid biosynthetic pathways suggested that this Porites holobiont is probably able to synthesize most of the common amino acids and that Symbiodinium is potentially able to provide essential amino acids to its host. We believe this to be the first molecular evidence of complementarity in amino acid metabolism between coral hosts and their symbionts. We successfully assembled genes originating from both the host coral and the symbiotic Symbiodinium to create a snapshot of the coral holobiont transcriptome. This dataset will facilitate a deeper understanding of molecular mechanisms of coral symbioses and stress responses.
Introduction
Coral reefs are estimated to harbor roughly one-third of all described marine species, and their productivity supports approximately one-quarter of marine fisheries. The major architects of coral reefs, the scleractinian corals, are anthozoan cnidarians that form obligate endosymbioses with photosynthetic dinoflagellates of the genus Symbiodinium. The unicellular symbionts are harbored in the host coral's gastrodermal (endodermal) tissue in intracellular vacuoles known as symbiosomes [1], which are thought to originate from the plasma membrane of host cells during the initial acquisition of symbionts by a phagocytic process [2]. Although many of the details of the interaction between the host and Symbiodinium remain to be explored, this association enables the massive rates of calcification that distinguish reef-building corals from other anthozoans, such as sea anemones and zoanthids.
Coral reefs face a range of environmental changes, including ocean acidification, seawater temperature increases, and declines in coral abundance. Extensive loss of reef habitats is one of the most pressing environmental issues of our time [3]–[5]. Recently, increasing instances of “coral bleaching” have been observed. In most cases, coral bleaching is a breakdown of the mutualism between the coral and the photosynthetic dinoflagellate, resulting from a stress response to environmental perturbation. The integrity of the coral holobiont – a complex symbiosis between the coral animal, its endosymbiotic zooxanthellae, and an associated community of microorganisms is essential for maintaining coral health [6]. The molecular mechanisms underlying the collapse of symbiosis and the bleaching response are complex and are still little understood. Bleaching is currently viewed as a host response to a compromised symbiont that is analogous to the innate immune responses that occur in other host–microbe interactions. Expulsion or elimination of the symbiont from host tissues is thought to involve a variety of mechanisms, including exocytosis, host cell detachment, and host cell apoptosis [7].
Genomic information for cnidarians has been accumulating; the genomes of two non-symbiotic cnidarians, the anemone Nematostella vectensis (Anthozoa) [8], and a hydra, Hydra magnipapillata (Hydrozoa) [9], have been sequenced. The genome of a scleractinian coral, Acropora digitifera, was decoded using next-generation sequencing (NGS) technology [10]. The latter genome was estimated to comprise 420 Mbp and contains about 23,700 predicted protein-coding genes. Recently NGS-based transcriptome datasets have become available for several anthozoan cnidarians including several coral species: Acropora millepora [11], [12], A. palmata [13] and Pocillopora damicornis [14], and a sea anemone Aiptasia pallida [15]. In addition, transcriptomes of two cultured strains of Symbiodinium have been reported [16] and the nuclear genome of S. minutum was decoded [17]. In order to better understand the molecular interactions, it is worthwhile to simultaneously capture the molecular states of all organisms comprising a coral holobiont.
The massive stony corals of the genus Porites are common, important reef builders in the Indo-Pacific Ocean. More than 80 named species and numerous unclassified forms have been identified [18]. In contrast to Acropora species, Porites corals generally transmit their symbionts directly from parents to offspring (vertical transmission) rather than acquiring them anew from the environment (horizontal transmission) in each generation [19]. Porites species have thicker tissues and appear more robust to thermal stress than other corals, such as Acropora, that have thinner tissues [20]. Thus Porites corals have been used for comparative analyses of stress responses [21]. Laboratory studies indicate that acidified seawater reduces calcification of Porites corals [22]–[24]. Geochemical tracers, such as oxygen isotope ratios, strontium-calcium ratios, and heavy metal concentrations in growth rings of the CaCO3 Porites skeleton have been used to monitor changes in sea surface temperature, salinity, and/or marine pollutants [25]–[28]. By such means, Porites taxa have been used to reconstruct past environmental changes so as to better understand tropical climate systems and to predict climate change [29], [30]. Nevertheless, molecular information about Porites corals is still limited. To address this deficiency and to improve the utility of Porites as an environmental indicator, we sequenced the transcriptome of Porites australiensis and its symbiotic algae (Figure? 1) using next generation technology (Illumina HiSeq2000) and constructed a transcriptome dataset that contains a large proportion of a symbiotic alga.
Figure 1. The scleractinian coral, Porites australiensis.

(A) A P. australiensis colony used in this study. The diameter is approximately 20 cm. (B) High magnification photo of the P. australiensis. Dot circle indicates one polyp and tiny brown particles are symbiotic algae, dinoflagellates, Symbiodinium.
Materials and Methods
RNA isolation, Transcriptome sequencing and assembly
The coral sample used in this study was collected at Sesoko island, Okinawa, Japan, under the Okinawa prefecture permission (Number: 20–69). A small Porites australiensis colony that has been maintained in the Sesoko research station at the University of the Ryukyus for five years (Figure? 1) was used in this study. A fragment (2 cm diameter) from the coral was snap frozen in liquid nitrogen and pulverized with an iron mortar and pestle. Total RNA was isolated using an RNeasy RNA extraction kit (QIAGEN). Total RNA was then fragmented into about 200 bp lengths and an RNA-seq library was prepared using a TruSeq RNA Sample Prep Kit v2 (Illumina). cDNA normalization using a duplex-specific nuclease was also performed. The library was sequenced (100 bp paired-end reads) using the Illumina HiSeq 2000 platform. Library preparation, cDNA normalization, and sequencing were outsourced to Hokkaido System Science Corporation, Japan. Low quality bases (Phred quality value, QV≥20) were trimmed from the raw data and read pairs of at least 80 bp were retained using SolexaQA [31]. Possible PCR duplicates that originated during library preparation were removed with ConDeTri [32]. Contamination with TruSeq adapter sequences was removed by ea-utils (http://code.google.com/p/ea-utils). Subsequently high-quality paired end reads were assembled with Velvet/Oases software [33], [34] using different K-mer sizes [35], [45], [55] and merged by K-mer 27 using Oases [34]. For each word size, the longest isoform of each locus was selected. In addition, in order to recover low-expression genes and to produce longer assemblies, different coverage cut-off values (5, 8, 10 and 12) were also applied. Contigs originating from different coverage cutoff values were concatenated and redundant sequences and transcript-variants were removed by CDHIT-EST [35] using default parameters. Contigs over 200 bp length were retained. Additionally, contigs containing translated Open Reading Frames (ORFs) in which 95% of the amino acids were identical were considered duplicates. In such cases, one of the duplicates was removed using CDHIT [35].
Separation of host or symbiont-originated sequences
To distinguish whether contig sequences originated from host coral or symbiont Symbiodinium, nucleotide sequences of the assembled contigs were aligned to the genome sequences of both coral A. digitifera [10] and dinoflagellate S. minutum [17] by BLASTN, and contigs that show nucleotide similarities with A. digitifera or S. minutum were identified. Contigs aligned with the both genomes were not annotated in this study. Contigs that aligned only to the A. digitifera genome were annotated as “Porites contigs” while those that aligned only to the S. minutum genome were annotated as “Symbiodinium contigs”, respectively. Different e-value cutoffs (1e−1, 1e−2, 1e−3, 1e−4, 1e−5 and 1e−10) were examined and a cutoff that maximized the number of Porites or Symbiodinium contigs was adopted (Table S3). We also applied the same analyses for reported coral transcriptome data from A. millepora [12], A. palmata [13], and P. damicornis [14], in order to investigate proportions of Symbiodinium sequences in each dataset.
Annotation of transcriptome assembly
Assembled transcriptome data were annotated as follows: 1) by BLAST homology searches against public protein databases: NCBI non-redundant protein sequences (NR) and Swiss-Prot [36], 2) by assignment of Gene Ontology (GO) terms [37], 3) by mapping to pathways using the KEGG annotation service KAAS [38], and 4) conserved protein domain searches with Pfam [39]. BLASTX homology searches were conducted against the Swiss-Prot and NCBI NR protein databases and an e-value cutoff of 1e−5 was applied. For GO annotation, GO IDs were assigned based on the UniProt IDs of the best matches in the Swiss-Prot database [36]. Then Generic GO slim terms were assigned using map2slim.pl in go-perl (http://search.cpan.org/~cmungall/go-perl/). For KAAS pathway annotation and analysis, we used the bi-directional best-hit (BBH) method to query the set of organisms representative for ‘eukaryotes’ as suggested on the KAAS website, using default settings. To screen and identify conserved protein domains, we used the Pfam database (Pfam-A.hmm, release 24.0; http://pfam.sanger.ac.uk) [39]. Translated amino acid entries matching conserved domains were identified using HMMER searches (hmmer3) [40]. In order to avoid eliminating Symbiodinium- or coral-specific rapidly evolving domains, we used an e-value cutoff of 1e−3, as proposed by Kawashima et al. [41]. For assessing completeness of the Porites contigs, mutual best-hit blast analyses (TBLAST, BLASTX, 1e−5) were performed against the sea anemone Nematostella vectensis proteome [8] in order to identify possible orthologs between Porites and Nematostella. We used Nematostella as a reference proteome in order to compare number of orthologous pairs between A. digitifera [10] and Nematostella with those of Porites and Nematostella. Alignment coverage of each Porites contig across full-length amino acid sequence of its possible Nematostella ortholog were investigated based on the BLAST search. For Symbiodinium contigs, S. minutum proteome dataset [17] was used for identifying orthologous pairs.
Estimation of polymorphism in Porites
For estimating the polymorphism rate of P. australiensis, high-quality, trimmed Illumina reads (see above) were re-mapped against possible host-originated contigs (Porites contigs) using the Burrows-Wheeler Aligner (BWA) [42] and SNPs (single nucleotide polymorphisms) in each contigs were detected. Because BWA allows gaps in alignments, we set the maximum indel size to 5 bp (bwa aln -n 0.05; since the maximum read length is 100 bp) in order to detect small indels. SNPs and small indels were called using SAMtools software packages [43]. To ensure reliable and high quality variant calling, SNPs and small indels were called only for positions with a minimal mapping quality (-Q) of 25, a coverage value (-d) of 10, and a maximum read depth (-D) of 200 using the varFilter command in the SAMtools package.
Identification of the symbiotic Symbiodinium type
Illumina sequence data that originated from the Symbiodinium internal transcribed spacer 2 (ITS-2) region of the nuclear ribosomal array were identified using DNA sequences of Symbiodinium-specific ITS-2 primers used for genotyping: itsD and ITS-2rev2 [44]. Sequences reads containing these primer sequences were detected using BLAST (BLASTN) and 100% matched sequences were retrieved. Then these were subsequently assembled into a contig using Phrap [45] with default parameters. Sequence variations within the assembled ITS-2 sequence were checked with BWA [42] and SAMtools [43] as mentioned above.
Results and Discussion
Sequencing and assembly of the transcriptome
Total RNA was isolated from a single colony of P. australiensis (Figure? 1A), and then a normalized cDNA library was prepared and sequenced with the Illumina HiSeq2000 system. We obtained 71 million paired-end sequences (14.3 Gbp, Table S1). Raw sequence data were submitted to the DDBJ Sequence Read Archive (DRA) under accession number DRA000906 (BioProject ID: PRJDB731). Subsequently, 45 million quality-trimmed read pairs (QV≥20, both paired read ≥80 bp and PCR duplicates and TruSeq adapter sequences removed, approximately 9 Gbp, Table S1) were assembled using Velvet/Oases software (Figure? 2, Table? 1). Contigs of <200 bp were discarded because the RNA was fragmented into approximately 200-bp lengths during library preparation (see Material and Methods) and these short contigs were most likely to be truncated. After exclusion of redundant sequences and transcript variants, we obtained 74,997 contigs without ambiguous sequence gaps, totaling 94.73 Mbp (Figure? 2, Table? 2). Although the size distribution was weighted toward smaller contigs, there were 31,199 contigs with lengths >1,000 bp, 1,952 contigs >5,000 bp, and 198 contigs >10,000 bp, respectively (Figure? 3A). The maximum contig length reached 54,796 bp; the average was 1,263 bp, and the N50 size was 2,037 bp (Table? 2), indicating that the assembly is of comparable or better quality than extant anthozoan transcriptome assemblies using NGS platforms (Table S2). Assembled sequences have been submitted to the DDBJ/EMBL-Bank/GenBank Transcriptome Shotgun Assembly (TSA) Database under accession numbers FX435232-FX505330 and FX799345-FX804242.
Figure 2. Flowchart of transcriptome sequencing, assembly, and separation of host or symbiont sequences.
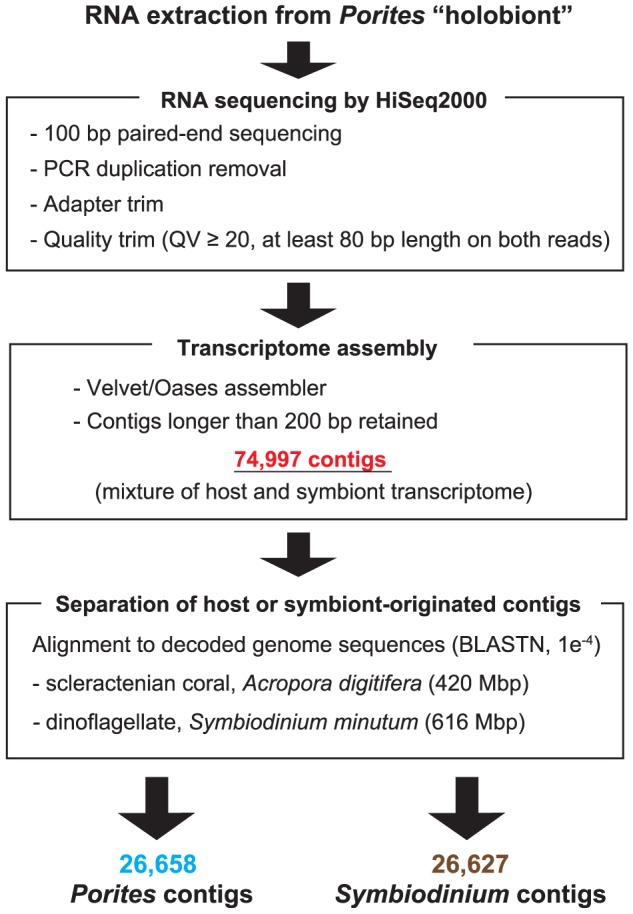
Table 1. Summary of the Porites australiensis “holobiont” transcriptome assembly.
| Number of unique sequences | 74,997 |
| Total basepair (Mbp) | 94.73 |
| Average (bp) | 1,263 |
| N50 size (bp) | 2,037 |
| Maximum contig length (bp) | 54,796 |
| Number of Porites cotigs | 26,658 (35.5%) |
| Total basepair (Mbp) | 44.57 |
| Average (bp) | 1,672 |
| N50 size (bp) | 2,960 |
| Maximum contig length (bp) | 44,581 |
| Number of Symbiodinium contigs | 26,627 (35.5%) |
| Total basepair (Mbp) | 32.01 |
| Average (bp) | 1,202 |
| N50 size (bp) | 1,571 |
| Maximum contig length (bp) | 12,901 |
Table 2. Summary of annotations of the transcriptome assembly.
| Number of sequence | Number of hit genes by BLAST search | Pfam | KEGG | ||||
| NCBI NR (BLASTX, 1e−5) | Swiss-Prot (BLASTX, 1e−5) | Number of domain | Number of annotated genes | Number of KO | Number of annotated genes | ||
| All | 74,997 | 33,935 (45.2%) | 25,893 (34.5%) | 6,081 | 30,002 (40%) | 5,001 | 7,452 (9.9%) |
| Porites contigs | 26,658 | 14,206 (53.3%) | 11,582 (43.4%) | 4,528 | 11,857 (44.5%) | 3,633 | 4,264 (16%) |
| Symbiodinium contigs | 26,627 | 13,813 (62.4%) | 10,240 (38.5%) | 3,569 | 13,513 (50.7%) | 1,352 | 1,854 (15.3%) |
Figure 3. Analyses of the Porites australiensis “holobiont” transcriptome assembly.

(A) Length distribution of transcripts in the transcriptome assembly. Yellow boxes indicate all contigs. Blue and brown lines indicate Porites contigs and Symbiodinium contigs, respectively. (B) Distribution of GC percentages of the assembled contigs. Black line: all contigs, blue dot line: Porites contigs, brown dot line: Symbiodinium contigs. (C) Comparison of proportions of Symbiodinium sequences between the scleractinian coral transcriptome assemblies. Each sequence was aligned to A. digitifera and S. minutum genome sequences by BLASTN (1e−5). Note that the high percentage of A. digitifera sequences shared with the A. millepora and A. palmata datasets occur because these three corals are congeneric.
Dissecting the “holobiont” transcriptome
Because total RNA was isolated from an adult P. australiensis colony (Figure? 1A), the transcriptome assembly contains genes from both corals and their zooxanthellae symbionts (Figure? 1B). When the GC content distribution of all contigs in the assembly was analyzed, two clear peaks of approximately 40% and 53% were detected (Figure? 3B), suggesting that these peaks possibly originated from the host (P. australiensis) and the symbiont (Symbiodinium sp.), respectively. The GC % of A. digitifera exons is about 40% [10] and that of a sea anemone, Nematostella, is similar [8], suggesting that a GC content of about 40% may be typical for anthozoan cnidarians. Therefore the 40% peak probably reflects P. australiensis genes. Taking into consideration the mean GC % of exons in the Symbiodinium minutum genome is about 50% [17], the secound peak might corresponds to the contigs from Symbiodinium.
Next we tried to identify contigs in the assembly that originated with Porites or Symbiodinium. We found that nucleotide alignment to both the Acropora and Symbiodinium genomes by BLASTN effectively separated them. In this study, we adopted an e-value cutoff 1e−4 (Table S3). 30,446 sequences hit against the A. digitifera genome and 30,415 sequences against S. minutum genome (3,788 sequences sequences overlapped) respectively (Table S3). We subsequently removed overlapping sequences, and annotated 26,658 sequences as “Porites contigs” and 26,627 sequences as “Symbiodinium contigs”. Annotated contigs comprised about 70% of all contigs and GC distributions of Porites and Symbiodinium contigs clearly matched the two peaks detected in the whole assembly (Figure? 3B), indicating that we effectively separated Porites and Symbiodinium genes in the assembly. These annotated contigs were used for further analyses (Table? 2, Figure? 3, 4).
Figure 4. Amino acid metabolism in Porites holobiont.

(A) Schematic drawing of amino acid exchange between a coral polyp and symbiotic Symbiodinium. (B) Summary of amino acid biosynthesic capabilities of Porites (host) and Symbiodinium (symbiont) inferred from KEGG annotation. Numbers indicate how many amino acids can be produced by Porites or Symbiodinium, and numbers in blankets show numbers of essential amino acids. Blue circle indicates Porites and brown circle indicates Symbiodinium, respectively. (C) Detail annotations of amino acid biosynthetic pathways in the Porites holobiont. Arrows indicate enzymes involved in each amino acid biosynthetic pathway. Enzymes that are not annotated as Porites or Symbiodinium are shown with a black line, and those not detected in the transcriptome dataset are shown with dotted lines. Blue indicates enzymes identified in Porites contigs. Brown indicates enzymes detected in Symbiodinium contigs. Brown arrows boxed in blue lines indicate enzymes detected in both Porites and Symbiodinium. Amino acids that can be produced by Symbiodinium are boxed in brown and those that can be produced by Porites are boxed in blue, respectively. Amino acids boxed in dotted lines indicate that biosynthetic capabilities are not clear.
In most transcriptome analyses of anthozoans [12], [13], [15], aposymbiotic materials were used for sequencing in order to minimize Symbiodinium contamination. Thus, we suspect that Symbiodinium sequences in those transcriptome assemblies have been under-detected. It has also been reported that RNA of adult P. damicornis colonies containing Symbiodinium was extracted, sequenced on a 454 platform and assembled, but that few Symbiodinium sequences had been isolated [14]. In contrast with previous studies, a large proportion of Symbiodinium sequences (at least 26,627 contigs, 35% of total assembled sequences) were in the transcriptome assembly (Figure? 3C). We confirmed that coral transcriptome data reported to date contained very few Symbiodinium-originated sequences (Figure? 3C). Therefore this study represents the first simultaneous transcriptome profile of both counterparts of coral holobiont.
Transcriptome annotation
For transcriptome annotation, BLAST homology searches against protein databases (NCBI NR and Swiss-Prot) were performed. Among 74,997 contigs, 33,935 (45.2%) have significant similarities (BLASTX, 1e−5) with NCBI NR and 25,893 (34.5%) with Swiss-Prot database entries, respectively. As mentioned above, 30,446 sequences showed similarity to the A. digitifera genome and 30,415 sequences to the S. minutum genome, respectively (Table? 2). Taken together, we were able to annotate 60,516 (80.7%) sequences using public protein databases (NCBI NR and Swiss-Prot) and A. digitifera [10] and S. minutum [17] genomic data. The remaining 14,481 sequences (total 7.12 Mbp) could not be annotated by BLAST and their average length (491.7 bp) was much shorter than that of the total assembly. These may be fragmented regions of other contigs, assembly artifacts, or bacterial contaminants. Some show similarities to coral microsatellite sequences deposited in the NCBI nucleotide (NT) database (data not shown).
Conserved protein domains in the contigs were identified with hmmer3 [40] using the Pfam domain database [39]. We identified 6,081 Pfam domains in 30,002 contigs (Table? 2). We were able to assign 5,001 KEGG ORTHOLOGY (KO) IDs to 7,452 contigs (Table? 2) using the KAAS BBH method [38].
Based on a BLAST search against the Swiss-Prot database, we were able to assign at least one GO term to the 11,048 Porites and 9,915 Symbiodinium contigs, respectively. These 20,963 contigs were grouped into 101 categories in the second hierarchy of the Generic GO slim. Figures S1, S2, S3 show GO annotation results for three GO sub-ontologies (cellular component, biological process, and molecular function). Since Porites corals are multicellular animals, cell death, neurological system process, cell proliferation, cell adhesion, and cell-cell signaling terms are more heavily represented than in Symbiodinium (2× more, Figure S1). DNA binding, enzyme binding, and transcription factor binding activity also seem to be higher in Porites (Figure S2). These may reflect the fact that 464 Porites contigs possess transcription factor domains (Table S4), whereas only 28 Symbiodinium contigs do (data not shown). On the other hand, transcripts of genes related to photosynthesis, transport, and generation of energy are more abundant among Symbiodinium contigs (Figure S1).
Estimating transcriptome completeness
About 44% of Porites contigs contain conserved protein domains and 4,528 unique Pfam domains were identified (Table? 2). In the gene models of the A. digitifera genome, unique 3,684 Pfam domains were detected [10], indicating that the Porites contigs probably contain a comparable variety of protein domains. In addition, we performed KEGG annotation and 3,633 unique KO IDs were identified among the Porites contigs (Table? 2). We detected 3,684 unique KO IDs in the A. digitifera proteome using the KAAS BBH method. A comparable number of KO IDs might indicate that, although RNA-seq was performed using a single RNA library, the majority of P. australiensis genes were successfully recovered in the transcriptome assembly. However it could be possible that those numbers in Porites contigs are overestimated due to incomplete clustering of transcript variants in the assembly.
An increasing number of studies have recently identified genes that encode transcription factors and signal transduction molecules in corals and have determined their roles in cnidarian evolution [46]–[50]. Therefore, for further assessing the completeness of the Porites transcriptome, the repertoire of transcription factors and signal transduction molecules was compared with those of other cnidarians whose genomes have been decoded (Table S4, 5). We detected comparable numbers of transcription factors and signaling molecules among the Porites contigs to what we previously found in the A. digitifera genome [10], e.g. Porites: 89 homeobox, 24 HMG box and 15 wnt genes; A. digitifera: 97 homeobox, 26 HMG box and 15 wnt genes (Table S4, 5). Next we identified orthologous gene pairs between Porites contigs and Nematostella by mutual-best-hit blast analysis; 7,825 pairs were detected. The similarities of the A. digitifera and Nematostella genomes resulted in 8,416 orthologous pairs, suggesting that Porites contigs represent the majority of the gene repertoire for Porites australiensis. 6,324 orthologous genes out of 7,825 have more than 80% alignment coverage across Nematostella orthologs, suggesting that most of the Porites contigs recover full-length open reading frames.
On the other hand, 3,569 Pfam domains and 1,352 KO IDs were detected among Symbiodinium contigs (Table? 2), while 4,035 Pfam domains and 2,622 KO IDs were detected in the S. minutum gene model [17], respectively. We also identified 28 Symbiodinium contigs containing transcription factor domains (data not shown). It has been reported that in transcriptome assemblies from two cultured strains of Symbiodinium, 156 and 87 sequences, respectively, contained at least one known transcription factor domain [16]. The S. minutum genome was found to contain 32 transcription factor genes [17]. We detected 15,914 orthologous pairs between the Symbiodinium contigs and S. minutum genome. In S. minutum genome, 23,487 orthologous groups were identified by compared with 150 genomes [17]. This might reflect that not all genes were expressed at the sampling point.
Polymorphism rate in Porites
Since total RNA was extracted from a single Porites colony, it is possible to estimate the polymorphism rate of P. australiensis within an individual. High quality Illumina reads were re-mapped against the reference Porites contigs using BWA software [42], and high quality SNPs and small indels (∼5 bp) within the Porites colony were identified using SAMtools [43]. Among the 44,569,031 nucleotides of the 26,658 Porites contigs, 425,728 SNPs and 11,788 small indels (total 20,213 bp) were detected. Taken together, the polymorphism rate between haplotypes, including small indels (less than 5 bp) and SNPs in the exons of Porites transcripts is estimated to be 1.0%, which is slightly higher than that observed in other cnidarian genomes (A. digitifera [0.4%, Shinzato and Mungpakdee, unpublished data], Nematostella [0.65%, 8], and Hydra [0.69%, 9].
Symbiont type
The genus Symbiodinium displays tremendous taxonomic diversity with nine divergent lineages. Clades A–I have been described in Symbiodinium based on nuclear ribosomal DNA (rDNA) and chloroplast 23S rDNA [51]. Each clade contains multiple genetic types, often resolved using the internal transcribed spacer 2 (ITS-2) regions [44], [52]. We failed to detect assembled sequences of nuclear rDNA or chloroplast 23S rDNA in our dataset. Accordingly, the ITS2 region-containing sequence reads were identified and assembled, and then we managed to reconstruct an ITS-2 region (341 bp) of the symbiotic Symbiodinium. BLAST homology searches against the NCBI NT database revealed that this sequence shows high similarity (340/341 bp identity) with clade C Symbiodinium sp. C15 (GenBank: JN558044). We also checked the sequence variation within the ITS2 region by BWA mapping and SAMtools, but no variation was detected (data not shown), indicating that the C15 type, a type common in Porites species across the Pacific [53], is the major Symbiodinium type in the P. australiensis colony used in this study.
Amino acid biosynthesis pathways in Porites holobiont
Bacteria, plants, and many fungi are able to synthesize all of the 20 amino acids commonly found in proteins, whereas all animals studied to date either lack the ability to synthesize one or more of these amino acids, or else they are unable to synthesize quantities sufficient to meet their metabolic needs. These amino acids are termed “essential” and must be obtained from the diet. For vertebrates, eight or more amino acids are essential; threonine, valine, methionine, leucine, isoleucine, phenylalanine, lysine, and tryptophan are required by all vertebrates, while arginine and/or histidine are also essential in some cases [54]. Although amino acid biosynthesis occurs via conserved pathways and is relatively well-characterized [55], amino acid exchange in cnidarian photoautotrophic symbioses is poorly understood. The issue of amino acid requirements is complicated by the presence of symbiotic algae [56], [57]. For example, examination of the amino acid biosynthetic capacity of the sea anemone Aiptasia pulchella – a facultative host of Symbiodinium sp. – suggested that seven amino acids (histidine, isoleucine, leucine, lysine, phenylalanine, tyrosine, and valine) are synthesized by the symbiotic algae and translocated to the sea anemone, and that methionine and threonine are likely to be synthesized by A. pulchella itself [57]. A genome-wide survey of amino acid biosynthetic pathway components in A. digitifera revealed that Acropora corals may be able to synthesize ten non-essential amino acids, but not cysteine [10], however amino acid metabolism in coral holobionts is still unclear (Figure? 4A). We reconstructed the amino acid biosynthetic pathways in the Porites holobiont based on KEGG IDs (Figure? 4B, C). In this study we took amino acid biosynthetic pathways missing one or two enzymes into consideration. Most of enzymes involved in amino acid biosynthetic pathways were detected in the transcriptomes of Porites and Symbiodinium, while four enzymes cannot be detected in the lysine anabolic pathway (Figure? 3C).
Interestingly many enzymes involved in essential amino acid biosynthesis are detected only in Symbiodinium contigs (Figure? 3B, Figure? 3C left side). Overrepresentation of “transport” GO terms in Symbiodinium contigs (Figure S1) implies that Symbiodinium transport essential amino acids to host cells. In contrast, enzymes for non-essential amino acid pathways were detected in both Symbiodinium and Porites (Figure? 3B, Figure? 3C, right side). Glutamic acid, glutamine, aspartic acid, and alanine are probably produced in both Porites and Symbiodinium. According our prediction, asparagine is synthesized in Symbiodinium while cysteine is synthesized in Porites (Figure? 3). We have shown previously that Acropora corals lack an essential enzyme for cysteine biosynthesis, cystatione ß-synthase [10], however, Porites seems to posses it (Figure? 3C), indicating that it does not depend upon Symbiodinium for cysteine biosynthesis and might account for its greater resilience to environmental stresses. An interesting example is the methionine biosynthesis pathway. Half of the enzymes reside in the Porites and Symbiodinium contigs, respectively, suggesting that methionine might be produced by intimate cooperation between host and symbiont in coral holobionts. While transcriptome sequencing was based upon a single RNA-seq library in this study, we were able to recover a surprisingly large gene repertoire for amino acid biosynthesis. Our data appear to provide the first direct molecular evidence of complementarity and syntrophy between coral hosts and their symbionts in amino acid metabolism.
Conclusions
We sequenced a Porites australiensis holobiont and demonstrated that decoded genomic data of an Acropora coral [10] and a Symbiodinium minutum [17] greatly assisted the characterization of assembled contigs from a mixture of RNAs from different organisms. In this study we were able distinguish about 70% of contigs as host or symbiont. The assembled sequences contain a wide variety of genetic information from both the coral and its symbiont, Symbiodinium, including genes for most enzymes in all amino acid biosynthetic pathways. When genome sequences of P. australiensis and its symbiotic Symbiodinium are available, the annotation could be further improved. The Porites holobiont transcriptome dataset allows us to utilize Porites australiensis, an abundant coral species from the Indo-Pacific, to reveal molecular mechanisms of coral symbiosis and coral stress responses. Our approach provides an opportunity to simultaneously analyze coral-symbiont interactions on transcriptomic level. Furthermore, analyzing the molecular bases of Porites calcification may enable researchers to improve the accuracy of future climate change prediction.
Supporting Information
Analysis of GO term enrichment of the “biological process” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Analysis of GO term enrichment of the “cellular component” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Analysis of GO term enrichment of the “molecular function” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Summary of the sequencing data.
(PDF)
Summary of published anthozoan transcriptome assemblies. References. 1. Moya A, Huisman L, Ball EE, Hayward DC, Grasso LC, Chua CM, Woo HN, Gattuso JP, Foret S, Miller DJ: Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO2-driven acidification during the initiation of calcification. Mol Ecol 2012, 21:2440–2454, 2. Polato NR, Vera JC, Baums IB: Gene discovery in the threatened elkhorn coral: 454 sequencing of the Acropora palmata transcriptome. PLoS One 2011, 6:e28634. 3. Traylor-Knowles N, Granger BR, Lubinski TJ, Parikh JR, Garamszegi S, Xia Y, Marto JA, Kaufman L, Finnerty JR: Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 2011, 12:585. 4. Lehnert EM, Burriesci MS, Pringle JR: Developing the anemone Aiptasia as a tractable model for cnidarian-dinoflagellate symbiosis: the transcriptome of aposymbiotic A. pallida. BMC Genomics 2012, 13:271.
(PDF)
Results of nucleotide sequence alignment of the assembled sequences with different e-value settings of BLASTN against Acropora digitifera and Symbiodinium minutum genome sequences. We selected e-value cut-off as 1e−4 in this study as the number of Porites contigs is the largest.
(PDF)
Comparison of the number of genes with transcription factor-related domains of Porites australiensis , Acropora digitifera , Nematostella vectensis and Hydra magnipapillata .
(PDF)
Comparison of the number of genes with signaling molecule-related domains of Porites australiensis , Acropora digitifera , Nematostella vectensis and Hydra magnipapillata .
(PDF)
List of Porites contigs (tab-separated text; contig names and NCBI accessions).
(ZIP)
List of Symbiodinium contigs (tab-separated text; contig names and NCBI accessions).
(ZIP)
Annotation files for all contigs (tab-separated text; contig name, NCBI accession, Swiss-Prot blast result, Pfam ID, KEGG ID).
(ZIP)
Acknowledgments
We thank members of Marine Genomics Unit, at OIST, for allowing us use their facilities, and Dr. Akira Iguchi (Sesoko Station, University of the Ryukyus) for helping with the preparation of coral samples. We thank Dr. Konstantin Khalturin (OIST) for critical reading of the manuscript. We thank Dr. Steven Aird (OIST) for editing the manuscript.
Funding Statement
This study was supported by the Interdisciplinary Collaborative Research Program of the Atmosphere and Ocean Research Institute, the University of Tokyo and by The Canon Foundation and in part by KAKENHI (24241071 and 25660172). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Yellowlees D, Rees TA, Leggat W (2008) Metabolic interactions between algal symbionts and invertebrate hosts. Plant Cell Env 31: 679–694 Available: http://www.ncbi.nlm.nih.gov/pubmed/18315536. [DOI] [PubMed] [Google Scholar]
- 2. Hohman TC, McNeil PL, Muscatine L (1982) Phagosome-lysosome fusion inhibited by algal symbionts of Hydra viridis. J? Cell Biol 94: 56–63 Available: http://www.ncbi.nlm.nih.gov/pubmed/7119017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carpenter KE, Abrar M, Aeby G, Aronson RB, Banks S, et al. (2008) One-third of reef-building corals face elevated extinction risk from climate change and local impacts. Science (80-) 321: 560–563 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18653892. [DOI] [PubMed] [Google Scholar]
- 4. Hoegh-Guldberg O, Mumby PJ, Hooten AJ, Steneck RS, Greenfield P, et al. (2007) Coral reefs under rapid climate change and ocean acidification. Science (80-) 318: 1737–1742 Available: http://www.ncbi.nlm.nih.gov/pubmed/18079392. [DOI] [PubMed] [Google Scholar]
- 5. Hughes TP, Baird AH, Bellwood DR, Card M, Connolly SR, et al. (2003) Climate change, human impacts, and the resilience of coral reefs. Science (80-) 301: 929–933 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12920289. [DOI] [PubMed] [Google Scholar]
- 6. Bourne DG, Garren M, Work TM, Rosenberg E, Smith GW, et al. (2009) Microbial disease and the coral holobiont. Trends Microbiol 17: 554–562 Available: http://www.ncbi.nlm.nih.gov/pubmed/19822428. [DOI] [PubMed] [Google Scholar]
- 7. Weis VM (2010) The susceptibility and resilience of corals to thermal stress: adaptation, acclimatization or both? Mol Ecol 19: 1515–1517 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20456235. [DOI] [PubMed] [Google Scholar]
- 8. Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, et al. (2007) Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science (80-) 317: 86–94 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17615350. [DOI] [PubMed] [Google Scholar]
- 9. Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, et al. (2010) The dynamic genome of Hydra. Nature 464: 592–596 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, et al. (2011) Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476: 320–323 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21785439. [DOI] [PubMed] [Google Scholar]
- 11. Meyer E, Aglyamova G V, Wang S, Buchanan-Carter J, Abrego D, et al. (2009) Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 10: 219 Available: http://www.ncbi.nlm.nih.gov/pubmed/19435504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moya A, Huisman L, Ball EE, Hayward DC, Grasso LC, et al. (2012) Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO(2)-driven acidification during the initiation of calcification. Mol Ecol 21: 2440–2454 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22490231. [DOI] [PubMed] [Google Scholar]
- 13. Polato NR, Vera JC, Baums IB (2011) Gene discovery in the threatened elkhorn coral: 454 sequencing of the Acropora palmata transcriptome. PLoS One 6: e28634 Available: http://www.ncbi.nlm.nih.gov/pubmed/22216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Traylor-Knowles N, Granger BR, Lubinski TJ, Parikh JR, Garamszegi S, et al. (2011) Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 12: 585 Available: http://www.ncbi.nlm.nih.gov/pubmed/22126435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lehnert EM, Burriesci MS, Pringle JR (2012) Developing the anemone Aiptasia as a tractable model for cnidarian-dinoflagellate symbiosis: the transcriptome of aposymbiotic A. pallida. BMC Genomics 13: 271 Available: http://www.ncbi.nlm.nih.gov/pubmed/22726260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bayer T, Aranda M, Sunagawa S, Yum LK, Desalvo MK, et al. (2012) Symbiodinium transcriptomes: genome insights into the dinoflagellate symbionts of reef-building corals. PLoS One 7: e35269 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22529998. Accessed 29 November 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shoguchi E, Shinzato C, Kawashima T, Gyoja F, Mungpakdee S, et al. (2013) Draft Assembly of the Symbiodinium minutum Nuclear Genome Reveals Dinoflagellate Gene Structure. Curr Biol Available: http://www.ncbi.nlm.nih.gov/pubmed/23850284. [DOI] [PubMed] [Google Scholar]
- 18.Appeltans W Boxshall GA, De Broyer C, de Voogd NJ, Gordon DP, Hoeksema BW, Horton T, Kennedy M, Mees J, Poore GCB, Read G, Stöhr S, Walter TC, Costello MJ BP (2012) World Register of Marine Species.
- 19. LaJeunesse TC, Thornhill DJ, Cox EF, Stanton FG, Fitt WK, et al. (2004) High diversity and host specificity observed among symbiotic dinoflagellates in reef coral communities from Hawaii. Coral Reefs 23: 596–603 Available: <Go to ISI>://000226095100021. [Google Scholar]
- 20. Loya Y, Sakai K, Yamazato K, Nakano Y, Sambali H, et al. (2001) Coral bleaching: the winners and the losers. Ecol Lett 4: 122–131 Available: <Go to ISI>://000167731300005. [Google Scholar]
- 21. Fitt WK, Gates RD, Hoegh-Guldberg O, Bythell JC, Jatkar A, et al. (2009) Response of two species of Indo-Pacific corals, Porites cylindrica and Stylophora pistillata, to short-term thermal stress: The host does matter in determining the tolerance of corals to bleaching. J? Exp Mar Bio Ecol 373: 102–110 Available: <Go to ISI>://000266897400004. [Google Scholar]
- 22. Anthony KRN, Kline DI, Diaz-Pulido G, Dove S, Hoegh-Guldberg O (2008) Ocean acidification causes bleaching and productivity loss in coral reef builders. Proc Natl Acad Sci U S A 105: 17442–17446 Available: <Go to ISI>://000260981800051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iguchi A, Ozaki S, Nakamura T, Inoue M, Tanaka Y, et al. (2012) Effects of acidified seawater on coral calcification and symbiotic algae on the massive coral Porites australiensis. Mar Environ Res 73: 32–36 Available: <Go to ISI>://000300818900005. [DOI] [PubMed] [Google Scholar]
- 24. Ohde S, Hossain MMM (2004) Effect of CaCO3 (aragonite) saturation state of seawater on calcification of Porites coral. Geochem? J 38: 613–621 Available: <Go to ISI>://000226353600012. [Google Scholar]
- 25. Beck JW, Edwards RL, Ito E, Taylor FW, Recy J, et al. (1992) Sea-Surface Temperature from Coral Skeletal Strontium Calcium Ratios. Science (80-) 257:: 644–647 Available: <Go to ISI>://A1992JF85200028. [DOI] [PubMed] [Google Scholar]
- 26. Gagan MK, Ayliffe LK, Beck JW, Cole JE, Druffel ERM, et al. (2000) New views of tropical paleoclimates from corals. Quat Sci Rev 19: 45–64 Available: <Go to ISI>://000084425500006. [Google Scholar]
- 27. Inoue M, Tanimizu M (2008) Anthropogenic lead inputs to the western Pacific during the 20th century. Sci Total Environ 406: 123–130 Available: <Go to ISI>://000260941400012. [DOI] [PubMed] [Google Scholar]
- 28. Shen GT, Boyle EA, Lea DW (1987) Cadmium in Corals as a Tracer of Historical Upwelling and Industrial Fallout. Nature 328: 794–796 Available: <Go to ISI>://A1987J762900054. [Google Scholar]
- 29. Cobb KM, Charles CD, Cheng H, Edwards RL (2003) El Nino/Southern Oscillation and tropical Pacific climate during the last millennium. Nature 424: 271–276 Available: http://www.ncbi.nlm.nih.gov/pubmed/12867972. [DOI] [PubMed] [Google Scholar]
- 30.Solomon S, Intergovernmental Panel on Climate Change., Intergovernmental Panel on Climate Change. Working Group I. (2007) Climate change 2007: the physical science basis: contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge; New York: Cambridge University Press. Available: http://www.loc.gov/catdir/enhancements/fy0806/2007282362-d.html. [Google Scholar]
- 31. Cox MP, Peterson DA, Biggs PJ (2010) SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11: 485 Available: http://www.ncbi.nlm.nih.gov/pubmed/20875133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smeds L, Kunstner A (2011) ConDeTri–a content dependent read trimmer for Illumina data. PLoS One 6: e26314 Available: http://www.ncbi.nlm.nih.gov/pubmed/22039460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18349386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28: 1086–1092 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22368243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16731699. [DOI] [PubMed] [Google Scholar]
- 36. Consortium U (2011) Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res 39: D214–9 Available: http://www.ncbi.nlm.nih.gov/pubmed/21051339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 Available: http://www.ncbi.nlm.nih.gov/pubmed/10802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–5 Available: http://www.ncbi.nlm.nih.gov/pubmed/17526522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, et al. (2006) Pfam: clans, web tools and services. Nucleic Acids Res 34: D247–51 Available: http://www.ncbi.nlm.nih.gov/pubmed/16381856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14: 755–763 Available: http://www.ncbi.nlm.nih.gov/pubmed/9918945. [DOI] [PubMed] [Google Scholar]
- 41. Kawashima T, Kawashima S, Tanaka C, Murai M, Yoneda M, et al. (2009) Domain shuffling and the evolution of vertebrates. Genome Res 19: 1393–1403 Available: http://www.ncbi.nlm.nih.gov/pubmed/19443856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 Available: http://www.ncbi.nlm.nih.gov/pubmed/19451168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 Available: http://www.ncbi.nlm.nih.gov/pubmed/19505943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pochon X, Garcia-Cuetos L, Baker AC, Castella E, Pawlowski J (2007) One-year survey of a single Micronesian reef reveals extraordinarily rich diversity of Symbiodinium types in soritid foraminifera. Coral Reefs 26: 867–882 Available: <Go to ISI>://000251579300016. [Google Scholar]
- 45. Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8: 195–202 Available: http://www.ncbi.nlm.nih.gov/pubmed/9521923. [DOI] [PubMed] [Google Scholar]
- 46. Shinzato C, Iguchi A, Hayward DC, Technau U, Ball EE, et al. (2008) Sox genes in the coral Acropora millepora: divergent expression patterns reflect differences in developmental mechanisms within the Anthozoa. BMC Evol Biol 8: 311 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19014479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Jong DM, Hislop NR, Hayward DC, Reece-Hoyes JS, Pontynen PC, et al. (2006) Components of both major axial patterning systems of the Bilateria are differentially expressed along the primary axis of a “radiate” animal, the anthozoan cnidarian Acropora millepora. Dev Biol 298: 632–643 Available: <Go to ISI>://000241180300024. [DOI] [PubMed] [Google Scholar]
- 48. Kusserow A, Pang K, Sturm C, Hrouda M, Lentfer J, et al. (2005) Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433: 156–160 Available: http://www.ncbi.nlm.nih.gov/pubmed/15650739. [DOI] [PubMed] [Google Scholar]
- 49. Dubuc TQ, Ryan JF, Shinzato C, Satoh N, Martindale MQ (2012) Coral Comparative Genomics Reveal Expanded Hox Cluster in the Cnidarian-Bilaterian Ancestor. Integr Comp Biol Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22767488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simionato E, Ledent V, Richards G, Thomas-Chollier M, Kerner P, et al. (2007) Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol Biol 7 Available: <Go to ISI>://000244937200001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pochon X, Gates RD (2010) A new Symbiodinium clade (Dinophyceae) from soritid foraminifera in Hawai'i. Mol Phylogenet Evol 56: 492–497 Available: <Go to ISI>://000278589500047. [DOI] [PubMed] [Google Scholar]
- 52. Stat M, Bird CE, Pochon X, Chasqui L, Chauka LJ, et al. (2011) Variation in Symbiodinium ITS2 sequence assemblages among coral colonies. PLoS One 6: e15854 Available: http://www.ncbi.nlm.nih.gov/pubmed/21246044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. LaJeunesse TC, Loh WKW, van Woesik R, Hoegh-Guldberg O, Schmidt GW, et al. (2003) Low symbiont diversity in southern Great Barrier Reef corals, relative to those of the Caribbean. Limnol Oceanogr 48: 2046–2054 Available: <Go to ISI>://000185433700030. [Google Scholar]
- 54. Furst P, Stehle P (2004) What are the essential elements needed for the determination of amino acid requirements in humans? J? Nutr 134: 1558S–1565S Available: http://www.ncbi.nlm.nih.gov/pubmed/15173430. [DOI] [PubMed] [Google Scholar]
- 55. Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H (2000) Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407: 81–86 Available: http://www.ncbi.nlm.nih.gov/pubmed/10993077. [DOI] [PubMed] [Google Scholar]
- 56. Swanson R, Hoegh-Guldberg O (1998) Amino acid synthesis in the symbiotic sea anemone Aiptasia pulchella. Mar Biol 131: 83–93 Available: <Go to ISI>://000073613500010. [Google Scholar]
- 57. Wang JT, Douglas AE (1999) Essential amino acid synthesis and nitrogen recycling in an alga-invertebrate symbiosis. Mar Biol 135: 219–222 Available: <Go to ISI>://000083839200002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of GO term enrichment of the “biological process” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Analysis of GO term enrichment of the “cellular component” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Analysis of GO term enrichment of the “molecular function” category for Porites and Symbiodinium contigs. GO terms containing at least 100 sequences are shown. The Y-axis represents proportions of contigs in each category of GO-assigned contigs (Porites: 9806, Symbiodinium: 9147). Blue bars indicate Porites contigs. Red bars indicate Symbiodinium contigs.
(PDF)
Summary of the sequencing data.
(PDF)
Summary of published anthozoan transcriptome assemblies. References. 1. Moya A, Huisman L, Ball EE, Hayward DC, Grasso LC, Chua CM, Woo HN, Gattuso JP, Foret S, Miller DJ: Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO2-driven acidification during the initiation of calcification. Mol Ecol 2012, 21:2440–2454, 2. Polato NR, Vera JC, Baums IB: Gene discovery in the threatened elkhorn coral: 454 sequencing of the Acropora palmata transcriptome. PLoS One 2011, 6:e28634. 3. Traylor-Knowles N, Granger BR, Lubinski TJ, Parikh JR, Garamszegi S, Xia Y, Marto JA, Kaufman L, Finnerty JR: Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 2011, 12:585. 4. Lehnert EM, Burriesci MS, Pringle JR: Developing the anemone Aiptasia as a tractable model for cnidarian-dinoflagellate symbiosis: the transcriptome of aposymbiotic A. pallida. BMC Genomics 2012, 13:271.
(PDF)
Results of nucleotide sequence alignment of the assembled sequences with different e-value settings of BLASTN against Acropora digitifera and Symbiodinium minutum genome sequences. We selected e-value cut-off as 1e−4 in this study as the number of Porites contigs is the largest.
(PDF)
Comparison of the number of genes with transcription factor-related domains of Porites australiensis , Acropora digitifera , Nematostella vectensis and Hydra magnipapillata .
(PDF)
Comparison of the number of genes with signaling molecule-related domains of Porites australiensis , Acropora digitifera , Nematostella vectensis and Hydra magnipapillata .
(PDF)
List of Porites contigs (tab-separated text; contig names and NCBI accessions).
(ZIP)
List of Symbiodinium contigs (tab-separated text; contig names and NCBI accessions).
(ZIP)
Annotation files for all contigs (tab-separated text; contig name, NCBI accession, Swiss-Prot blast result, Pfam ID, KEGG ID).
(ZIP)