

Abstract
The recognition that few human diseases are thoroughly addressed by mono-specific, monoclonal antibodies (mAbs) continues to drive the development of antibody therapeutics with additional specificities and enhanced activity. Historically, efforts to engineer additional antigen recognition into molecules have relied predominantly on the reformatting of immunoglobulin domains. In this report we describe a series of fully functional mAbs to which additional specificities have been imparted through the recombinant fusion of relatively short polypeptides sequences. The sequences are selected for binding to a particular target from combinatorial libraries that express linear, disulfide-constrained, or domain-based structures. The potential for fusion of peptides to the N- and C- termini of both the heavy and light chains affords the bivalent expression of up to four different peptides. The resulting molecules, called zybodies, can gain up to four additional specificities, while retaining the original functionality and specificity of the scaffold antibody. We explore the use of two clinically significant oncology antibodies, trastuzumab and cetuximab, as zybody scaffolds and demonstrate functional enhancements in each case. The affect of fusion position on both peptide and scaffold function is explored, and penta-specific zybodies are demonstrated to simultaneously engage five targets (ErbB2, EGFR, IGF-1R, Ang2 and integrin αvβ3). Bispecific, trastuzumab-based zybodies targeting ErbB2 and Ang2 are shown to exhibit superior efficacy to trastuzumab in an angiogenesis-dependent xenograft tumor model. A cetuximab-based bispecific zybody that targeting EGFR and ErbB3 simultaneously disrupted multiple intracellular signaling pathways; inhibited tumor cell proliferation; and showed efficacy superior to that of cetuximab in a xenograft tumor model.
Keywords: antibody engineering, multi-specific antibody
Introduction
Exquisite target specificity, bivalent binding, innate effector function and inherent in vitro and in vivo stability have allowed monoclonal antibodies (mAbs) to be successfully exploited for the treatment of human disease. Ironically, the mono-specificity of mAbs that has fueled their therapeutic success may limit their application across a broad spectrum of diseases in which there are often multiple and redundant mechanisms that promote or progress the disease. Combination therapies that comprise the co-administration of a mAb with small molecule drugs1 or mixtures of mAbs2 have partially addressed this limitation, but such combinations remain challenged by the additive costs of and time for parallel discovery and development. The ability to extend the functionality of mAbs to include multiple specificities and valencies, while retaining mAb-like production and stability, affords the opportunity to address the inherent complexity of human diseases in ways that are not possible with conventional mAbs.
In recent years, a number of multi-specific and multi-valent biomolecules have been engineered from immunoglobulin (Ig) or non-Ig sub-domains, modified full-length antibodies or heterodimeric mAbs. Sub-domain approaches (e.g., dAbs,3 Adnectins4 and affibodies5) are inherently mono-specific and mono-valent, but can be concatenated to form low-order, multi-specific and multi-valent biomolecules (e.g., TandAbs6). These formats display in vivo half-lives that are substantially less than those of conventional mAbs, and thus require further engineering, such as pegylation or fusion to an Fc domain, to extend half-life. Modified antibody formats (e.g., two-in-one,7 DVD-Ig,8 antigen-binding CH3 domains9 and chemically programmed antibodies10) are built upon a full-length antibody scaffold and therefore retain many of the structural and functional properties of conventional mAbs. As a consequence of their design, however, this group remains largely bi-valent and bispecific. Heterodimeric mAbs (e.g., triomabs,11 knobs-into-holes,12 Crossmabs13 and electrostatic matching14), can often yield thermodynamically stable proteins with inherent Fc-mediated effector function, but are also limited to bispecificity and mono-valent interactions with their targets.
We sought to develop a platform of antibody-based therapeutics with greater degrees of specificity and valency, without compromising the drug-like properties of the parental antibody upon which they were built. In comparison to antibodies, peptides and small, domain-based proteins (e.g., affibodies) face developmental challenges as independent therapeutics due in part to their inherently shorter in vivo half-lives, mono-valency and mono-functionality. However, these deficiencies may be ameliorated through recombinant fusion to an antibody scaffold. We have previously described the use of target-specific peptides to impart bispecificity and enhanced efficacy to adalimumab (Humira®) in an in vivo model of arthritis.15 In this report we expand on several aspects of these molecules, which are termed zybodies. Since the peptides may be recombinantly fused to the N- or C-termini of the heavy and light chains, we investigate the effects of fusion to each of the four positions. In addition to linear and disulfide-constrained peptides, we explore the use of small, domain-based proteins and characterize a series of penta-specific zybodies that can simultaneously engage ErbB2, EGFR, IGF-1R, Ang2 and integrin αvβ3. Finally, we expand the platform for use in oncology indications by developing zybodies based on two clinically significant mAbs, trastuzumab (Herceptin®) and cetuximab (Erbitux®). We demonstrate that these zybodies simultaneously disrupt multiple intracellular signaling pathways; inhibit tumor cell proliferation; and, as compared with the parental antibodies, show superior efficacy in xenograft tumor models.
Results
Zybody composition
A zybody is a full-length mAb to which modular recognition domains (MRDs), are fused recombinantly via the N- and C-termini of the mAb heavy and light chains (Fig. 1A). MRDs are relatively small polypeptides sequences (typically less than 60 amino acids) selected for target specificity from combinatorial libraries. We chose sequences representing small, domain-based structures such as knottins16 or affibodies,5 as well as unstructured (linear) or disulfide-constrained peptides. The MRD sequences described in this report are presented in Table 1. Depending on the number of MRDs fused to the mAb scaffold, zybodies may be from bi- to penta-specific, with each of the specificity represented bivalently (Fig. 1B). Additional valencies can be achieved by fusion of a single MRD to multiple termini. The arming of a conventional mAb with up to four additional specificities requires unique nomenclature that defines the mAb scaffold, MRD location and corresponding specificities as illustrated in Figure 1C.
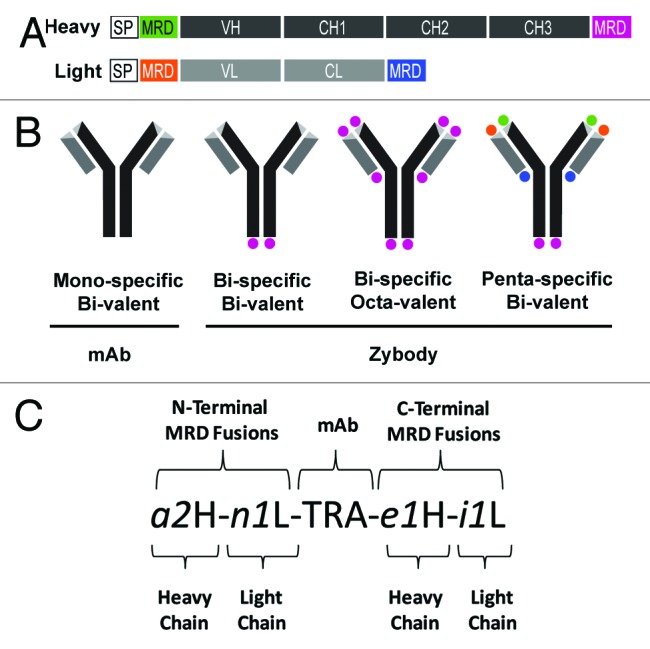
Figure 1. Schematic diagram of zybody assembly and nomenclature. (A) Schematic representation of immunoglobulin heavy (H) and light (L) chain genetic fusions that constitute a penta-specific zybody with four unique MRDs. Mouse γ and κ chain signal peptides (SP) were fused to the heavy and light chains, respectively. Signal peptides, Ig variable (V) and constant (C) domains, and MRDs are encoded in one contiguous open reading frame. (B) A schematic representation of a mono-specific antibody scaffold along with three exemplary zybodies, illustrating varying degrees of specificity and valency. (C) The zybody nomenclature identifies the antibody scaffold by a capitalized three-letter code (e.g., TRA for trastuzumab, ADA for adalimumab, CET for cetuximab, PAL for palivizumab). MRDs are indentified with an italicized, lowercase alphanumeric code (e.g., a2). The letter of the MRD code indicates the MRD target (e.g., i for IGF-1R, e for EGFR, n for integrin αvβ3, a for Ang2, c for ErbB3), followed by a unique numeric identifier and either an H (heavy) or an L (light), indicating the chain to which it is fused. The terminus to which the MRD is fused can be inferred from the position of the MRD identifier relative to the scaffold.
Table 1. Modular recognition domains (MRDs).
| Target | MRD ID | Structure | Reference | Sequence |
|---|---|---|---|---|
| Ang2 | a1 | Disulfide Constrained | Zyngenia | LWDDCYFFPNPPHCYNSP |
| Ang2 | a2 | Linear | Zyngenia | DEHQTNFLPLDQDEALLYEEFILQQGLE |
| IGF1R | i1 | Affibody | Li, 2010 | VDNKFNKEGFYAAIEILALPNLNRKQSTAFISSLEDDPSQSANLLAEAKKLNDAQAPK |
| EGFR | e1 | Affibody | Gostring, 2010 | VDNKFNKEMWAAWEEIRNLPNLNGWQMTAFIASLVDDPSQSANLLAEAKKLNDAQAPK |
| ErbB3 | c1 | Affibody | Kronqvist, 2011 | VDNKFNKERYSAYYEIWQLPNLNVRQKAAFIGSLQDDPSQSANLLAEAKKLNDAQAPK |
| αvβ3 | n1 | Knottin | Kimura, 2009 | GCPQGRGDWAPTSCKQDSDCRAGCVCGPNGFCG |
MRDs used in zybody construction were adapted from the indicated references.
Production and analytical characterization
The initial steps to validate the zybody platform were to: (1) establish the ability of the mAb scaffold to accommodate MRD fusions at its each terminus; (2) define the number of specificities that could be added without altering scaffold function; and (3) explore the diversity of MRD structures amenable to the platform. Zybodies and the parent scaffold mAbs described here are readily produced in HEK293F suspension cells that are transiently transfected with equimolar ratios of independent heavy and light chain cDNA expression constructs,17 and typically yielded protein levels that ranged from 50 to 250 μg/ml. Larger scale bioreactors (10 L) seeded with stably transfected CHO cell lines routinely achieve 1 g/l under non-optimized conditions (Fig. S1). Affinity purification on protein A yielded proteins that have SDS PAGE and analytical size-exclusion chromatography migration patterns of the anticipated size and homogeneity (Fig. S2A and S2B). Furthermore, as measured by differential scanning calorimetry (DSC) and 30-d accelerated (at 50°C) stability studies, the zybodies displayed in vitro stability profiles that are comparable to those of the parental trastuzumab (Fig. S2C and S2D). These data demonstrate that zybodies are readily expressed using conventional mAb production processes and that the fusion of MRDs does not significantly change mAb stability.
Binding characteristics
To assess the effects of the position of the MRD on binding and function of both MRD and scaffold mAb, four trastuzumab-based, bispecific zybodies were produced (TRA-a1H, TRA-a1L, a1H-TRA and a1L-TRA). In these zybodies, the same Ang2 antagonizing MRD (a1) was fused to the C- or N-terminus of the heavy or light chain. The a1 MRD was identified and affinity matured through phage display of short (10–18 amino acid), disulfide-constrained combinatorial peptide libraries and was subsequently fused to the antibody scaffold via peptide linkers (5 to 12 residues) composed of glycine and serine residues.
Analysis by surface plasmon resonance (SPR) of MBP-a1, a C-terminal fusion of a1 with the bacterial maltose binding protein (MBP) revealed that the a1 MRD has a monovalent affinity for Ang2 of 22.20 (± 0.08) nM (Table S2). Trastuzumab-based zybodies, containing the same MRD, exhibited 6 to 17-fold tighter binding to Ang2, likely demonstrating the avidity advantage introduced by the bivalent expression of the MRD. Fusion of a1 to the C-terminus of the light chain (TRA-a1L) resulted in a 2–3-fold increase in KD compared with zybodies modified at the other three positions. The reduced affinity of the C-terminal light chain fusion is consistent with other MRDs that we have explored at this position, including the affibodies i1 and e1, but may also reflect sub-optimal linker design. A similar affinity ranking of the MRDs were confirmed for all the four bispecific zybodies in direct binding ELISA (Fig. 2A, left).

Figure 2. Characterization of zybody binding. (A) The affect of position of the MRD on Ang2 binding was measured for trastuzumab (TRA) and four bispecific, bi-valent trastuzumab-based zybodies containing an Ang2-binding MRD, a1. Zybodies were assessed for their ability to bind to Ang2 (left), ErbB2-Fc (center) and FcγRIIIa (right). (B) Simultaneous binding to cell surface ErbB2 and soluble Ang2 was assessed by FACS after incubating SKBR3 cells with (a) anti-human Igκ FITC, (b) TRA-a1H and anti-human Igκ FITC, (c) TRA-a1H and Ang2-biotin, (d) TRA-a1H, anti-human Igκ FITC and Ang2-biotin. Ang2-biotin binding was detected using streptavidin-PE.
The affect of the position of the MRD on the ability of the trastuzumab scaffold to bind to ErbB2 was also assessed. SPR analysis showed that TRA-a1H, TRA-a1L and a1H-TRA bound ErbB2 with affinities comparable to that of trastuzumab, whereas the N-terminal, light chain fusion, a1L-TRA, displayed a modest 2-fold increase in the KD (Table S2). Studies by ELISA using immobilized ErbB2-Fc confirmed the modest decrease in binding affinity of a1L-TRA relative to the zybodies (Fig. 2A, middle).
A critical functional attribute of IgG1 antibodies is their ability to affect antibody-dependent cell cytotoxicity (ADCC) through direct binding to FcRγIIIa (CD16). To determine if the fusion of an Ang2-binding MRD to the termini of trastuzumab altered FcRγIIIa binding, the bispecific zybodies were assayed for direct binding to immobilized FcRγIIIa in a standard ELISA. As shown in Figure 2A right, comparable FcRγIIIa engagement was observed for trastuzumab and each of the four bispecific zybodies. Overall, the target-binding analyses of the bispecific zybodies indicate that each of the four termini of a mAb scaffold are acceptable fusion points for MRDs and that such fusions have little or no effect on the binding of the parental mAb to either its target or to FcRγIII.
To determine whether zybodies were able to bind cell surface and soluble targets simultaneously, SK-BR3 cells (which express ErbB2, but not Ang2 or its cellular receptor, Tie-2) were incubated with TRA-a1H and Ang2. Bound zybody was visualized by flow cytometric analyses using FITC-labeled mouse anti-human Igκ and/or PE-labeled Ang2 (Fig. 2B). Cell-bound zybody was readily detected with the individual fluorochromes (Fig. 2B, b and c) and the homogenous double-staining of cells (Fig. 2B, d) shows the simultaneous binding of zybody to cell surface ErbB2 and soluble Ang2.
To explore both the number and structural diversity of MRDs that can be simultaneously incorporated into a zybody, a trastuzumab-based penta-specific zybody (a2H-n1L-TRA-e1H-i1L) was constructed with antagonistic MRDs specific for Ang2 (a2), EGFR (e1), αvβ3 integrin (n1) and IGF-1R (i1). The MRDs were derived from two affibodies, engineered to target IGF-1R18 and EGFR,19 a knottin specific for αvβ3 integrin16 and an Ang2-binding peptide, identified through phage display of a randomized, unstructured peptide library. The zybody was captured on a SPR sensor chip by immobilized EGFR-Fc through its EGFR-binding MRD and then exposed sequentially to the soluble ErbB2-Fc, IGF-1R, Ang2 and αvβ3 integrin (Fig. 3A). The initial increase in mass caused by the binding of the penta-specific zybody was followed by four additional mass gains as each target was added. The sequential and additive increase in mass was indicative of the formation of a complex of the zybody engaged with all five targets. Furthermore, analysis by FACS demonstrated that this same penta-specific zybody simultaneously bound to A431 cells (via EGFR) and the soluble forms of the four other target molecules, namely ErbB2, IGF-1R, αvβ3 integrin and Ang2 (Fig. S4).
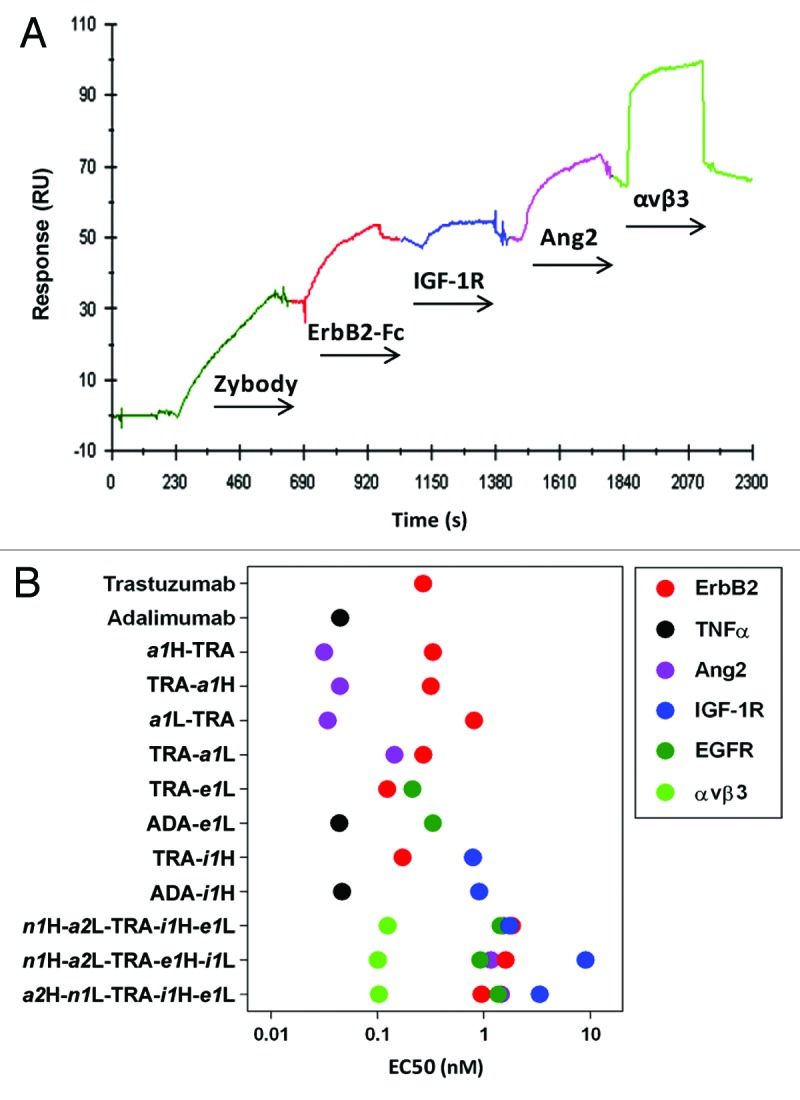
Figure 3. Binding of multi-specific zybodies. (A) SPR analysis demonstrated simultaneous binding of the penta-specific zybody, a2H-n1L-TRA-i1H-e1L, to five targets. EGFR-Fc (20 μg/ml) was immobilized on a CM5 sensor chip using the amine coupling procedure described in the Biacore 3000 manual. This was followed by sequential exposure to the zybody (30 μg/ml), ErbB2-Fc (20 μg/ml), IGF-1R (20 μg/ml), Ang2 (2 μg/ml) and integrin αvβ3 (5 μg/ml). Each ligand was injected for 4 min. (B) Summary of direct binding EC50 values for select zybodies and comparitor antibodies determined by ELISA. The full dilution curves are provided in Figure S3.
The binding characteristics of multi-specific zybodies were further explored with three additional penta-specific zybodies, each bearing the same specificities but in different configurations. Among these penta-specifics, binding was largely independent of MRD position (Fig. 3B). EC50 values for the binding to the specific targets clustered in nanomolar ranges that were generally higher than that observed for bispecifics based on the trastuzumab and adalimumab (ADA) scaffolds. The influence of MRD position was most overt with the low affinity IGF-1R-binding MRD (i1), which consistently displayed a lower affinity when fused to the C-terminus of the light chain. Overall, these data establish that a single zybody can engage up to five independent targets simultaneously through a diverse repertoire of recognition domains.
Zybodies target multiple therapeutic pathways within a cell population
A bispecific zybody (TRA-i1H) that targets ErbB2 and IGF-1R was evaluated to examine the biological consequences of simultaneously engaging two receptors. The model system compared the ability of TRA-i1H, trastuzumab and various control proteins to inhibit the proliferation of MCF-7 cells and a clone that was stably transfected with ErbB2, MCF-7ErbB2. MCF-7 cells express only very low levels of ErbB2 and are resistant to the anti-proliferative effects of trastuzumab.20 MCF-7ErbB2 cells express 10-fold higher levels of ErbB2 than the parental cell line, but these levels are still substantially less than those in the trastuzumab-sensitive cell line, SK-BR-3 (Table S1). As measured by SPR (Fig. 3A), ELISA (Fig. S3) or FACS (Fig. S4), the i1 MRD exhibits relatively poor binding to IGF-1R; however, in the context of an antibody scaffold that binds to target cells with high affinity, the i1 MRD acquires significant biological activity (Fig. 4). While we were unable to directly inhibit the binding of IGF to MCF-7 cells with any of the i1 zybodies (data not shown), an overnight pre-incubation of cells with TRA-i1H resulted in reduced IGF-1 binding to the ErbB2 overexpressing cells (Fig. 4A). Western blotting indicates that the bispecific zybody, TRA-i1H was superior to trastuzumab, the control zybody for the MRD (ADA-i1H) and the combination of trastuzumab and ADA-i1H in its ability to downregulate IGF-1R levels and inhibit IGF-1 mediated phosphorylation of Akt (Fig. 4B). Finally, TRA-i1H proved uniquely effective in inhibiting the proliferation of MCF-7ErbB2 cells (Fig. 4C). Combined, these data demonstrate that simultaneous engagement of distinct cellular receptors by a single bispecific protein induces biological responses that are not achieved with the same targeting moieties either on separate molecules or in combination.

Figure 4. A bispecific zybody enhances inhibition of IGF-induced binding, signaling and proliferation. (A) MCF-7 and MCF-7ErbB2 cells were treated with 63 nM of various combinations of trastuzumab (TRA) and bispecific zybodies that target IGF-1R (TRA-i1H or ADA-i1H). Twenty-four hours after treatment, the ability of cells to bind biotinylated IGF-1 was measured by FACS. (B) Cells treated as in (A), were further stimulated with IGF1 for 10 min and solubilized in lysis buffer. Immunoblotting indicates a reduction in IGF-1R levels and IGF1-induced phosphorylated Akt (pAkt) in MCF-7ErbB2 cells treated with TRA-i1H. Total Akt (tAkt) and β-tubulin levels were unchanged. (C) Inhibition of proliferation of MCF-7 and MCF-7ErbB2 cell was assayed after treatment for 96 h with the combinations described in (A).
An ErbB2/Ang2-targeting zybody inhibits tumor growth in vivo through two different mechanisms
The therapeutic potential of the trastuzumab-based zybodies was assessed to determine if their multi-target binding profiles modulated the biological activities in a way similar to that of the individual specificities. TRA-a1H and trastuzumab inhibited proliferation of SK-BR-3 cells at similar levels (Fig. 5A), thus indicating that the biological activity of the trastuzumab scaffold was retained and largely unaffected by the presence of Ang2. Additionally, the a1 MRD in TRA-a1H was a potent antagonist of Ang2 interaction with its receptor, Tie-2 (Fig. 5B).
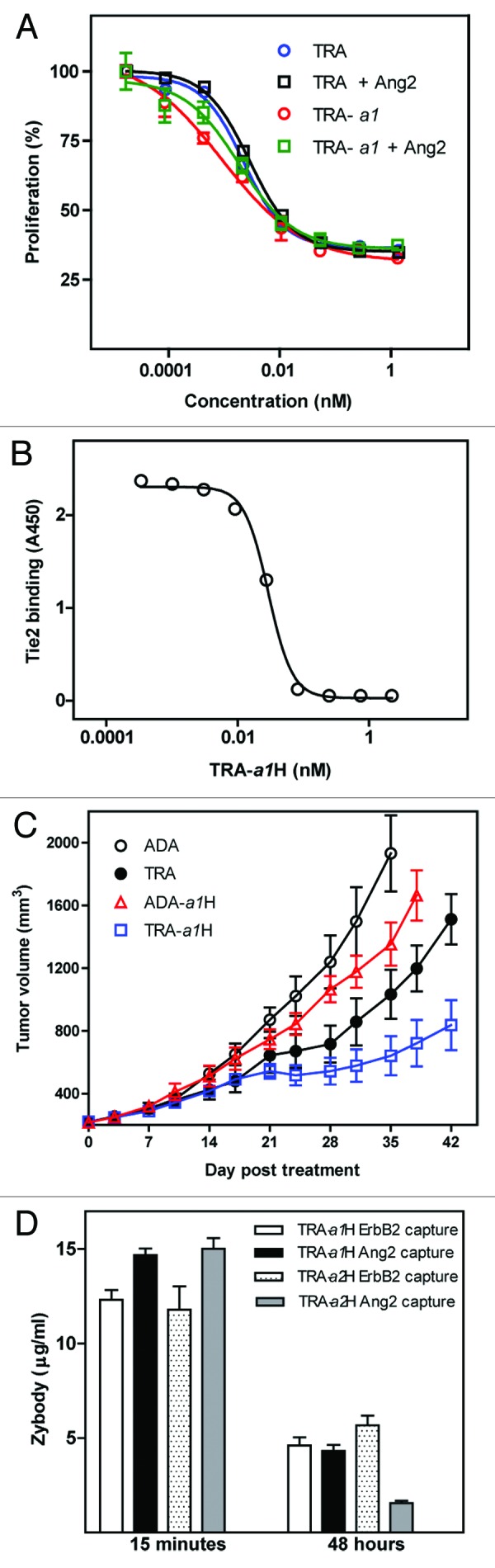
Figure 5. TRA-a1H in vitro and in vivo efficacy. (A) Inhibition of SKBR3 cell proliferation by trastuzumab (TRA) and TRA-a1H was measured in the presence and absence of Ang2. (B) The inhibition of Ang2 binding to Tie2 by TRA-a1H was measured by ELISA. Data represents mean and standard deviation of duplicate samples. (C) The in vivo efficacy of TRA-a1H was assessed in a xenograft tumor model sensitive to the independent inhibition of ErbB2 and Ang2. Mice with pre-established SK-OV-3 tumors were treated with adalimumab (ADA) as a negative control, trastuzumab (TRA), the bispecific zybodies targeting ErbB2 and Ang2 (TRA-a1H) or TNFα and Ang2 (ADA-a1H). P value for Mann Whitney t-test comparing TRA-a1H to trastuzumab at day 42 is 0.007. (D) The in vivo stability of individual target binding moieties on a zybody was measured in CD1 mice after a single injection of trastuzumab-based, bispecific zybodies containing either the a1 or the a2 MRD. Serum samples were collected at 15 min and 48 h and were assayed for mAb scaffold binding (ErbB2 capture) or MRD binding (Ang2 capture) in ELISA assays.
To determine whether the zybodies could simultaneously inhibit two therapeutic pathways in vivo, the anti-tumor activity of TRA-a1H and related control proteins were assessed in an ErbB2- and angiogenesis-dependent xenograft tumor model (SK-OV-3) (Fig. 5C). TRA-a1H was significantly more effective in inhibiting tumor growth than were trastuzumab (p = 0.007), adalimumab (an isotype control of irrelevant specificity), or the Ang2 antagonizing zybody ADA-a1H, which has the same a1 MRD but fused to the adalimumab heavy chain.
An understanding of the pharmacodynamic properties of multi-specific therapeutics, such as zybodies, requires pharmacokinetic assessment of each specificity contained in the molecule. The in vivo stability was determined by analysis of both the Ang2 and ErbB2 binding of the zybody present in serum from CD1 mice that received a single intravenous injection (1 mg/kg) of zybody. Serum samples were collected at 15 min and 48 h, and were assayed by ELISA. Following administration of TRA-a1H, scaffold- and MRD-mediated binding to their respective targets was similar at both time points (Fig. 5D). In contrast to TRA-a1H, TRA-a2H—which contains an MRD derived from a linear peptide library—lost significant ability to bind Ang2 over the same time period. The results indicate that it is possible to confer peptides with stability and pharmacokinetics equivalent to that of the mAb scaffold, but this is dependent on the sequence of the MRD itself.
A EGFR/ErbB3-targeting zybody inhibits tumor growth in vivo through two different receptors
In recent years, ErbB3 has emerged as an important heterodimerization partner of both ErbB2 and EGFR and has been implicated in the progression of several types of cancer.21 As such, ErbB3 has become a promising therapeutic target individually and in combination with ErbB222 or EGFR.23 We generated a zybody composed of the EGFR-targeting cetuximab and a peptide that binds ErbB3 and antagonizes the receptor’s ligand, HRG.24 This bispecific zybody (CET-c1H) was assessed for its ability to downregulate ligand-induced signaling, inhibit cell proliferation and tumor growth (Fig. 6). The pancreatic adenocarcinoma cell line, BxPC-3, expresses both EGFR and ErbB3 and exhibits an increased phosphorylation of these receptors in response to stimulation with TGFα and HRG1β respectively (Fig. 6A). Cetuximab and ADA-c1H (both mono-specific for EGFR and ErbB3, respectively) were able to inhibit phosphorylation of receptors, and downstream enzymes Erk and Akt, in a ligand-specific manner; however, the bispecific zybody CET-c1H efficiently inhibited intracellular signaling through the phosphorylation of both Erk and Akt by both TGFα and HRGβ (separately or combined). Inhibition of BxPC-3 cell proliferation was significantly enhanced by treatment with CET-c1H compared with either of the mono-specific agents used separately or combined (Fig. 6B). In this instance, anti-respiratory syncytial virus palivizumab (PAL) was used as an irrelevant scaffold on which to build the MRD control, PAL-c1H. The simultaneous targeting of EGFR and ErbB3 with CET-c1H in vivo (Fig. 6C) was significantly more effective in inhibiting the growth of human tumor xenografts of BxPC-3 than were cetuximab (p = 0.045), the isotype control palivizumab or the (mono-specific) ErbB3 antagonizing MRD, c1 fused to palivizumab (PAL-c1H).

Figure 6. Cetuximab-based zybodies inhibit signaling, cell proliferation and tumor growth in vivo. (A) BxPC-3 cells were pretreated for 3.5 h at 37°C in serum-free media with 63 nM of various combinations of cetuximab (CET), the cetuximab zybody containing and ErbB3 MRD (CET-c1H) or the MRD control zybody (ADA-c1H). Cells then received TGFα or HRG1β for 10 min prior to lysis, western blotting and detection of phosphorylated Erk (pERK), phosphorylated Akt (pAKT), phosphorylated EGFR (pEGFR), pErbB3. (B) Inhibition of proliferation of BxPC-3 cells was assayed after treatment for 96 h with the indicated proteins. Palivizumab (PAL) was used as an irrelevant scaffold (specific for respiratory syncytial virus) on which to build the MRD control, PAL-c1H. (C) The in vivo efficacy of CET-c1H was assessed in a xenograft tumor model sensitive to the independent inhibition of EGFR and ErbB3. Mice with pre-established BxPC-3 tumors were treated with cetuximab (CET) or the bispecific zybody targeting EGFR and ErbB3 (CET-c1H). Palivizumab (PAL) and PAL-c1H were used as the isotype and MRD controls, respectively. The P value for Mann Whitney t-test comparing CET-c1H to cetuximab at day 42 is 0.045.
Discussion
We describe here a multi-specific, multi-valent technology that enhances the activity of conventional mAbs without compromising their desirable drug-like properties. This approach is notable in that the target-binding elements (MRDs) are made up of short sequences derived from linear, disulfide-constrained and domain-based combinatorial libraries. Small peptides often make poor therapeutics themselves, in large part as a consequence of their short in vivo half-lives. We and others25,26 have shown that, in the context of an IgG-Fc or intact immunoglobulin, peptides can acquire in vivo stability profiles that are comparable to those of conventional mAbs. In this report, a disulfide-constrained MRD (a1) was shown to exhibit in vivo stability superior to that of a linear peptide (a2). While MRD stability is likely influenced by structure, we have observed a sufficient number of unstable, constrained or structured MRDs (not shown) to conclude that stability is ultimately defined by the specific sequence. In addition to potential gains in stability, the bi- or multi-valent presentation of the peptides in the context of the zybody may give rise to enhanced avidity. Furthermore, in zybodies, the binding activity of the parental scaffold affords further opportunity for increases in avidity through recognition of heterotypic antigens or epitopes. To our knowledge, this is the first report of a recombinant biomolecule with nanomolar specificity for up to 5 independent targets. This higher order of specificity and valency has not been demonstrated in formats such as two-in-one7 triomabs,11 knobs-into-holes,12 or Crossmabs;13 electrostatic matching,14 although application of the zybody concept could be used to extend these formats as well. Bio-therapeutics, particularly those derived from extensively engineered or non-human origins, carry the risk of immunogenicity, which is a significant safety and efficacy concern. In addition to desired agonistic or antagonistic properties, MRDs may also be selected for reduced immunogenic potential using a variety of in silico and in vitro assessments.27-30
In contrast to many other multi-specific antibody formats, the fusions of these peptides represent a relatively small increase in the overall mass of the antibody (less than 5% for a 30 amino acid peptide). Indeed, fusion of four such peptides, yielding a penta-specific zybody, would still constitute a smaller increase in mass than a bispecific antibody constructed using one Fv (e.g., DVD-Ig8). Small changes to the scaffold size and structure are likely the reason that the ease of manufacture, stability, original antigen-specificity and Fc receptor binding of the scaffold mAb are all retained. Furthermore, in preliminary studies (data not shown) we have observed that in concordance with FcR binding, the ADCC activity of a Herceptin-ang2 zybody is very similar to that of the scaffold mAb.
We previously reported on the use of zybodies to target two soluble factors;15 here, we demonstrate that zybodies can be very effective at simultaneously modulating the activity of two cell surface receptors (EGFR and ErbB3), or alternatively, the combination of receptor and soluble ligand (ErbB2 and Ang2). In addition, we have expanded the use of zybodies to potential applications in oncology. The clinical need for oncology therapies that target more than a single component of disease is underscored by the fact that many mAbs currently in use fail in a significant proportion of patients either because of acquired resistance or inherent target heterogeneity. For example, the overall response rate of patients to trastuzumab in metastatic breast cancers that overexpress ErbB2 is only 35%,31 implying one or more resistance mechanisms. Signaling cross-talk and receptor interactions have been reported between ErbB2 and IGF-1R32 and are implicated as a mechanism of resistance to trastuzumab.33,34 Expression of ErbB3 can also mediate solid tumor drug resistance, particularly following anti-ErbB2 or anti-EGFR therapies.21
Our data demonstrate that zybodies targeting combinations of ErbB2/IGF-1R or EGFR/ErbB3 can be potent inhibitors of signaling, cell proliferation and tumor growth.
In addition to the potential importance of simultaneously engaging multiple receptors, the ability to bind individual receptors through more than one epitope has revealed therapeutic opportunities. For example, trastuzumab and pertuzumab target non-overlapping epitopes on ErbB2, and are synergistic when used in combination.2 Likewise, the benefits of targeting multiple epitopes on an individual molecule have been demonstrated for IGF-1R35,36 and EGFR.37 Furthermore, recent studies have shown that multi-epitopic, anti-EGFR antibodies downregulate cell surface receptors through a mechanism unavailable to conventional (mono-epitopic) antibodies.38 The ability to engineer additional valencies or specificity into a zybody affords the opportunity to develop such multi-epitopic therapeutics.
The clinical efficacy of conventional mAbs when used as single agents is wholly dependent on the identification of single molecular entities that uniquely and exclusively modulate disease pathways. Multi-specific, multi-epitopic and multi-valent therapeutics with appropriate drug-like properties afford the opportunity to explore the biology of target constellations and interacting disease pathways, and thus bring new functionality to existing mAbs. The ability to assemble zybodies from a repertoire of independently developed MRDs and mAbs, combined with the benefits of robust stability and conventional production methods, should enable the design of novel therapeutics that will better address the complexity of human disease.
Materials and Methods
Cell Lines and proteins
Ang2, ErbB2-Fc, EGFR-Fc, Tie-2, αvβ3 integrin, IGF-1R and IGF1were obtained from R&D Systems. Trastuzumab (Herceptin®), adalimumab (Humira®), cetuximab (Erbitux®) and palivizumab (Synagis®) were obtained from Blue Door Pharma. BxPC-3, SKBR-3, SK-OV-3 and MCF-7 cells were obtained from American Type Culture Collection. MCF-7 ErbB2 cells were developed at Zyngenia and are described in Table S1.
Modular recognition domains (MRDs)
MRDs used in zybody construction were adapted from the sources indicated in Table 1. The linear and disulfide-constrained MRDs were developed at Zyngenia as follows. Naïve peptide libraries were generated using degenerate oligonucleotides synthesized in lengths of 10–12 codons. Oligonucleotides (Ella Biotec) were incorporated into a modified pCOMB phagemid vector using Kunkel mutagenesis. Three rounds of panning were performed using 100 nM, 75 nM and 50 nM Ang2-biotin (R&D Systems) with Tween20 concentrations of 0.1%, 0.2%, and 0.5%, respectively. Bound phage were eluted with 0.2 M glycine (pH 2.2), neutralized with 1 M Tris (pH 9.0) and recovered with TG1 cells and VCSM13 helper phage, then precipitated with PEG and resuspended in PBS, 0.5% BSA. Ang2 -binding peptides were identified by ELISA and the corresponding phagemid were sequenced. Phage were characterized for the ability to inhibit the interaction between Ang2 and Tie2 in an ELISA assay. Peptides were ranked based on ELISA activities and binding motifs characterized using ClustalW and Weblogo algorithms. The A9 peptide (DCYFYPNPPHCY) was selected for further maturation. A maturation library was produced using the following oligonucleotide (IDT Technologies): 5′ CAG GCG GCC GGT GGC GGT TCT NNS NNS NNS GAC TGT TAC TTC TAC CCG AAC CCG CCG CAT TGT TAC NNS NNS NNS GGC CAG GCC GGC CAG GGT GGT3′, where the underlined positions were randomized using doped nucleotide mixes with the parent nucleotide at 70% and remaining three nucleotides each at 10%. NNS codons were added to flank the core peptide to increase the Ang2 binding contacts. Three rounds of panning were performed essentially as described above, using antigen concentrations of 50 nM, 5 nM and 0.5 nM Ang2-biotin for the three rounds of selections, respectively. Phage recovered from the third round of panning were characterized by ELISA for binding for binding to Ang2.
Expression, purification analytical characterization
HEK293-F suspension cells (Invitrogen) were transiently transfected with equimolar amounts of pCEP4 heavy and light chain expression constructs. Culture volumes ranged from 20 to 1000 ml and were harvested between 4 and 6 d in culture by centrifugation and filtration (0.22 μm). Zybody titers were assessed by ELISA, using anti-human Fc (Abcam) capture and anti-human κ HRP (Abcam) detection. Zybodies were captured from filtered culture supernatants using Protein A mAb Select SuRe affinity resin (GE Healthcare) and were acid eluted (pH 3.0), followed by immediate neutralization. Zybody homogeneity was characterized using BEH200 size-exclusion chromatography column (Waters) with a mobile phase consisting of 20 mM sodium phosphate, 200 mM sodium chloride (pH of 7.0) at a flow rate of 0.4 ml/min. If aggregation levels exceeded 10%, monomeric zybodies were fractioned by size exclusion chromatography (Superdex 200, GE Healthcare) in 20 mM histidine, 140 mM NaCl (pH 6.0). Monomeric antibody fractions were pooled, dialyzed against PBS and stored at −80°C. Differential scanning calorimetry was performed using a capDSC (GE Healthcare). The temperature range scanned was 30–130°C using a scan rate of 60°C/hour. The protein melting point (Tm) is defined as a peak in the temperature-dependent specific heat capacity graph.
Direct binding ELISA
Microtiter plates were coated with the target proteins (30 ng/ml Ang2, 30 ng/ml ErbB2-Fc or 1 μg/ml FcRγIIIa (CD16) (R&D Systems) at 4°C overnight. After blocking (ELISA Blocker, Thermo Scientific) and washing (PBS, 0.1% Tween 20), serially diluted zybody was added to wells and incubated for 2 h at room temperature. Plates were washed again, followed by addition of anti-human HRP and incubation for 1 h at room temperature. After a final wash step, 3,3′,5,5′-tetramethylbenzidine reagent (KPL, Inc.) was added and absorbance was measured at 450 nm using a plate reader (SpectraMax).
Inhibition of Ang2 binding to Tie2
Microtiter plate wells were coated with 100 ng/ml Ang2 (R&D Systems) and blocked with ELISA Blocker (Thermo Scientific). Soluble Tie2 was added to Ang2-coated wells in the presence of various concentrations of zybody. Bound Tie2 was detected using a mouse-anti-Tie2 antibody (R&D Systems), followed by goat-anti-mouse Ig-HRP (eBioscience) and 3,3′,5,5′-tetramethylbenzidine reagent (KPL, Inc.). Absorbance was measured at 450 nm using a plate reader (SpectraMax).
Cell proliferation
MCF-7, MCF-7 ErbB2, SK-BR-3 and BxPC-3 cells were plated in a 96-well plates at 2 × 103 cells/well in corresponding growth medium (MCF-7 in DMEM, 10% FBS; SK-BR-3 in McCoys, 10% FBS; BxPC-3 in RPMI1640, 10% FBS). The following day, growth medium was replaced with reduced serum medium (MCF-7, DMEM, 5% horse serum; SK-BR-3, McCoys, 5% horse serum; BxPC-3, RPMI1640, 0.5%FBS). After 24 h, cells were treated with serial dilutions of proteins (starting with 10 or 30 µg/ml) made in corresponding reduced serum medium. After 96 h, cell viability was measured using Cell Titer-Glo (Promega) and an EnVision 2104 Multilabel reader (PerkinElmer).
Flow cytometry
MCF-7 and MCF-7ErbB2 cells were plated in a 24-well plate at 0.5 × 106 cells/well in DMEM, 10% FBS. After 24 h, cells were treated with 10 µg/ml antibodies overnight at 37°C. Cells were detached with trypsin and resuspended in FACS buffer (PBS, 0.1% BSA and 0.1% sodium azide) containing 100 ng/ml biotinylated IGF-1. After 20 min at room temperature, bound biotinylated IGF-1 was detected by flow cytometry (FACS Canto II, BD Bioscience) using streptavidin-PE (BD Biosciences).
Western blot analysis
MCF-7, MCF-7ErbB2 and BxPC-3 cells were plated in a 6-well plate at 0.5 × 106/well in DMEM, 10% FBS. After 24 h, cells were treated with 10 µg/mL zybodies in serum-free medium for 3 h (BxPC-3) or overnight (MCF-7 and MCF-7ErbB2) and then stimulated with the indicated ligands for 10 min at 37°C. Cells were washed and then solubilized in 200 μl of cell lysis buffer containing 1% Triton X 100 in 20 mM TRIS-HCl (pH 7.5), 0.15 mM NaCl, 1 μg/mL leupeptin, 1 μg/ml aprotinin, 1 mM sodium vanadate and 1 mM EDTA. Cell lysates were separated on 4–12% SDS-PAGE (Invitrogen) and transferred to PVDF membranes. Immunoblotting was performed with antibodies specific to phospho-Akt (R&D Systems), Akt (R&D Systems), phospho-ERK (R&D Systems), Erk (R&D Systems), IGF-1R (Cell Signaling), EGFR (Thermo Fisher), ErbB2 (Thermo Fisher), ErbB3 (Thermo Fisher) and β-tubulin (Santa Cruz Biotechnology) and detected with horseradish peroxidase conjugated secondary antibodies using enhanced chemiluminescence system (Invitrogen).
In vivo stability
A single, 1 mg/kg intravenous dose was administered to female 6–8 week old, CD1 mice (Harlan Laboratories). Serum samples were taken at 15 min and 48 h and then were assessed for direct binding to both scaffold-specific (e.g., ErbB2) and MRD-specific (e.g., Ang2) target proteins. Immulon 4HBX flat-bottom microtiter wells (Thermo Electron) were coated with 0.5 µg/ml of the target protein overnight at 4°C. After blocking (ELISA Blocker, Thermo Electron), serum samples diluted in 10% mouse serum were added and incubated at room temperature for 1 h. Captured zybodies were detected with anti-human κ chain HRP conjugated antibody (Abcam) and serum concentrations were calculated from zybody-specific standard curves.
Xenograft tumor models
Animals were obtained from Taconic Farms, Inc. and housed four per cage at the BioQual animal facility. All animal protocols were approved by, and performed in strict accordance with the Institutional Animal Care and Use Committee (IACUC). Female NOD/SCID mice, 8 weeks old were implanted with 5 × 106 SK-OV-3 or BxPC-3 cells in 50% Matrigel by subcutaneous injection of 100 μl of cell suspension into the upper right flank of each mouse. When tumors reached an average size of 220 mm3, mice were randomized into treatment groups with seven mice per group. In the SK-OV-3 study, all zybodies and control antibodies were administered intraperitoneally at 4 mg/kg, twice weekly. Animals in the BxPC-3 study received twice-weekly intraperitoneal injections at 30 mg/kg (first dose), 15 mg/kg (second and third dose) and 7.5 mg/kg (4th through 11th dose). In both studies, tumor length and width were measured twice per week using calipers, and tumor volumes were calculated by the formula ½ × length × width.2 Animals were sacrificed if tumor size exceeded 2,000 mm3.
Supplementary Material
Acknowledgments
We thank Anton Nguyen and Elvin Ng for their expert technical assistance.
Disclosure of Potential Conflicts of Interest
The authors are employees of Zyngenia and/or have an equity interest in the company.
Supplemental Materials
Supplemental materials may be downloaded here: www.landesbioscience.com/journals/mabs/article/23043
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/23043
References
- 1.Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. NeoALTTO Study Team Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379:633–40. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baselga J, Cortés J, Kim SB, Im SA, Hegg R, Im YH, et al. CLEOPATRA Study Group Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–19. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–90. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–51. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 5.Nord K, Nilsson J, Nilsson B, Uhlén M, Nygren PA. A combinatorial library of an α-helical bacterial receptor domain. Protein Eng. 1995;8:601–8. doi: 10.1093/protein/8.6.601. [DOI] [PubMed] [Google Scholar]
- 6.Reusch U, Le Gall F, Hensel M, Moldenhauer G, Ho AD, Little M, et al. Effect of tetravalent bispecific CD19xCD3 recombinant antibody construct and CD28 costimulation on lysis of malignant B cells from patients with chronic lymphocytic leukemia by autologous T cells. Int J Cancer. 2004;112:509–18. doi: 10.1002/ijc.20417. [DOI] [PubMed] [Google Scholar]
- 7.Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–4. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- 8.Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25:1290–7. doi: 10.1038/nbt1345. [DOI] [PubMed] [Google Scholar]
- 9.Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschläger M, Antes B, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel. 2010;23:289–97. doi: 10.1093/protein/gzq005. [DOI] [PubMed] [Google Scholar]
- 10.Gavrilyuk JI, Wuellner U, Salahuddin S, Goswami RK, Sinha SC, Barbas CF., 3rd An efficient chemical approach to bispecific antibodies and antibodies of high valency. Bioorg Med Chem Lett. 2009;19:3716–20. doi: 10.1016/j.bmcl.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219–25. [PubMed] [Google Scholar]
- 12.Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 13.Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA. 2011;108:11187–92. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–46. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanakaraj P, Puffer BA, Yao XT, Kankanala S, Boyd E, Shah RR, et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. MAbs. 2012;4:600–13. doi: 10.4161/mabs.21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura RH, Cheng Z, Gambhir SS, Cochran JR. Engineered knottin peptides: a new class of agents for imaging integrin expression in living subjects. Cancer Res. 2009;69:2435–42. doi: 10.1158/0008-5472.CAN-08-2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backliwal G, Hildinger M, Chenuet S, Wulhfard S, De Jesus M, Wurm FM. Rational vector design and multi-pathway modulation of HEK 293E cells yield recombinant antibody titers exceeding 1 g/l by transient transfection under serum-free conditions. Nucleic Acids Res. 2008;36:e96. doi: 10.1093/nar/gkn423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Lundberg E, Vernet E, Larsson B, Höidén-Guthenberg I, Gräslund T. Selection of affibody molecules to the ligand-binding site of the insulin-like growth factor-1 receptor. Biotechnol Appl Biochem. 2010;55:99–109. doi: 10.1042/BA20090226. [DOI] [PubMed] [Google Scholar]
- 19.Göstring L, Chew MT, Orlova A, Höidén-Guthenberg I, Wennborg A, Carlsson J, et al. Quantification of internalization of EGFR-binding Affibody molecules: Methodological aspects. Int J Oncol. 2010;36:757–63. doi: 10.3892/ijo_00000551. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty AK, Liang K, DiGiovanna MP. Co-targeting insulin-like growth factor I receptor and HER2: dramatic effects of HER2 inhibitors on nonoverexpressing breast cancer. Cancer Res. 2008;68:1538–45. doi: 10.1158/0008-5472.CAN-07-5935. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. 2007;97:453–7. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012;11:582–93. doi: 10.1158/1535-7163.MCT-11-0820. [DOI] [PubMed] [Google Scholar]
- 23.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20:472–86. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Kronqvist N, Malm M, Göstring L, Gunneriusson E, Nilsson M, Höidén Guthenberg I, et al. Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel. 2011;24:385–96. doi: 10.1093/protein/gzq118. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. 2010;10:575–85. doi: 10.1038/nrc2894. [DOI] [PubMed] [Google Scholar]
- 26.Shimamoto G, Gegg C, Boone T, Quéva C. Peptibodies: A flexible alternative format to antibodies. MAbs. 2012;4:586–91. doi: 10.4161/mabs.21024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–7. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 28.Perry LC, Jones TD, Baker MP. New approaches to prediction of immune responses to therapeutic proteins during preclinical development. Drugs R D. 2008;9:385–96. doi: 10.2165/0126839-200809060-00004. [DOI] [PubMed] [Google Scholar]
- 29.Bryson CJ, Jones TD, Baker MP. Prediction of immunogenicity of therapeutic proteins: validity of computational tools. BioDrugs. 2010;24:1–8. doi: 10.2165/11318560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 30.Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- 31.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26. doi: 10.1200/JCO.20.3.719. [DOI] [PubMed] [Google Scholar]
- 32.Jin Q, Esteva FJ. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:485–98. doi: 10.1007/s10911-008-9107-3. [DOI] [PubMed] [Google Scholar]
- 33.Pohlmann PR, Mayer IA, Mernaugh R. Resistance to Trastuzumab in Breast Cancer. Clin Cancer Res. 2009;15:7479–91. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. 2007;Annals of oncology: official journal of the European Society for Medical Oncology / ESMO18:977–84. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 35.Glaser S, Demarest S, Miller BR, Hariharan K, Ho S, Dong J, et al. Compositions that Bind Multiple Epitopes of IGF-1R United States Patent Application 20090130105 2009.
- 36.Dong J, Demarest SJ, Sereno A, Tamraz S, Langley E, Doern A, et al. Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol Cancer Ther. 2010;9:2593–604. doi: 10.1158/1535-7163.MCT-09-1018. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, et al. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588–97. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- 38.Spangler JB, Manzari MT, Rosalia EK, Chen TF, Wittrup KD. Triepitopic antibody fusions inhibit cetuximab-resistant BRAF and KRAS mutant tumors via EGFR signal repression. J Mol Biol. 2012;422:532–44. doi: 10.1016/j.jmb.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.