

Abstract
Multispecific antibody-like molecules have the potential to advance the standard-of-care in many human diseases. The design of therapeutic molecules in this class, however, has proven to be difficult and, despite significant successes in preclinical research, only one trivalent antibody, catumaxomab, has demonstrated clinical utility. The challenge originates from the complexity of the design space where multiple parameters such as affinity, avidity, effector functions, and pharmaceutical properties need to be engineered in concurrent fashion to achieve the desired therapeutic efficacy. Here, we present a rapid prototyping approach that allows us to successfully optimize these parameters within one campaign cycle that includes modular design, yeast display of structure focused antibody libraries and high throughput biophysical profiling. We delineate this approach by presenting a design case study of MM-141, a tetravalent bispecific antibody targeting two compensatory signaling growth factor receptors: insulin-like growth factor 1 receptor (IGF-1R) and v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (ErbB3). A MM-141 proof-of-concept (POC) parent molecule did not meet initial design criteria due to modest bioactivity and poor stability properties. Using a combination of yeast display, structured-guided antibody design and library-scale thermal challenge assay, we discovered a diverse set of stable and active anti-IGF-1R and anti-ErbB3 single-chain variable fragments (scFvs). These optimized modules were reformatted to create a diverse set of full-length tetravalent bispecific antibodies. These re-engineered molecules achieved complete blockade of growth factor induced pro-survival signaling, were stable in serum, and had adequate activity and pharmaceutical properties for clinical development. We believe this approach can be readily applied to the optimization of other classes of bispecific or even multispecific antibody-like molecules.
Keywords: bispecific antibodies, scFv, yeast display, library, high throughput screening, IGF-1R, ErbB3
Introduction
Monoclonal antibodies (mAbs) have significantly increased our ability to treat human disease; however, many pathologies are heterogeneous in nature and multiple points of pathway blockade are needed to obtain optimal therapeutic effect.1,2 Thus, bispecific or multispecific antibody-like molecules present a novel promising class of therapeutic agents.3 In the last two decades, more than 30 different bispecific formats have been reported. Despite significant successes in preclinical research, the engineering of clinic-ready multispecific antibodies has proven to be challenging, and only one trivalent molecule, catumaxomab, has demonstrated clinical utility.4 This challenge originates from the complexity of the design, where multiple parameters such as affinities, avidities, and pharmaceutical properties need to be optimized. Historically, these parameters were optimized sequentially, and researchers emphasized high affinities for mAbs and high stability for non-antibody scaffolds.5 Affinity and stability of antibody modules, however, are often coupled and, as illustrated in Figure 1, optimization of one parameter in isolation can result in de-optimization of another and vice versa. As a result, multiple iterative design cycles may be required to construct molecules with desired characteristics. This iterative approach is not streamlined and requires substantial commitment of resources and time. We present here a rapid prototyping method that combines modular design, yeast display of structure-focused scFv libraries and micro-scale assay analysis, to rapidly optimize bio-therapeutic molecules within one campaign cycle.

Figure 1. Rapid Prototyping approach streamlines antibody optimization for affinity and stability by reducing number of iterative steps.
We delineate this rapid prototyping method by presenting a case study on the optimization of an active and manufacturable tetravalent bispecific antibody (BsAb) targeting IGF-1R and ErbB3. Both IGF-1R and ErbB3 signal through the PI3K/AKT/mTOR signaling pathway,6,7 and have been implicated as mechanisms of resistance to therapies directed at blocking ErbB1 (cetuximab, erlotinib), ErbB2 (trastuzumab, lapatinib), and mTOR (everolimus).8 Since IGF-1R and ErbB3 are often co-expressed in a variety of cancer cell lines, we anticipate that concurrent inhibition of these survival signaling pathways would be required to achieve optimal tumor growth inhibition. To validate this hypothesis, we produced a proof-of-concept (POC) tetravalent, bispecific antibody co-targeting IGF-1R and ErbB3. The POC bispecific antibody was based on the bispecific format originally described by Morisson,9 and was generated by genetically fusing anti-IGF-1R scFvs to the C-terminus of an anti-ErbB3 IgG2 antibody. We chose this antibody-like format because of its high avidity, lack of spatial interference between the antigen binding entities, and presence of intact Fc. Therefore, this format should be able to engage both antigens simultaneously, bind to neonatal Fc receptor (FcRn), and possess effector functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC). This format is also amenable to scale-up using well-established expression and protein A based purification techniques.10-12 Even though this POC bispecific molecule showed promising activity in cell-based assays, it was not suitable for further development because of its modest activity and tendency to form aggregates in solution.
Given that both affinity and stability of multiple parts of the POC molecule had to be improved, we pursued a modular optimization approach based on the combination of structure-focused library design and yeast surface display. This modular approach is particularly important due to the vast design space approaching 1080 sequence variants in CDRs alone, which is not feasible to efficiently explore in iterative fashion.13 We selected yeast display as an engineering platform for two main reasons. First, in contrast to E. coli, the endoplasmic reticulum quality control mechanisms of yeast are closer to those of mammalian cells. This alleviates library expression bias and provides access to more diverse antibody sequences.14,15 Second, yeast displayed libraries can be screened by fluorescence-activated cell sorting (FACS), which provides quantitative affinity discrimination between clones and eliminates artifacts due to host expression biases.16-18 We used these qualities of yeast display in conjunction with a library-scale thermal challenge assay that couples stability and affinity of scFv modules19-21 to pursue their concurrent optimization within a single campaign.
Following the single selection campaign, selected scFvs were expressed in a high throughput manner for micro-scale profiling. Using micro-scale characterization techniques, these scFvs were assessed for binding affinity to antigens, inhibition of ligand-induced signaling in cancer cell lines, thermal unfolding transition temperature,21 and stability in serum. In these assays soluble anti-IGF-1R and anti-ErbB3 scFvs were superior to the original modules. Last, we combined a diverse subset of the optimized parts into 24 tetravalent bispecific antibodies, and characterized them by standard bioactivity and biophysical techniques. We demonstrate that optimized molecules meet our design criteria, comprise sufficient number of non-identical sequences, and have adequate activity and pharmaceutical characteristics to allow their therapeutic development. We believe that our approach is generalizable and can be used for rapid prototyping and optimization of multiple classes of antibody-like molecules.
Results
POC bispecific antibody
IGF-1R and ErbB3 signaling pathways have been implicated as potential escape mechanisms to multiple targeted cancer therapies.22,23 We have recently shown that the IGF-1R pathway is often co-activated with the ErbB3 pathway in a multitude of cancer cell lines (data not shown). Therefore, we expect that a bispecific antibody targeting both pathways may lead to effective tumor blockade and greatly expand the therapeutic scope and benefits of existing targeted therapies. To confirm this hypothesis, a POC bispecific (bs5F) was created by fusing in-house anti-IGF-1R scFvs (clone 5.7) to the C-terminal of an in-house anti-ErbB3 IgG2 (clone 2.3).
Activity of POC bispecific antibody in signaling inhibition and xenograft studies
We first determined the ability of the POC bispecific antibody to antagonize IGF-1R and ErbB3 by examining its ability to inhibit IGF-1 and Heregulin (HRG) induced signaling in BxPC-3 cells. Cells were pretreated for 1 h with different concentrations of antibody, and then stimulated with dual ligands (HRG + IGF-1). As shown in Figures 2A and 2B, the POC bispecific antibody inhibited the phosphorylation of both IGF-1R and ErbB3, with IC50 values of 14 nM and 2.3 nM, respectively. These results suggest that both arms of POC bispecific antibody were functionally active; however, even at a concentration of 1 μM, the signaling inhibition for pIGF-1R was incomplete (Fig. 2D).

Figure 2. Characterization of POC bispecific antibody. POC molecules inhibit phosphorylation of (A) ErbB3, (B) IGF-1R and (C) AKT in BxPC-3 pancreatic adenocarcinoma cell line. Cells were pretreated for 1 h with the different concentrations of the antibody, and stimulated with 30 ng/mL HRG and 100 ng/mL of IGF-1 for 10 min. The cells were lysed, and ELISA used to detect amounts of pErbB3, pIGF1R and pAKT. (D) Summary of signaling inhibition of POC antibody. (E) Activity of POC bispecific (bs5F) and comparator monospecific antibodies: 5.7 IgG2 directed at IGF1R and 2.3 IgG2 directed at ErbB3 in a subcutaneous BxPC-3 xenografts model.
AKT is a downstream protein kinase common to both ErbB3 and IGF-1R signaling cascades, and has been shown to play an important role in cancer cell survival. As shown in Figure 2C, the POC bispecific antibody also inhibited downstream signaling via phosphorylation of AKT, with an IC50 value of 12 nM; however, at 1 µM concentration, inhibition of ligand-induced signaling was incomplete. We believed that the incomplete AKT blockade was due to sub-optimal affinities of both the anti-IGF-1R and anti-ErbB3 arms. Therefore, affinity maturation for both anti-IGF-1R and anti-ErbB3 parts were needed to increase the therapeutic efficacy of this antibody.
Next, we determined the activity of the POC bispecific antibody in a subcutaneous BxPC-3 xenograft model, and compared its activity to comparator monospecific antibodies: clone 5.7 IgG2 (anti-IGF-1R) and clone 2.3 IgG2 (anti-ErbB3). As shown in Figure 2E, in comparison to control, BxPC-3 cells responded well to the 3 different treatments with POC bispecific antibody (bs5F) being the most active. This result suggests that both ErbB3 and IGF-1R signaling pathways play an important role in the in vivo growth of BxPC-3 cells; however, simultaneous blockade of both these pathways is needed for maximum activity. To confirm that POC bispecific was indeed inhibiting the target receptors, we also tested tumor lysates, and found that POC bispecific actively downregulated both IGF-1R and ErbB3 receptors throughout the course of this study (data not shown).
Stability of POC bispecific antibody
Figures 3A-C show the size exclusion chromatography (SEC) profiles of the POC bispecific and two comparator antibodies in 1X phosphate-buffered saline (PBS) following protein A purification. The POC bispecific antibody was aggregated (% monomers: 89 ± 2%), while both the comparator antibodies (clone 5.7 IgG2 and clone 2.3 IgG2) that make up this bispecific molecule were over 99% monomeric. As shown in Figure 3D, at a concentration of 5 mg/mL in PBS, the POC bispecific antibody was unstable and aggregated by over 14% after storage for a month at 4°C. Both comparator antibodies remained stable over time.

Figure 3. SEC profile of (A) POC bispecific, and (B and C) comparator monospecific antibodies. (D) Stability of POC antibody and comparator IgGs over 1 month at 4°C. All proteins were only protein A purified, and were at a concentration of 5 mg/mL in 1X PBS. Percent monomers were determined by injecting 50 μg of protein A purified sample on a TSKgel SuperSW3000 column (4.6mm ID x 30 cm) using 10 mM sodium phosphate (+ 450 mM NaCl, pH 7.1) as running buffer.
We hypothesized that the POC bispecific antibody was unstable because it had unoptimized scFv modules that lacked sufficient intrinsic stability. This phenomenon is well understood,24 and stabilization of scFvs by a variety of techniques such as linker optimization, disulfide bridge engineering, loop grafting into stable frameworks, co-variation analysis, structure-focused design and phage display were developed.25
Optimization path for POC bispecific
Because its activity and stability were suboptimal, the POC bispecific antibody was deemed unsuitable for further development. The modular approach was used to make a more manufacturable and active bispecific molecule (Fig. 4). Briefly, using a combination of structure-guided library design, yeast display, and micro-scale characterization technique, both the anti-IGF-1R and anti-ErbB3 scFvs were independently affinity matured and stabilized. In a high throughput (HTP) manner, the soluble forms of the best binding scFvs were produced and triaged. Next, the best anti-IGF-1R and anti-ErbB3 parts were combined to create optimized bispecific molecules.

Figure 4. Modular approach for optimizing POC bispecific antibody. Using a combination of yeast display and structure focused design, both anti-ErbB3 and anti-IGF1R modules were independently optimized in scFv format, and characterized using micro-scale characterization techniques. The improved modules were then combined in both orientations to create a diverse set of optimized bispecific antibodies.
Yeast display library design and screening
Optimized scFvs for both anti-IGF-1R and anti-ErbB3 were selected using a novel “2-in-1” covalent yeast display system. As illustrated in Figure 5, the same plasmid can be used either for displaying scFvs on the surface of yeast or secreting scFvs into solution. In this system, scFvs containing C-terminal Flag-tag are fused to an anchor protein that is expressed on the yeast surface; identical restriction sites were engineered on both termini of the gene encoding the anchor protein. For soluble expression, the covalent anchor is cleaved from the plasmid by restriction enzyme digestion. We found that the resulting linearized plasmids can be transformed into yeast without DNA purification or sub-cloning steps to produce soluble scFvs. On average, the soluble expression titers were 5 mg/L, which was sufficient to produce material for micro-scale analysis.
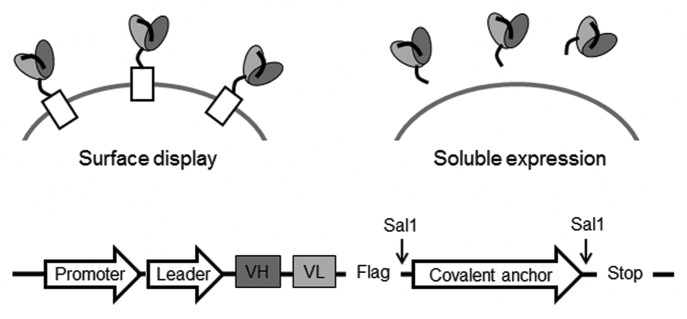
Figure 5. “2-in-1” covalent yeast display system. The same plasmid can be used for displaying scFvs on surface or secreting them in solution. For secreted expression, the covalent anchor is cleaved using Sal1 digestion, and the linearized vector transformed into yeast without additional DNA purification steps.
To enrich for scFvs with enhanced stability, a “thermal challenge assay” was developed.19,21Figure 6A shows FACS results of stable clone scFv1 and unstable clone scFv4. These two scFvs have similar binding and expression profiles that were not differentiated by FACS. However, after being heat-shocked at 55°C and 60°C, scFv4 lost significant binding activity as revealed by the fluorescence intensities in quadrant Q2, suggesting that the thermal challenge method can be used to differentiate unstable clones. In Figure 6B, four representative anti-ErbB3 scFvs with similar affinities but varying thermostability (Tm) were heated over a temperature gradient for 5 min, cooled and tested for residual binding to antigen. The ratio of resulting fluorescence intensities normalized for expression levels with and without heating was used for ranking. These results revealed that scFv1 is thermostable, whereas scFv4 is unstable as it lost > 50% of its initial binding at 55°C. The differentiation seen on yeast surface correlates well with the stability trend of soluble scFvs (data not shown), suggesting that this assay can be used to select scFvs for stability.

Figure 6. Development of library scale thermal challenge assay. (A) Flow cytometry dot plots of two representative anti-ErB3 scFvs: scFv1 (top) and scFv4 (bottom) binding to ErbB3-Fc before and after heat-shock at two different temperatures (55°C and 60°C). Anti-Flag-Alexa647 secondary was used to measure scFv expression level (x-axis), and anti-his-Alexa 488 secondary to measure binding of his-tagged ErbB3-Fc (y-axis). (B) Differentiation of four representative anti-ErbB3 scFvs using thermal challenge on yeast surface.
Our scFv library design and selection scheme is illustrated in Figure 7. The anti-ErbB3 scFv was derived from a phage-displayed human naïve scFv library; its variable genes belonged to IGH3–9 and IGL3–19 germline families. The anti-IGF-1R antibody was derived from a phage-displayed semi-synthetic human Fab library;26 its variable genes belonged to IGH2–23 and IGK1–12 germline families. Briefly, anti-ErbB3 and anti-IGF-1R scFvs were expressed in VH-VL orientation connected by a glycine-serine based linker. The library templates were created from wild-type by mutating atypical amino acids (germlining) in frameworks, as well as introducing structure- and knowledge-based stabilizing mutations. Unique restriction sites were introduced on frameworks through silent mutations for shuffling CDRs (Fig. 7A). Based on homology modeling, site-specific diversity was introduced in CDR-H1/H2, CDR-H3 and CDR-L2/L3 to create sub-libraries Lib1, Lib2 and Lib3, respectively. These sub-library fragments were then co-transformed with parental vector V1, V2 and V3, respectively, via homologous recombination known as “gap-repair” into yeast (Fig. 7B). After two to three rounds of selection in parallel, the selected sub-libraries (Lib1*, Lib2*, Lib3*) were isolated, PCR amplified and re-assembled using batch recombination to create full library LibF (Fig. 7C) LibF was subjected to additional rounds of selection under more stringent conditions. In the last round, the library was heat-shocked at 52.5°C to eliminate unstable binders, and the library was washed in the presence of excess parental IgG to remove weak scFv binders (Fig. 7D). The top binders were sorted and plated to form colonies for further characterization of individual clones.

Figure 7. Scheme of library design and selection for scFvs. (A) Template scFvs were sub-cloned into our yeast display vector containing FLAG tag and linker. Silent mutations were introduced on frameworks to create unique restriction sites: S1, S2, S3, S4 and S5 for shuffling of CDRs (open bars). (B) Three sub-libraries Lib1, Lib2 and Lib3 encoding designed mutagenesis on CDR-H1/H2, CDR-H3 and VL CDRs (filled black bars) were made in parallel from oligos. To eliminate WT CDR background, stuffer DNA (filled gray bars) was sub-cloned to generate vector V1, V2 and V3, which were then double digested and co-transformed with correlated sub-libraries into yeast through homologous recombination. (C) After two to three rounds of selection in parallel, the selected sub-libraries Lib1*, Lib2* and Lib3* encoding CDRs (stripped bars) were isolated from yeast and PCR amplified. (D) Full library (LibF) was assembled using batch recombination via overlapping PCR. After additional rounds of selection, the full library was selected under more stringent selection pressure: (1) heat-shocked to eliminate unstable binders, (2) bound to low concentration of antigen, (3) washed in the presence of 50-fold excess of parental IgG to compete off weaker binders. The top antigen binders were collected using FACS sorting.
Enrichment in the libraries is highlighted in Figure 8. Compared with libraries prior to selection (Fig. 8A and 8C), all sub-libraries (Lib1, Lib2, Lib3) restored antigen binding activities after selections (Fig. 8B and 8D), and the batch recombined full library (LibF) showed further improvement. For anti-ErbB3 scFvs, Lib2 of CDR-H3 shows the most improvement over wild-type clone (WT+). After batch recombined selected sub-libraries and additional round of selection, the full library (LibF) binding was further improved. The significance of batch re-assembly approach is better highlighted in the anti-IGF-1R campaign, where each sub-library (Lib1, Lib2, Lib3) had moderate enrichment after selections (Fig. 8D) compared with pre-selection (Fig. 8C), but did not show better than wild-type clone (WT+). After batch recombination and two additional rounds of selection, however, the full library (LibF) showed significant improved binding over the wild type clone.

Figure 8. Flow cytometry analysis of antigen binding activity of libraries before and after selections. (A and B) ErbB3 binding for three sub-libraries (Lib1, Lib2, Lib3) and full library (LibF), before and after selections, in comparison to the wild-type clone (WT+) and background (buffer). (C and D) IGF-1R binding of libraries before and after selections, in comparison to the wild-type and background. Cells were labeled with anti-Flag-Alexa 647 for surface expression (x-axis), and anti-his6-Alexa 488 for binding of IGF-1R-his6 (y-axis).
Next, the individual clones were heat shocked at 60°C and tested for residual binding to antigen using FACS. As shown in Figure 9, the majority of scFvs that retained binding after thermal challenge showed 5–50-fold improved affinity over wild-type in both anti-ErbB3 (data not shown) and anti-IGF-1R (Fig. 9) campaigns and are diverse in their sequences (data not shown).

Figure 9. High throughput thermal challenge assay of scFvs on yeast surface. Randomly selected IGF-1R binders from the final round of library selection (gray) and wild-type clone (black) were heat-shocked at 60°C for 5 min, chilled on ice, bound with 20nM IGF-1R-His6 and labeled with anti-Flag-Alexa 647 for expression and anti-his6-Alexa 488 for IGF-1R binding. The residual binding after heat-shock was measured on FACS. Mean fluorescent intensity (MFI) of antigen binding was normalized to MFI of expression.
Triage of soluble optimized anti-IGF-1R and anti-ErbB3 scFvs
Using a combination of “2-in-1” yeast display plasmid and high throughput expression and purification techniques, the optimized scFvs were expressed in soluble form and triaged using different micro-scale characterization techniques that require only microgram quantities of proteins.
Activity of anti-IGF-1R and anti-ErbB3 scFvs
First, we used ELISA to confirm that scFvs retained their binding affinity upon conversion from yeast surface display form to soluble form. Most scFvs bound tightly to the antigen with the best showing up to 30-fold improvement in affinity (Figs. S1 and S2).
Next, we determined the ability of optimized anti-IGF-1R and anti-ErbB3 scFvs to block IGF-1 or HRG induced signaling. These experiments were performed by incubating BxPC-3 cells with different concentrations of scFvs for 1 h followed by stimulation with either IGF-1 or HRG. Figures 10A and 10B show representative inhibition curves for pIGF-1R and pAKT, respectively. Figure 11 summarizes the signaling inhibition IC50 values and remaining percentage inhibition for all anti-IGF1R scFvs and anti-ErbB3 scFvs. In comparison to wild type, optimized scFvs had lower IC50 values, and inhibited higher fraction of ligand induced upstream and downstream signaling at highest dose (1 μM) suggesting that they were more active. In fact, two anti-IGF-1R scFvs (P4 and M57) showed 45-fold improvement in IC50 (for pIGF-1R) over the wild type scFv (Fig. S1). For both anti-IGF1R and anti-ErbB3 scFvs, there was good correlation between pAKT and pIGF-1R/pErbB3 IC50 and inhibition values (Fig. 11); however, it is interesting to note that the scFvs that showed highest affinity in ELISA were not the most active in signaling inhibition studies (Figs. S1 and S2). This could either be due to (1) the conformation of recombinant IGF-1R or ErbB3 coated on ELISA plate for binding assay could be different than the native form expressed on the cell surface; or (2) these scFvs may bind slightly different epitopes that could affect their ability to block ligand binding or signaling.
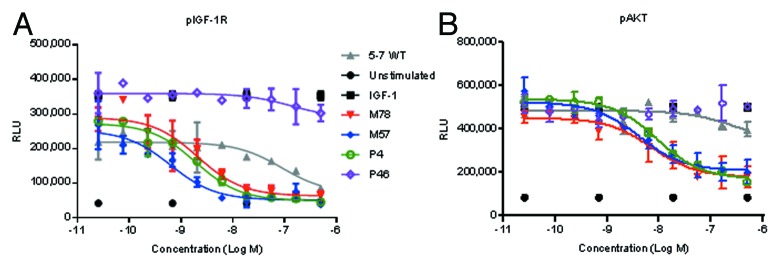
Figure 10. Activity of representative anti-IGF-1R scFvs in inhibiting IGF-1 induced phosphorylation of (A) IGF-1R, and (B) AKT. BxPC-3 cells were pretreated for 1 h with indicated concentrations of scFvs, and stimulated for 15 min with 100 ng/mL IGF-1. Cells were lysed, and ELISA was used to quantify the amounts of phosphorylated proteins.

Figure 11. Signaling inhibition properties of (A and C) anti-IGF-1R scFvs and (B and D) anti-ErbB3 scFvs (filled diamonds), in comparison to wild-type (black open diamond). Clones marked with black diamonds were chosen as optimized modules for reassembling into optimized bispecific antibodies.
Stability of anti-IGF-1R and anti-ErbB3 scFvs
Because these scFv modules can become parts of a clinical bispecific molecule, their stability in serum is critical for retaining maximum in vivo activities and pharmacokinetics (PK) profiles. Therefore, an assay was developed to indirectly assess whether scFvs undergo proteolytic cleavage or aggregation in serum. In this assay, scFvs were incubated in mouse serum at 37°C for 3 d, and tested for binding to IGF-1R or ErbB3 using the ELISA binding assay. Absorbance values at the inflection point of binding curve were normalized to Day 0 to determine residual binding after serum incubation.
We also used differential scanning fluorimetry (DSF) as an orthogonal technique to assess the stability of the optimized scFvs.27,28 The DSF and serum stability data for all anti-IGF-1R and anti-ErbB3 scFvs are summarized in Figure 12. Some anti-IGF-1R scFvs did not retain their initial binding, suggesting that they were unstable in mouse serum, but the majority of the tested variants retained greater than 90% of initial binding. On the other hand, half of the anti-ErbB3 scFvs lost over 50% of their initial binding, suggesting those scFvs were unstable in mouse serum. There were, however, a few stable anti-ErbB3 scFvs (M27, M20, B72, P6, and B60) that retained over 80% of initial binding (Fig. S2). As shown in Figure 12, consistent with the promise of the thermal challenge screening applied during library selection, the majority of both anti-IGF-1R and anti-ErbB3 scFvs was stable and had comparable or higher Tm than wild type.
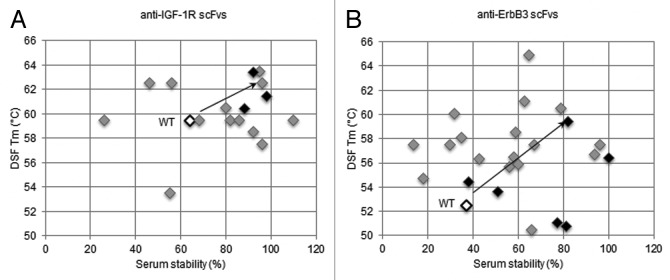
Figure 12. Overview of serum and thermal stabilities for (A) anti-IGF-1R scFvs and (B) anti-ErbB3 scFvs (filled diamonds), in comparison to wild-type (black open diamond). In serum stability assay, scFvs were incubated in mouse serum at 37°C for 3 d and then tested for residual binding to antigen (IGF-1R or ErbB3) using ELISA. For DSF, melting temperature (Tm) was calculated by taking the maximal value of first derivative of the resulting fluorescence data with respect to temperature. Clones marked with black diamonds were chosen as optimized modules for re-assembly into re-engineered bispecific antibodies.
Construction of re-engineered (optimized) bispecific antibodies
As illustrated in Figure 4, the best anti-IGF-1R and anti-ErbB3 scFvs were selected and combined in both orientations to create multiple re-engineered bispecific antibodies. ScFvs that were strong inhibitors of signaling but were unstable in mouse serum or had low Tm were considered only for the Fab side of re-engineered bispecific molecules. Similarly, scFvs that were stable but moderate inhibitors of signaling were fused only to the C-terminal side of anti-ErbB3 or anti-IGF1R IgGs. Two unstable anti-ErbB3 scFvs (M1.3 and c8) were also chosen in this orientation due to their divergent sequences. ScFvs that were both stable and strong inhibitors of signaling were combined in both orientations. All scFvs containing residues that can cause potential CMC liabilities, e.g., solvent exposed methionine residues, unpaired cysteines, deamidation sites and N-linked glycosylation sites were not considered for conversion into bispecific molecules. The Fab side of the molecule was created by fusing VH and VL domains from scFv to N-terminal of CH1 and CL domains, respectively.
Characterization of re-engineered (optimized) bispecific antibodies
Figure 13 shows the activity of representative optimized bispecific antibodies in inhibiting phosphorylation of IGF-1R, ErbB3, and AKT in BxPC-3 cells in response to dual ligand stimulation. In comparison to the POC version, all the optimized bispecific antibodies were more active as they had lower IC50 and greater percentage of inhibition of pIGF-1R, pErbB3 and pAKT (Table 1). The majority of optimized bispecific antibodies displayed between 10–25-fold improvements in pAKT signaling inhibition. For molecules in the αIGF-1R-Fc-αErbB3 orientation, P4-G1-M1.3 and M78-G1-M1.3 were most active at inhibiting pAKT. For molecules in the αErbB3-Fc- αIGF-1R orientation, M7-G1-M78 and M7-G1-P4 were most active at inhibiting pAKT.

Figure 13. Activity of representative optimized bispecific antibodies in inhibiting IGF-1 and HRG induced phosphorylation of (A) IGF-1R, (B) ErbB3 and (C) AKT. BxPC-3 cells were pretreated for 1 h with indicated concentrations of scFvs, and stimulated for 15 min with 30 ng/mL HRG + 100 ng/mL IGF-1. Cells were lysed, and ELISA used to quantify the amounts of phosphorylated proteins.
Table 1. Properties of the optimized bispecific antibodies.
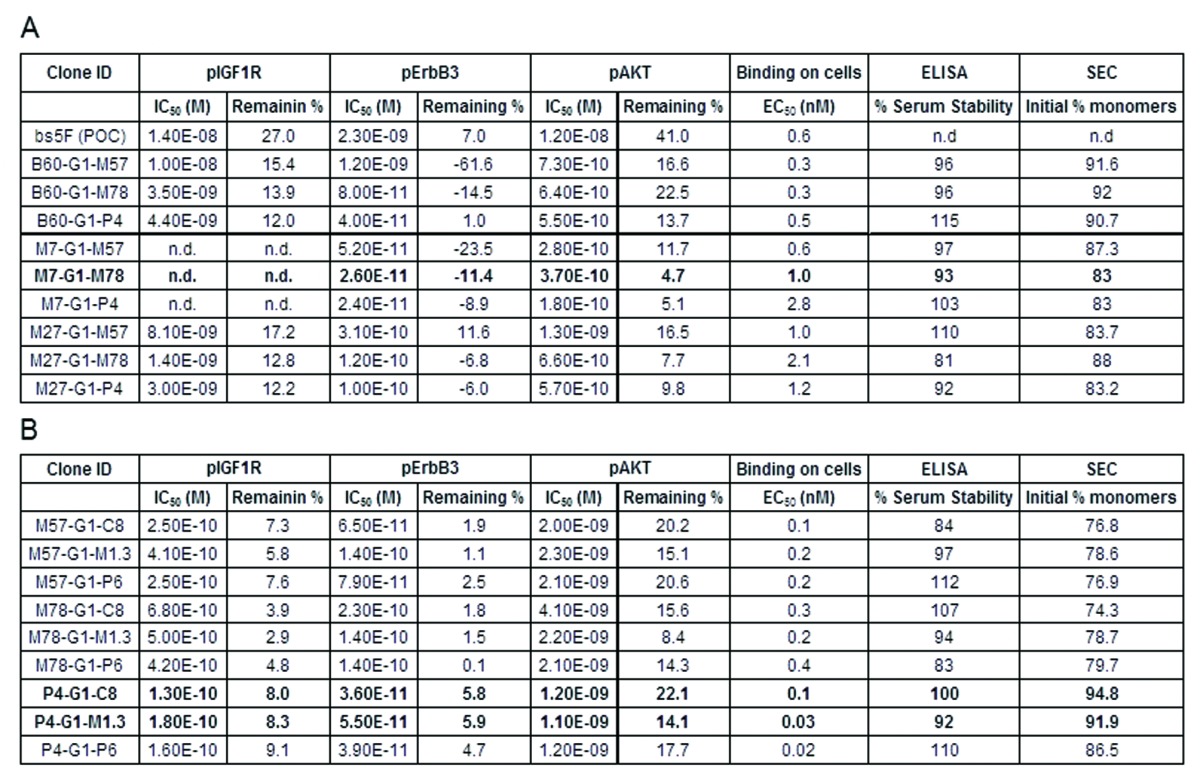
(A) Bispecific antibodies in αErbB3-Fc-αIGF1R orientation. (B) Bispecific antibodies in αIGF1R-Fc-αErbB3 orientation. Initial % monomers were measured by injecting 50 μg of protein A purified sample on a TSKgel SuperSW3000 column (4.6mm ID x 30 cm) using 10 mM sodium phosphate (+ 450 mM NaCl, pH 7.1) as running buffer. Clones in bold were chosen as therapeutic lead candidates.
The binding affinity of optimized bispecific antibodies to BxPC-3 cells was also measured using FACS. The binding data demonstrated that the majority of optimized bispecific antibodies had 2–6-fold improved affinity in EC50 compared with the POC bispecific, with two molecules (P4-G1-P6, P4-G1-M1.3) displaying 20–30-fold improvement. Similarly to our observations made with scFvs, there was no clear correlation between FACS binding data and signaling inhibition.
As shown in Table 1, after incubation in serum, the majority of optimized bispecific antibodies retained more than 80% of their initial binding. It is notable that some of them were more stable in mouse serum than their scFv modules, as both P4-G1-M1.3 and M57-G1-M1.3 retained more than 90% of their initial binding. This could either be due to production-related differences of O-linked glycosylation patterns in scFv, or due to the steric inhibition of degradation provided by the IgG part of the molecule.
For a molecule to be considered a therapeutic lead, it is critical that it can be produced in substantial amounts with high purity and high yield. Typically, purification losses are higher and yields are lower with aggregation-prone proteins. Therefore, the initial monomer percentage is a good indicator of the manufacturability of a protein. As can be seen in Table 1B, the bispecific antibodies in αIGF1R-Fc-αErbB3 orientation, P4-G1-c8 and P4-G1-M1.3, had the highest monomer percentage; on the other hand, in the αErbB3-Fc-αIGF1R orientation (Table 1A), the B60 series was the best.
Based on these combined results, we decided to select three therapeutic lead candidates: P4-G1-C8, P4-G1-M1.3 and M7-G1-M78 for further pre-clinical development. To mitigate risks that could be a function of structural class, we selected at least one molecule from each orientation. P4-G1-M1.3 was selected because it was the most active molecule in αIGF-1R-Fc-αErbB3 orientation and had high initial percent monomers. In the other orientation, of the two most active molecules, M7-G1-M78 was selected over M7-G1-P4, to maintain sequence diversity from P4-G1-M1.3. P4-G1-c8 was also selected because it had the highest initial percent monomers, and our sequence analysis (data not shown) predicted it to have the most favorable CMC profile.
Figure 14 shows the stability of the three lead candidates at 4°C. The material for this study was produced using stable CHO cell lines and was only protein A purified. In comparison to the POC bispecific (Fig. 3D), all three lead candidates were more stable and aggregated by less than 0.1% in a month. These three molecules were then further analyzed based on multiple biological assays, xenograft studies and PK studies (data not shown) to select the clinical development lead for the MM-141 project.
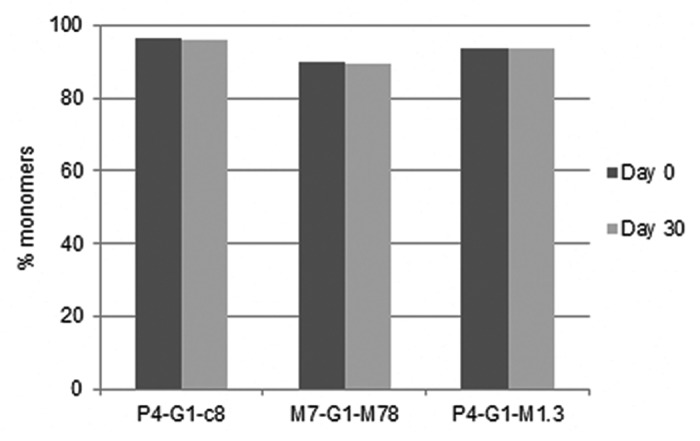
Figure 14. Stability of representative optimized bispecific antibodies over 1 month at 4°C in research formulation buffer (6 mg/mL in 20 mM histidine, 150 mM NaCl, pH6.5). The proteins were produced using stably transfected CHO cells and were only protein A purified.
Discussion
Despite the substantial advances in our understanding of antibody structure-function relationships, engineering of bispecific antibody-like molecule remains difficult. There are multiple contributing factors: the complexity of the sequence space for an antibody with multiple non-identical binding sites that preclude efficient iterative design, coupling of activity and pharmaceutical properties that require their concurrent optimization, and the need to design bispecific antibodies to be superior to monospecific antibodies and antibody combination therapies.29 We previously described a systems approach to therapeutic design of bispecific antibodies that addresses the latter challenge and allows fine-tuning of antibody-like molecules to a specific biological problem.30-32 Here, we present a rapid prototyping approach that facilitates the efficient engineering of bispecific antibodies with improved activity, stability and manufacturability. We use a well-understood tetravalent antibody format where the antigen binding sites of different specificity are located on the opposite ends of the molecule.9,24,33 We considered variable domains to be modular and pursued their concurrent parallel optimization using structure-focused, position-specific libraries that were screened in a “2-in-1” yeast display platform. The scFvs selected for activity in functional assays, serum and thermal stability are then combined in high throughput fashion into full-length bispecific antibodies that are transiently expressed in HEK293 cells and are profiled for activity and stability. We illustrate the power of this approach by describing the engineering of MM-141, a tetravalent bispecific antibody targeting redundant IGF-1R and ErbB3 signaling.
Affinity and stability are the key characteristics of antibody modules, and many approaches have been undertaken to improve these parameters. Random mutagenesis using error-prone PCR is a common strategy that does not require added library design efforts.34-36 However, this method can introduce uncontrolled mutations that are not essential for the function, which raise concerns about added immunogenicity and undesirable post-translational modifications. Light chain shuffling is often used when an initial library is available and no additional design efforts are needed to improve the affinity and diversity of binders;37-38 however, this approach is biased by the properties of the original library and cannot be reliably applied to optimize stability. In contrast, multiple antibody properties can be improved by precise structure-focused antibody design.39-40 All of these methods are iterative in nature, where the best variants from each round are combined in the final design and much of the originally sampled sequence diversity is discarded. This linear optimization is not ideal in the situations where optimization of multiple parameters is needed to improve properties of the molecule.
We took a hybrid approach where we used structural analysis to design targeted CDR-specific antibody libraries and yeast display techniques to select the binders on the basis of affinity and stability. To rapidly explore a large number of diverse sequence variants from individual libraries, we used a batch recombination technique.41-42 The advantages of the batch recombination approach are highlighted by the optimization of anti-IGF-1R scFv where best binders from individual libraries did not show improved binding, but after batch recombination and selection, we recovered variants with significantly improved affinity.
Our library screening approach integrated concurrent optimization of stability and affinity. To allow for efficient miniaturization, we utilized the antibody triage scheme that couples scFv thermostability with affinity measurement.43 Yeast display has been used to improve protein stability such as single chain T-cell receptor (scTCR),19 but in our experience the differentiation at surface expression level is not sufficient to discriminate between scFvs of varying stability. Because the viability of yeast drops rapidly when the temperature is elevated above 50°C, we introduced a soft thermal challenge that preserves sufficient yeast survival rate. While a strong heat shock at 79°C on a yeast library was recently reported,44 this method requires DNA isolation, PCR amplification and re-transfection for subsequent rounds of selection, decreasing the overall throughput and introducing potential for a mutation bias. Our mild 52.5°C challenge leads to unfolding of unstable yeast-fused scFv that refold during the recovery phase, therefore, the selected clones have a combination of higher thermal stability and faster refolding rates, which makes them resistant to aggregation. It has been reported that a transient heat denaturation method can be used to select aggregation-resistant domain antibodies that have refolding advantages,45 suggesting that antibody modules that unfold reversibly may have better colloidal stability. For stability profiling of soluble scFv modules, we utilized differential scanning fluorescence (DSF)27 and serum stability analyzed by antigen-binding ELISA. Both of these assays require microgram quantities of protein and can be performed using standard laboratory equipment.
We next combined the selected sets of antigen-binding modules with the preferred Fc scaffold into the full-length bispecific molecules and profiled them for affinity, bioactivity, thermostability, stability in serum, high expression yield and homogeneity. We observed that our modularity assumption holds true with one notable exception: one of the most active scFvs (M1.3) showed suboptimal stability in serum that does not present itself in the context of a full bispecific molecule. In fact, bispecific antibodies containing this module (P4-G1-M1.3 and M57-G1-M1.3) were stable in serum, and retained full binding to the antigens following three-day incubation. This observation highlights the importance of capturing and maintaining sufficient sequence and property diversity through lead selection to mitigate the risk of false negatives due to unanticipated interactions within the molecule.
The rapid prototyping approach described here offers several advantages over the conventional, iterative antibody optimization approach. First, multiple parameters such as affinities and stability can be optimized within a single design cycle, resulting in substantial savings of time and resources in advancing biotherapeutic molecules from biological proof-of-concept molecule to the clinical development candidate. Second, the modular nature of this approach allows the flexibility to fine-tune the molecular properties (such as affinities and avidities) of each module independently to a specific biological design objective. Third, this approach advances a sufficient number of therapeutic candidates with sufficient diversity in sequence and properties mitigating the risk of failure due to unanticipated late-stage challenges, such as poor PK properties, enhanced immunogenicity, or poor manufacturability. Fourth, this approach can easily be extended to optimize other multi-functional antibody molecules in a single design cycle by swapping out modules from one molecule to another.
Combination small molecule cytotoxic therapy has become a standard strategy in cancer treatment and we anticipate that combinations of antibodies will be commonly used in the clinic. In fact, the combination of trastuzumab and pertuzumab with docetaxel chemotherapy has recently been approved by the Food and Drug Administration (FDA) for the treatment of patients with HER2-positive metastatic breast cancer.46 Tetravalent bispecific antibodies simultaneously targeting two antigens can be designed to be superior to monospecific antibody combinations due to its higher avidity and ability to cross-link two distinct receptors on cells. We describe here the rapid prototyping approach toward engineering of tetravalent bispecific antibody molecules for activity and manufacturability and illustrate its success by presenting a case study of MM-141, a novel co-inhibitor of IGF-1R and ErbB3. We believe that such an approach will be generally applicable to optimization of multispecific antibody-like molecules leading to their increased use in treating human diseases.
Materials and Methods
Materials
pCEP4 vector, 293F cells, F17 media, HAMS-F12 media, RPMI medium, L-glutamine, Opti-MEM I, Lipofectamine LTX, pluronic F-68, 5000X sypro orange solution were purchased from Invitrogen. Linear Polyethylene imine (PEI, MW: 25 kDa) was purchased from Polysciences. 1× PBS was ordered from Lonza. CHO K1 cells were obtained from ATCC and suspension adapted in-house. Hyclone SFM4CHO media, TMB substrate, and fetal bovine serum (FBS) were purchased from Fisher Scientific. Library oligos were custom-synthesized at Yale University. iProof High-Fidelity DNA Polymerase was purchased from Bio-Rad. YPD plates, CM glucose minus tryptophan plates were purchased from Teknova. EZ-Link Sulfo-NHS-LC-Biotin, Fluorescence Biotin Quantitation Kit, DTT were purchased from Pierce. Streptavidin MicroBeads, LS Columns (MACS columns) were purchased from Miltenyi. M2 anti-Flag antibody, M2 Anti-Flag affinity gel, disodium phosphate, monosodium phosphate, dextrose, galactose, raffinose lithium acetate (LiAc), salmon sperm DNA (ssDNA), PEG3500, Tween-20, anti-FLAG-HRP antibody, mouse serum and glucose were purchased from Sigma. Alexa Fluor 647 and 488, Goat-anti-human IgG (Fc specific) were purchased from Life Technologies. Yeast extract, peptone, yeast nitrogen base and casamino acids were purchased from VWR. Zymoprep yeast plasmid miniprep kit was purchased from Zymo Research. IGF-1R-his, ErbB3-Fc and anti-His6 mAb were purchased from R&D Systems. Reacti-bind plates (96-well) and protein-free blocking buffer were purchased from Pierce. Anti-Fc-HRP antibody was purchased from Jackson Labs. TSKgel SuperSW3000 column was purchased from Tosoh Bioscience. Anti-AKT antibody was purchased from Millipore. BxPC-3, DU145 and A549 cell lines were obtained from ATCC. Anti-Fc-DyLight 649 secondary antibody was purchased from Abcam. Growth factor reduced matrigel was purchased from BD biosciences. All solutions were prepared in 18MΩ DI water.
Expression and purification of POC and re-engineered bispecific antibodies
All the bispecific antibodies were expressed either using transient transfection in 293F cells, or stable transfection in CHO K1 cells. All the genes were synthesized at DNA 2.0 and subcloned into either pCEP4 for transient transfection, or pMP10K (in-house proprietary vector) for stable transfection using standard microbiology techniques. The ratio of heavy chain and light chain DNA was kept 1:1 in all transfections. Unless otherwise stated, all bispecific antibodies were produced using the transient system.
Transient transfection
Cell culture media was prepared by adding 20 mL of 200 mM L-glutamine and 10 mL of 10% pluronic F-68 to 1 L of F17 media. For transient transfections, 450 mL of 293F cells were grown in cell culture media (37°C, 5% CO2) to a density of 1.5–2.0 million/mL in a baffled shake flask. On the day of transfection, 500 ug of total DNA and 1 mL of PEI solution (1 mg/mL) were premixed in 50 mL of cell culture media, briefly vortexed, incubated for 15 min, and added to cell culture. After a week, the cells were harvested by centrifugation (4000 g, 20 min), and the supernatant filtered using a 0.22 μm filter.
Stable transfection
Suspension adapted CHO K1 cells were grown in Hyclone Media supplemented with 8 mM L-glutamine to a density of 2 million/mL. On the day of transfection, the cells were resuspended in a serum-free media (Opti-MEM I) at a density of 80,000 cells/mL. The cells (500 uL) were then transfected with 1 ug of total DNA (including 10 ng of pNeo vector, an in-house vector carrying the geneticin selection marker) using 2.75 uL of lipofectamine in a 24-well plate. After 3 h, 1 mL of recovery media (HAMS-F12 + 10% FBS) was added, and the transfected cells allowed to recover for 48 h. The cells were then expanded into a 96-well plate, and the selection marker geneticin was added to the recovery media at 500 ug/ml. After four more days, the media was replaced with serum free Hyclone media (supplemented with L-glutamine), and the transfected cells allowed to adapt. After a week, the selected cells had formed colonies, and the wells were tested for the highest producing clone with an immunoblot. The highest-expressing clones were expanded to a 24-well plate, then to a T-25 flask, and eventually to a shake flask. The highest expressing clones were confirmed with SDS-PAGE, and scaled up to the desired volume. The cells were harvested by centrifugation (6000 g, 30 min) when the viability fell below 80%, and the supernatant filtered using a 0.22 μm filter.
Protein A chromatography
The filtered supernatant was loaded on a protein A column pre-equilibrated with PBS, and washed with high conductivity buffer (PBS+ 500 mM NaCl) to reduce non-specific interactions. The bound protein was eluted using 0.1 M acetic acid (pH 2.9), neutralized with 1M Tris base, and dialyzed against 1X PBS overnight.
Xenograft efficacy study
Female athymic nude mice were obtained from Charles River Laboratories at 4 to 5 weeks of age and were allowed to acclimatize for at least 72 h prior to use. To initiate tumor growth, on Day 0, mice were injected subcutaneously on the flank with 5x106 BxPC-3 cells, propagated in vitro by serial culture, in a 1:1 mixture of growth factor reduced matrigel and PBS. Mice were randomized into study groups and drug administration was initiated when the mean tumor volume reached 170 mm3. All antibodies were administrated by intraperitoneal injection, q3d, in 0.2 ml/ mouse of PBS. Control mice received only PBS injections. Tumor growth was monitored twice a week by measuring length (L) and width (W) using digital Vernier calipers, and tumor volume (mm3) was calculated using the formula π/6(L x W2). Student’s t-test was used to calculate statistical significance of the data set. Tumors were harvested 24 h after the final dose, and profiling was performed (by western blot) to assess effects of POC bispecific antibody on downregulation of IGF-1R and ErbB3.
Library design: Identification of library positions for stabilizing mutations and amino acid diversification
Sequence-based and structure-focused methods were used to identify candidate residues for mutagenesis and diversification. In brief, sequence-based methods included residue frequency, consensus, and canonical structure analysis methods to find residues in the VH and VL that occur rarely in natural antibody repertoire. Databases of VH and VL protein sequences derived from NCBI Entrez Protein database were used to establish the individual amino acid frequencies at each residue position of natural antibody repertoire. The consensus analysis was performed in reference to sets of VH and VL consensus sequences defined by Kabat.47 Canonical analysis was performed in reference to canonical structural residues critical to CDR conformation.48 The amino acids in scFvs in which frequencies in equivalent positions of natural antibody repertoire were less than 10% or that violated consensus sequences or canonical structures were considered candidates for mutations and were further analyzed in the context of structural models. The initial homology models of scFvs were built by MolIDE49 using independent zero-gap templates for VH and VL domains and energy-refined using SCWRL50-52 or using Chiron.53 The potential impact of the candidate mutations to most frequent, canonical or consensus residues on affinity was conducted visually in PyMOL54 with the assumption that the antigen binding site encompasses Chothia CDR1 and CDR3 loops of VH and VL domains. The potential impact of the candidate mutations on stability was conducted by assessing the steric clashes using PyMOL or energy differences using Eris.55 In addition, positions where scFv stabilizing mutations were previously described56,57 and mutations that eliminate potential CMC liabilities58 were analyzed using a similar approach. The candidate mutations that met pre-defined selection criteria were incorporated into scFv libraries. A combination of statistical and structural analysis was used to select residues in CDR loops that are amenable to diversification. First, we used the information on the amino acid selection bias during the initial lead generation and excluded the sites where strong “non-naïve” consensus has emerged. Next, we used structural analysis using PyMOL to select 15–18 positions per scFv that met the following criteria: (1) they were proximal to the positions where strong “non-naïve” consensus has emerged; (2) they were evenly distributed along the scFv paratope. Finally, these mutations were combined in yeast libraries in a manner that minimized the need for long oligo synthesis and the number of sequential subcloning steps.
Library design
Assembly of sub-libraries
Three sub-libraries Lib1, Lib2 and Lib3 with targeted mutagenesis on CDR-H1/H2, CDR-H3 and CDRL2/L3, respectively, were designed for each module. For anti-ErbB3 module, CDRs were diversified using spiked oligonucleotides such that each position retained 25–50% wild-type amino acids.59 For anti-IGF-1R module, NNS degenerated codons were used to diversify CDRs. Double-stranded DNA libraries were synthesized by using 20 pmol of library oligos as template and high fidelity PCR polymerase iProof (BioRad). The resulting DNA fragments were purified by 1% agarose gel, and the sub-library assembled using overlapping PCR of purified fragments. The DNA was then amplified by PCR, and purified using phenol extraction and ethanol precipitation. Precipitated DNA sub-libraries were dissolved in 10 mM Tris pH8.0, and the DNA concentration quantified directly on 2% E-gel, due to UV260 absorbance interference from free dNTPs that can also co-precipitate in PCR.
Construction of sub-library vectors: v1, v2 and v3
Parental scFv was synthesized in VH-ASTGGGGSGGGGSGGGGSGGGGS-VL with C-terminal Flag-tag, and unique restriction sites (S1-S5) were created by introducing silent mutations on frameworks. The synthesized scFv was sub-cloned into an in-house proprietary yeast display vector pMYD1000. For each vector, CDRs (CDRH1/H2 for v1, CDRH3 for v2 and CDRL2/L3 for v3) were replaced with stuffer DNA to remove wild-type background from libraries. The stuffed vectors (v1, v2, v3) were double digested with designed restriction enzymes (NEB), precipitated with ethanol and dissolved in 10 mM Tris pH 8.0 to a final concentration of 1μg/uL.
Transformation of libraries into yeast
PCR assembled and amplified fragments of each sub-libraries (Lib1, Lib2 and Lib3) were co-transformed with digested vectors v1, v2 and v3 respectively into yeast strain EBYZ (zeocine resistant strain of EBY100), using the electroporation method descripted previously.60 Small samples of transformed culture were serially diluted, plated on CM glucose minus tryptophan plates (Teknova), and incubated at 30°C for 3 d, and the colonies were counted to estimate library size.
Growth and induction of libraries
Transformed yeast libraries were grown in 100 mL of SD-CAA growth media (2.0% glucose, 0.67% yeast nitrogen base, 0.5% casamino acids, 0.54% disodium phosphate, 0.74% monosodium phosphate, 25 ug/mL zeocin), overnight at 30°C and 200 rpm. The OD (600 nm) of overnight culture was measured, 50 mL of SD-CAA media inoculated at a density of 1 OD600 / mL, and incubated at 30°C for an additional 6 h to dilute untransformed yeast in the culture. 2x109 cells or ~20x library size of cells were centrifuged at 3000 g and resuspended in SG-CAA induction media (2.0% galactose, 0.67% yeast nitrogen base, 0.5% casamino acids, 0.54% disodium phosphate, 0.74% monosodium phosphate, 25 ug/mL zeocin) at a density of 1 OD600/mL, and incubated at 18°C (200 rpm) for 2 d. The cells were centrifuged and resuspended in binding buffer (1X DPBS +0.5% BSA) for selection.
Sub-library selection
For each sub-library, first round of enrichment was performed on MACS cell separation system using streptavidin coated magnetic beads as descripted previously.13,61-63 Briefly, 2x109 induced cells were resuspended in 5 mL of binding buffer, and incubated with either 500 nM of biotinylated IGF-1R-His or 100 nM of ErbB3-Fc-His for 1 h at room temperature. The cells were centrifuged, washed three times with binding buffer and resuspended in 1 mL of binding buffer. 200 uL of Streptavidin MicroBeads were added to the cells, and incubated on ice for 15 min, with gentle inversion every 2 min. The cells were pelleted by centrifugation (2500 g, 5 min), washed three times, and resuspended in 10 mL of ice-cold binding buffer and loaded onto a pre-equilibrated LS column placed in the magnetic field. Once loading was complete, the column was washed with 10 mL of binding buffer. The bound cells were eluted with 5 ml of growth media, and the elution was repeated. The OD of the eluted cells was measured to estimate total number of cells eluted, eluted cells were grown and induced as described earlier for the next round of selection. For subsequent rounds of FACS sorting, the induced cells were incubated with decreasing concentrations of antigen, washed three times, and incubated with a 200 uL solution of 2 μg/mL Alexa647 labeled M2 anti-FLAG antibody and 2 μg/mL Alexa 488 labeled anti-His6 antibody for 30 min followed by three washes. The cells were then resuspended in binding buffer and sorted on BDAria. Typically, the top 0.1–0.5% of antigen binding cells was collected. To eliminate clones that displayed the non-specific binding, yeast surface displayed scFvs were screened against assay detection reagents as well as Fc-tagged and HHHHHH-tagged extracellular domain of Epidermal Growth Factor Receptor.
Assembly of LibF
After three rounds of selection (one round on MACS, two rounds on FACS), plasmid DNA of selected sub-libraries was extracted from yeast using Zymoprep kit. The DNA fragments of interest were PCR amplified; gel purified and re-assembled using overlapping PCR in batch to create a full library (LibF), which was then purified in a 1% agarose gel. The LibF was then PCR amplified, phenol extracted and ethanol precipitated as described earlier.
Thermo-challenge selection of LibF
LibF was selected one round for anti-ErbB3 or two rounds for anti-IGF-1R. For the final round of selection, each LibF was heat shocked at 52.5°C for 5 min, chilled on ice immediately for 5 min, and incubated with either 0.5 nM of ErbB3-Fc-his or 2 nM of IGF-1R-his for 30 min at room temperature. The cells were centrifuged at 2000 g, washed, incubated with parental IgGs for 1 h at a concentration 50 fold over antigen concentration (25 nM for clone E3B IgG2 or 100 nM for clone 5.7 IgG2) to compete off weak binders. The cells were then labeled with anti-Flag-Alexa647 and anti-His6-Alexa488 and sorted as described earlier. The sorted yeast cells were plated on tryptophan negative selection plate, and incubated at 30°C for 3 d. Yeast cells were scraped, and plasmid DNA isolated according to manufacturer’s protocol using Zymoprep kit. The DNA was transformed into DH5α E. coli competent cells, and the resulting colonies submitted for plasmid mini-prep and DNA sequencing.
“Cook & Bind” thermo challenge screening on yeast surface
Yeast colonies were grown in 1 mL of SD-CAA growth media in a 48 deep-well plate at 30°C and 200 rpm overnight to saturate. 0.25ml of cells (density about 3.0–5.0 OD600/ml) were transferred into 1 mL of SG-CAA induction media in a 48 deep-well plate at a density of 0.5–1 OD600/mL, and incubated at 18°C, 200 rpm for 2 d. The cells were harvested by centrifugation (3000 g, 5 min), washed and resuspended in binding buffer. Twenty uL (~5e5 cells) of yeast solution was heat shocked at different temperatures for 5 min, cooled on ice, and incubated with either 2 nM of ErbB3-Fc-his or 20 nM of IGF1R-his for 1 h at room temperature (22°C ± 2°C) in a 96-well plate. The cells were spun and washed three times, then incubated with 100 μL solution of 2 ug/mL anti-Flag-Alexa647 and 2 ug/mL anti-His6-Alexa488 secondary antibodies for 30 min. Cells were washed and resuspended in FACS buffer. Samples were read using a Becton Dickinson’s FACS Calibur, the resulting anti-His6 MFI (mean fluorescence intensities) of antigen binding were normalized for expression level (anti-Flag MFI), and the data plotted and analyzed using GraphPad PRISM.
Expression and purification of anti-ErbB3 and anti-IGF1R scFvs
All the scFvs had a c-terminal flag tag, and were expressed using a proprietary in-house vector pMYD1000, which also carries a gene for tryptophan synthesis that was used as a selection marker. To express scFvs in soluble form, plasmid DNA was digested with Sal1 enzyme to cleave the covalent fusion gene, and the resulting linear DNA was transformed into yeast cells.
Transformation
All the scFvs were transformed using a modified version of Gietz’s protocol. Briefly, a EBYZ colony (tryptophan deficient strain) was grown in 5 mL of YPD media (1.0% yeast extract, 2.0% peptone, 2.0% glucose, 25 ug/mL zeocin) overnight at 30°C and 200 rpm. The OD (600 nm) of the overnight culture was measured, and 50 mL of warm 2X YPD media was inoculated at a density of 0.25 OD600/mL. The culture was incubated at 30°C (200 rpm) until the cell density reached ~1 OD (takes ~5 h). The cells were harvested at 3000 g, and washed with 30 mL of sterile water. The cells were centrifuged again, resuspended in 100 mM LiAc solution at a density of 2 x 107 /mL, and 50 ul (for each transformation) were transferred to a 1.5 mL microfuge tube. The cells were pelleted by centrifugation at 2000 g, and supernatant carefully removed. The cells were mixed with 360 μL of transformation mix [240 μl of 50% PEG (w/v), 36 ul of 1M LiAc, 50 ul of ssDNA at 2 mg/mL, X μL of DNA (1 μg), and 34-X μL of water]. The resulting mixture was vortexed, and heat shocked in a water bath at 42°C for 50 min. The transformed cells were pelleted at 3000 g for 2 min, resuspended in YPD media and incubated for 0.5 h at 30°C. The cells were centrifuged again, resuspended in SD-CAA media and plated on selective CM glucose plates (minus tryptophan). The plates were incubated at 30°C for 3 d to allow colony formation.
Expression of scFvs
For each scFv, 100 mL of 2× SD-CAA growth media (2.0% glucose, 1.34% yeast nitrogen base, 1% casamino acids, 0.54% disodium phosphate, 0.74% monosodium phosphate, 25 ug/mL zeocin) was inoculated with a transformed colony. The culture was incubated in a shaker (30°C, 200 rpm) until the cell density reached saturation (~4 OD). The cells were centrifuged at 3000 g, resuspended in YPG induction media (1 yeast extract, 2% peptone, 2% galactose, 0.54% disodium phosphate, 0.74% monosodium phosphate, 25 ug/mL zeocin) and incubated at 20°C (200 rpm) for 3 d. The cells were harvested by centrifugation (3000 g, 10 min), and the supernatant filtered using a 0.22 μm filter.
Anti-flag chromatography
All the scFvs were purified using M2 anti-flag affinity resin in a high throughput manner using a vacuum manifold. The filtered supernatant was loaded on an anti-flag column pre-equilibrated with 1X PBS, and washed with high conductivity buffer (PBS+ 500 mM NaCl) to reduce non-specific interactions. The bound protein was eluted using 0.1 M acetic acid (pH 2.9), and neutralized with 1 M Tris base.
Signaling inhibition studies
Single ligand stimulation
Thirty-five thousand BxPC-3 cells were plated in RPMI media (+10% FBS) overnight at 37°C. The following day, cells were starved in RPMI media containing 0.5% serum, and incubated overnight at 37°C. Cells were pretreated for 1 h with indicated concentrations of scFvs, and stimulated for 15 min with either 100 ng/mL IGF-1, or 135 ng/mL HRG1b1-ECD. Cells were washed with PBS, and lysed in M-PER buffer supplemented with protease and phosphatase inhibitors. The resulting lysate was used for pErbB3, pIGF-1R and pAKT signaling inhibition ELISA.
Dual ligand stimulation
Thirty-five thousand BxPC-3 cells were plated in RPMI media (+10% FBS) overnight at 37°C. The following day, cells were starved in RPMI media containing 0.5% serum, and incubated overnight at 37°C. Cells were pretreated for 1 h with indicated concentrations of scFvs or bispecific antibodies, and stimulated for 15 min with 30 ng/mL HRG1b1-ECD + 100 ng/mL IGF-1. Cells were washed with PBS, and lysed in M-PER buffer supplemented with protease and phosphatase inhibitors. The resulting lysate was used for pErbB3, pIGF-1R and pAKT signaling inhibition ELISA.
Signal inhibition - ELISA analysis
ELISA for pErbB3 and pIGF-1R are from commercial sources (R and D Systems). For the pAKT ELISA assay, plates were coated with anti-AKT (Millipore), blocked with PBS+2%BSA, incubated with lysates and standards and detected with a biotinylated anti-pAKT (Ser473) and streptavidin-HRP. ELISA pico chemiluminescent substrate was added and plates read on a Perkin-Elmer Envision plate reader. All IC50 curves and calculated IC50 values were generated in Graphpad Prism. Percent inhibition was calculated using the following formula: 100*((fit.max-min.observed)/(fit.max-untreated control)).
Differential Scanning Fluorescence (DSF)
DSF assay was performed using IQ5 Real Time Detection System (Bio-Rad). Briefly, 19 ul of scFv solution (15 μM) was mixed with 1 μl of 20X sypro orange solution, and added to a 96-well plate. The plate was heated from 20°C to 90°C at a rate of 1C/min and the resulting fluorescence data collected. The data was transferred to GraphPad prism for analysis, and Tm was calculated by taking the maximal value of first derivative of the resulting fluorescence data with respect to temperature.
Binding by FACS for bispecific antibodies
Cell culture
BxPC-3 cells were maintained in RPMI media supplemented with 10% FBS, penicillin/streptomycin, and L-glutamine. On the day of FACS, the media was removed, and the cells washed with PBS. The cells were detached from tissue culture plates by incubating with 0.25% trypsin-EDTA for 5 min at 37°C. The detached cells were neutralized with serum containing RPMI media, spun down at 1000 g, and resuspended in FACS buffer (1X PBS + 2% Serum + 0.1% Azide). The cells were pipetted up and down, and passed through a cell strainer to reduce amount of aggregates. The cells were again spun down at 1000 g, and resuspended in FACS buffer at a density of 1 x 106 cells / mL. In a 96-well conical bottom plate, 50ul of cell suspension was aliquoted per well to give 50,000 cells / well.
FACS binding
Starting at a concentration of 2 μM, ten 3-fold dilutions of bispecific antibodies and one blank were prepared in FACS buffer, and 50 μL was mixed with 50 μL of cell suspension (50,000 cells) in a 96-well plate. The plates were incubated at room temperature with gentle agitation for 2 h. The plates were spun at 1000 g for 5 min, the supernatant removed, and the resulting cell pellets washed three times in FACS buffer. The cells were then incubated with 50 μL of anti-Fc-DyLight 649 secondary antibody (diluted 1:100 in FACS buffer) for 1 h. Cells were washed again three times with FACS buffer, and resuspended in 100 μL fixing buffer (PBS with 1% paraformaldehyde, 2% FBS). The samples were transferred to a 96-well plate, and kept in the dark at 4°C until use. Samples were read using a Becton Dickinson’s FACS Calibur, and median fluorescent intensities (MFI) were determined using FlowJo. One site - total binding model was used to determine EC50 values with GraphPad PRISM.
Binding of scFvs (or bispecific antibodies) to soluble IGF-1R-his or ErbB3-his
Reacti-bind plates (96-well) were coated with either 50 µL of ErbB3-His or IGF-1R-His (2 μg/mL in PBS), and incubated overnight at 4°C. The next day, the plates were washed with PBS-T (PBS + 0.05% Tween-20), blocked for 1 h at room temperature with 100 μL of blocking buffer and washed again with PBS-T. Plates were incubated with 50 μL of scFvs (or bispecific antibodies) at room temperature for 2 h, and then washed with PBS-T. Antibody concentrations started at 500 nM (in PBS-T), and included ten additional 2-fold dilutions and one blank (PBS-T only). Plates were then incubated with 50 μL of anti-FLAG-HRP antibody (or anti-Fc-HRP antibody for bispecific antibodies) diluted 1:20,000 in PBS-T for 1 h at room temperature, and washed again with PBS-T. The plates were incubated with 100 μL of TMB substrate for 5–10 min at room temperature and the reaction was stopped by adding 100 μL of Stop solution. The absorbance was measured at 450 nm, and the resulting data analyzed using GraphPad Prism.
Stability of re-engineered bispecific antibodies or scFvs in mouse serum
Re-engineered bispecific antibodies or scFvs were incubated in mouse serum at a final concentration of 2.5 μM for 0 d or 3 d at 37°C. The samples (2.5 μM) were diluted 1:5 in mouse serum to obtain a starting concentration of 500 nM. Ten additional samples were prepared by diluting the resulting 500 nM solution 2-fold in PBS-T (containing 20% mouse serum). All the samples were then assayed for binding to either IGF-1R-His or ErbB3-His using the colorimetric ELISA binding assay described above. Absorbance values (450 nm) were normalized to 0 d at the inflection point of each binding curve to determine residual binding after incubating in serum for 3 d at 37°C.
Size exclusion chromatography of bispecific antibodies
50 μg of sample was injected on a TSKgel SuperSW3000 column (4.6mm ID x 30 cm) using 10 mM sodium phosphate (+ 450 mM NaCl) as running buffer. All measurements were performed on Agilent 1100 HPLC, which was equipped with an auto sampler, a binary pump and a diode array detector. Data was analyzed using Chemstation software.
Supplementary Material
Acknowledgments
We thank Marco Muda, Stephen Sazinsky, Melissa Geddie, Brian Harms, Petra Loesch and Seth Fidel for valuable discussions and editing inputs in this manuscript. We appreciate Gege Tan for excellent experimental assistance. We thank Erin Soley, Sharlene Adams, Jose Varghese, Daryl Drummond and Gavin MacBeath for their scientific insights.
Glossary
Abbreviations:
- scFv
single chain variable fragment
- CDR
complementary determining region
- IGF-1R
type I insulin-like growth factor receptor
- ErbB3
v-erb-b2 erythroblastic leukemia viral oncogene homolog
- HRG
heregulin
- POC
proof of concept
- FACS
fluorescence-activated cell sorting
- HTP
high throughput
- SEC
size exclusion chromatography
- DSF
differential scanning fluorimetry
Supplementary Material
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/mabs/article/23363/
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/23363
References
- 1.Chen LF, Cohen EEW, Grandis JR. New strategies in head and neck cancer: understanding resistance to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2010;16:2489–95. doi: 10.1158/1078-0432.CCR-09-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gossage L, Eisen T. Targeting multiple kinase pathways: a change in paradigm. Clin Cancer Res. 2010;16:1973–8. doi: 10.1158/1078-0432.CCR-09-3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marvin JS, Zhu Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol Sin. 2005;26:649–58. doi: 10.1111/j.1745-7254.2005.00119.x. [DOI] [PubMed] [Google Scholar]
- 4.Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36:458–67. doi: 10.1016/j.ctrv.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17:871–9. doi: 10.1158/1078-0432.CCR-10-2621. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) J Natl Cancer Inst. 2001;93:1852–7. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 9.Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997;15:159–63. doi: 10.1038/nbt0297-159. [DOI] [PubMed] [Google Scholar]
- 10.Cohen-Solal JFG, Cassard L, Fridman WH, Sautès-Fridman C. Fc gamma receptors. Immunol Lett. 2004;92:199–205. doi: 10.1016/j.imlet.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, et al. Complement Activation Determines the Therapeutic Activity of Rituximab In Vivo J. Immunol. 2003;171:1581–7. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 12.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–9. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 14.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–7. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 15.Bowley DR, Labrijn AF, Zwick MB, Burton DR. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng Des Sel. 2007;20:81–90. doi: 10.1093/protein/gzl057. [DOI] [PubMed] [Google Scholar]
- 16.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–73. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo A, Ueda M. Yeast cell-surface display--applications of molecular display. Appl Microbiol Biotechnol. 2004;64:28–40. doi: 10.1007/s00253-003-1492-3. [DOI] [PubMed] [Google Scholar]
- 18.Pepper LR, Cho YK, Boder ET, Shusta EV. A decade of yeast surface display technology: where are we now? Comb Chem High Throughput Screen. 2008;11:127–34. doi: 10.2174/138620708783744516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol. 2000;18:754–9. doi: 10.1038/77325. [DOI] [PubMed] [Google Scholar]
- 20.Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol. 1999;292:949–56. doi: 10.1006/jmbi.1999.3130. [DOI] [PubMed] [Google Scholar]
- 21.Orr BA, Carr LM, Wittrup KD, Roy EJ, Kranz DM. Rapid method for measuring ScFv thermal stability by yeast surface display. Biotechnol Prog. 2003;19:631–8. doi: 10.1021/bp0200797. [DOI] [PubMed] [Google Scholar]
- 22.Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoël MJ, Fartoux L, Venot C, Bladt F, et al. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res. 2009;15:5445–56. doi: 10.1158/1078-0432.CCR-08-2980. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70:1204–14. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 24.Michaelson JS, Demarest SJ, Miller B, Amatucci A, Snyder WB, Wu X, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTbetaR. MAbs. 2009;1:128–41. doi: 10.4161/mabs.1.2.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jäger M, Gehrig P, Plückthun A. The scFv fragment of the antibody hu4D5-8: evidence for early premature domain interaction in refolding. J Mol Biol. 2001;305:1111–29. doi: 10.1006/jmbi.2000.4342. [DOI] [PubMed] [Google Scholar]
- 26.Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23:344–8. doi: 10.1038/nbt1067. [DOI] [PubMed] [Google Scholar]
- 27.Lavinder JJ, Hari SB, Sullivan BJ, Magliery TJ. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J Am Chem Soc. 2009;131:3794–5. doi: 10.1021/ja8049063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King AC, Woods M, Liu W, Lu Z, Gill D, Krebs MR. High-throughput measurement, correlation analysis, and machine-learning predictions for pH and thermal stabilities of Pfizer-generated antibodies. Protein Sci. 2011;20:1546–57. doi: 10.1002/pro.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald J, Lugovskoy A. Rational engineering of antibody therapeutics targeting multiple oncogene pathways. MAbs. 2011;3:299–309. doi: 10.4161/mabs.3.3.15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012;11:582–93. doi: 10.1158/1535-7163.MCT-11-0820. [DOI] [PubMed] [Google Scholar]
- 32.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 33.Dong J, Sereno A, Snyder WB, Miller BR, Tamraz S, Doern A, et al. Stable IgG-like bispecific antibodies directed toward the type I insulin-like growth factor receptor demonstrate enhanced ligand blockade and anti-tumor activity. J Biol Chem. 2011;286:4703–17. doi: 10.1074/jbc.M110.184317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daugherty PS, Chen G, Iverson BL, Georgiou G. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain Fv antibodies. Proc Natl Acad Sci U S A. 2000;97:2029–34. doi: 10.1073/pnas.030527597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, et al. Impact of single-chain Fv antibody fragment affinity on nanoparticle targeting of epidermal growth factor receptor-expressing tumor cells. J Mol Biol. 2007;371:934–47. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T, et al. Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol. 2002;9:933–42. doi: 10.1016/S1074-5521(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 37.Schier R, Bye J, Apell G, McCall A, Adams GP, Malmqvist M, et al. Isolation of high-affinity monomeric human anti-c-erbB-2 single chain Fv using affinity-driven selection. J Mol Biol. 1996;255:28–43. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 38.Brockmann EC, Akter S, Savukoski T, Huovinen T, Lehmusvuori A, Leivo J, et al. Synthetic single-framework antibody library integrated with rapid affinity maturation by VL shuffling. Protein Eng Des Sel. 2011;24:691–700. doi: 10.1093/protein/gzr023. [DOI] [PubMed] [Google Scholar]
- 39.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–6. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan JL, Arndt JW, Hanf K, Li G, Hall J, Demarest S, et al. Structural understanding of stabilization patterns in engineered bispecific Ig-like antibody molecules. Proteins. 2009;77:832–41. doi: 10.1002/prot.22502. [DOI] [PubMed] [Google Scholar]
- 41.Mabry R, Lewis KE, Moore M, McKernan PA, Bukowski TR, Bontadelli K, et al. Engineering of stable bispecific antibodies targeting IL-17A and IL-23. Protein Eng Des Sel. 2010;23:115–27. doi: 10.1093/protein/gzp073. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Keck ZY, Saha A, Xia J, Conrad F, Lou J, et al. Affinity maturation to improve human monoclonal antibody neutralization potency and breadth against hepatitis C virus. J Biol Chem. 2011;286:44218–33. doi: 10.1074/jbc.M111.290783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel. 2010;23:549–57. doi: 10.1093/protein/gzq028. [DOI] [PubMed] [Google Scholar]
- 44.Traxlmayr MW, Faissner M, Stadlmayr G, Hasenhindl C, Antes B, Rüker F, et al. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta. 2012;1824:542–9. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22:1161–5. doi: 10.1038/nbt1000. [DOI] [PubMed] [Google Scholar]
- 46.Baselga J, Cortés J, Kim SB, Im SA, Hegg R, Im YH, et al. CLEOPATRA Study Group Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–19. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu TT, Kabat EA, Bilofsky H. Some sequence similarities among cloned mouse DNA segments that code for lambda and kappa light chains of immunoglobulins. Proc Natl Acad Sci U S A. 1979;76:4617–21. doi: 10.1073/pnas.76.9.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol. 1996;263:800–15. doi: 10.1006/jmbi.1996.0617. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Canutescu AA, Dunbrack RL., Jr. SCWRL and MolIDE: computer programs for side-chain conformation prediction and homology modeling. Nat Protoc. 2008;3:1832–47. doi: 10.1038/nprot.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canutescu AA, Dunbrack RL., Jr. MollDE: a homology modeling framework you can click with. Bioinformatics. 2005;21:2914–6. doi: 10.1093/bioinformatics/bti438. [DOI] [PubMed] [Google Scholar]
- 51.Canutescu AA, Dunbrack RL., Jr. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–72. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–14. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins. 2011;79:261–70. doi: 10.1002/prot.22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeLano WL. The case for open-source software in drug discovery. Drug Discov Today. 2005;10:213–7. doi: 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
- 55.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nat Methods. 2007;4:466–7. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 56.Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 57.Ewert S, Huber T, Honegger A, Plückthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325:531–53. doi: 10.1016/S0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- 58.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–52. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, et al. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol. 2007;25:107–16. doi: 10.1038/nbt1269. [DOI] [PubMed] [Google Scholar]
- 60.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23:155–9. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 61.Feldhaus MJ, Siegel RW. Yeast display of antibody fragments: a discovery and characterization platform. J Immunol Methods. 2004;290:69–80. doi: 10.1016/j.jim.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 62.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–70. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 63.Feldhaus M, Siegel R. Flow cytometric screening of yeast surface display libraries. Methods Mol Biol. 2004;263:311–32. doi: 10.1385/1-59259-773-4:311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.