

Abstract
Ouabain-induced hypertension in rodents provides a model to study cardiovascular changes associated with human hypertension. We examined vascular function in rats after long-term treatment with ouabain. Systolic blood pressure (SBP) was measured by tail-cuff plethysmography in male Sprague-Dawley rats treated with ouabain (Oua, ~25 μg·day−1) or placebo for 8 weeks. Blood pressure increased in ouabain-treated animals, reaching 30% above baseline SBP after 7 weeks. At the end of treatment, vascular responses were studied in mesenteric resistance arteries (MRA) by wire myography. Contraction to potassium chloride (KCl) in intact and denuded arteries showed greater sensitivity in Oua-treated animals. Contraction to phenylephrine (PE) and relaxation to acetylcholine (ACh) were similar between groups with a lower response to sodium nitroprusside (SNP) in Oua-treated arteries. Sensitivity to endothelin-1 (ET-1) was higher in Oua-treated arteries. Na+-K+ ATPase activity was decreased in MRA from Oua-treated animals, whereas protein expression of the Na+-K+-ATPase α2 isoform was increased in heart and unchanged in mesenteric artery. Pre-incubation with indomethacin (10−5M) or L-NAME (10−4M) abolished the differences in KCl response and Na+-K+ ATPase activity. Changes in MRA are consistent with enhanced vascular smooth muscle cell reactivity, a contributor to the increased vascular tone observed in this model of hypertension.
Keywords: sodium pump, blood pressure, glycosides, contraction, vascular smooth muscle cells
INTRODUCTION
Alterations in vascular smooth muscle influencing contractility can lead to increased vascular tone and hypertension (1). Ouabain is an endogenous cardiotonic steroid with important roles in health and disease, regulating from cardiovascular function to metabolism and cell growth (2). This adrenocortical hormone (3) displays central and peripheral actions and ouabain plasma levels are increased in patients with essential hypertension (4) or congestive heart failure (5). The sodium pump (Na+-K+ ATPase) plays a vital role in the regulation of membrane potential in smooth muscle and the heart (6). The catalytic subunit of this enzyme, α, has three isoforms detected in rat: α1 is relatively insensitive, whereas α2 and α3 are relatively sensitive to cardiac glycosides such as ouabain. Like other glycosides (7), ouabain inhibits Na+-K+ ATPase activity by binding to the α2 subunit. It has been proposed that this inhibition alters intracellular calcium (Ca+2) homeostasis in smooth muscle cells, thus enhancing the responses to contractile agonists (8-11).
Although some models of ouabain treatment display no changes in blood pressure (12-14), rodent models subjected to long-term treatment with exogenous ouabain have been extensively used to investigate the effects of increased ouabain levels on blood pressure regulation (15-19). In this rodent model, blood pressure rises after 1 week of ouabain treatment and stabilizes after 5 weeks, resulting in approximately 30% increase in systolic blood pressure (SBP). Increased sympathetic activity and impaired baroreflex have been observed in models of ouabain-induced hypertension (18;20).
Alterations observed in vascular responses after ouabain treatment depend in part on the protocol (length of treatment and doses used) and the particular circulation being analyzed. For example, the contractile response to KCl has been reported to be similar in aorta, superior mesenteric or tail arteries (21), but greater in renal vessels (22) from ouabain-treated animals. Similarly, responses to α-adrenergic agonists have been reported to be higher (22) or lower (21) in vessels of ouabain-treated animals. It has been suggested that the lower adrenergic response reported in the aorta, but not in small resistance arteries (23), is the result of increased vasodilatory mechanisms induced by ouabain treatment (24-26), whereas no differences in vasodilatory responses have been reported in renal arteries (22). Recently, in rats treated for 5, 10 or 20 weeks with ouabain (8 μg·kg−1·day−1), no differences in vasodilatory responses were observed in MRA and a greater contraction to noradrenaline was observed only after 20 weeks of treatment (27).
The endothelin system plays an important role in blood pressure regulation (28). It is composed of peptide ligands, with endothelin-1 (ET-1) being the predominant form expressed in the vasculature, and receptors (ETA and ETB) ubiquitously expressed throughout the cardiovascular system (28;29). Evidence indicates that ouabain treatment activates the brain (30) and vascular (25) endothelin system. Di Filippo et al 2003, showed that four weeks of ouabain treatment increases ET-1 levels and decreases ETA receptor expression in the brain (30). At the vascular level, ouabain treatment increases ETA receptor expression with no changes in ETB receptor expression (25). In ouabain-treated animals, the administration of an endothelin ETA receptor blocker prevents the development of hypertension (25;30). Na+-K+ ATPase inhibition by ouabain plays an important role in controlling vascular tone (31), therefore, functional vascular Na+-K+ ATPase activity has been studied in different models of hypertension (32;33). In hypertensive rats, changes in Na+-K+ ATPase isoform expression have been detected in the aorta (34); whereas, in hypertensive patients, expression of the α2 isoform in cardiac tissue is increased (35). In ouabain-treated rats, no differences in vascular Na+-K+ ATPase activity have been reported in conductance vessels, however the sensitivity to inhibition by ouabain was increased in the aorta and decreased in tail arteries (21).
Our study addresses the effects of ouabain treatment over 8 weeks (25 μg·kg−1·day−1) on rat cardiovascular responses. Blood pressure was recorded and at the end of treatment small mesenteric resistance arteries (MRA) were studied using wire myography to determine: 1) contraction to potassium chloride, phenylephrine and ET-1; 2) relaxation to acetylcholine and sodium nitroprusside and 3) functional Na+-K+ ATPase activity. Protein expression of the Na+-K+ ATPase α2 subunit was also measured in heart tissue and main mesenteric artery in ouabain-treated animals.
MATERIALS & METHODS
Animal Preparation
12-week-old male Sprague-Dawley (SD) rats were obtained from Charles River Laboratory (Wilmington, MA). The animals were housed at a constant room temperature, humidity and light cycle (12:12h light dark), with a global 18% protein extruded rodent chow (Harlan Laboratories, Indianapolis, IN) and water available ad libitum. All experiments were performed in accordance with the guidelines of the Wake Forest University School of Medicine Institutional Animal Care and Use Committee.
Blood Pressure Measurements
Indirect systolic, diastolic, and mean arterial blood pressure and heart rate measurements were taken daily via tail-cuff plethysmography (Columbus Instruments, Columbus, OH), as previously described (36). Ten blood pressure readings per animal were recorded, from which the average was calculated for a single daily value over the course of 9 weeks: 1 week of baseline measurements and 8 weeks of ouabain treatment measurements.
Ouabain Pellet Implantation
After 1 week of baseline blood pressure measurements, the rats were divided into ouabain-treated and sham groups. The animals were anesthetized with 3% isoflurane, and a small incision was made on the back of the neck where a 1.5mg 60-day ouabain slow-release pellet (~25 μg of ouabain/day, Innovative Research, Sarasota, FL) was implanted subcutaneously in the ouabain-treated group (n=6). The control group (n=5) had a sham surgery performed in the same manner that the ouabain pellet was implanted.
Tissue Collections
After 8 weeks of treatment, animals were painlessly euthanized by asphyxiation with carbon dioxide (CO2) followed by a cardiectomy. Heart ventricles and main mesenteric artery were dissected, cleaned and stored at −80°C until further use. For reactivity studies mesenteric arcades were carefully removed and third-branch mesenteric arteries were isolated and cleaned of fat and connective tissue while kept in cold Krebs-Henseleit Buffer (KHB) containing (in mmol/l): NaCl 118, KCl 4.47, NaHCO3 25, KH2PO4 1.2, MgSO4 1.2, CaCl2 ·2H20 2.5, glucose 5.5.
Western Blotting
Collected heart tissues were homogenized and microsomes were isolated. Main mesenteric arteries were homogenized in lysis buffer (Hepes 50 mM, Nacl 150 mM, Triton X-100 1%, EDTA 1 mM, cocktail of protease inhibitors), and centrifuged at 2000xg for 5min, protein concentration was determined in supernatant using the Bradford method. For heart tissue, 100 μg of protein from control (n=5) and ouabain-treated (n=5) were used. For the mesenteric artery, 18 μg of total protein extract from arteries in control (n=4) and ouabain-treated (n=4) and 10 μg of total rat brain as positive control were used. Proteins were separated by 7.5% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat milk in Tris-buffered solution and incubated overnight at 4°C with primary rabbit polyclonal antibody anti-Na+-K+ ATPase α2 at 1:1000 dilution (Millipore, Billerica, MA). Anti-rabbit HRP-conjugated antibody (1:3000 dilution) was used as secondary antibody. The immunocomplexes were detected using an enhanced chemiluminescence system and ChemiDocTM XRS Software (Bio-Rad). Optical density relative to a standard sample, in arbitrary units (a.u.), was calculated using ImageJ Software (NIH, Bethesda, MD).
Vascular Reactivity Experiments
Arterial segments, maximum of 2mm in length, were mounted between an isometric force transducer (Kistler Morce DSC 6, Seattle, WA, USA) and a displacement device on a myograph (Multi Myograph, Model 620M Danish Myo Technologies, Aarhus, Denmark) using two stainless steel wires (diameter 40 μm), as described previously (37;38). The myograph organ bath (5 ml) was filled with KHB maintained at 37°C and aerated with 95% O2/5% CO2. In a subgroup of control (n=4) and Oua-treated (n=4) animals the endothelium was destroyed by passing a human hair through the lumen as described by Pulgar and Figueroa (37). The vessels were washed and incubated for 30 min before the normalization procedure was performed. Each arterial segment was stretched in a stepwise manner and the internal circumference and corresponding wall tension at each stretch were calculated and plotted to produce a resting wall tension–internal circumference curve for that particular artery using the DMT Normalization Module (ADInstruments). Arterial segments were normalized to 0.9·L100, with L100 being the internal circumference the vessels would have if they were exposed to a transmural pressure of 100 mm Hg, as previously described (39). Optimal diameters (OD) were calculated as OD=0.9·L100/π. MRA OD showed no differences between control and ouabain-treated animals (260±9 vs 255±10 μm, p>0.05). After obtaining the optimal diameter, a 30-min equilibration period preceded the addition of test substances.
Response to Potassium Chloride (KCl)
After equilibration, in order to test the viability of the arterial preparations and determine responses to non-receptor mediated contraction, MRA were exposed successively to increasing concentrations of potassium (K+) in KHB. Arterial segments were exposed to nine different concentrations of K+ (6.25–75 mM), with each dose being maintained for 2 min and then washed with KHB before the subsequent concentration was introduced. In parallel experiments, different arterial segments were denuded or pre-incubated for 15 min with the nitric oxide synthase inhibitor L-NAME (10−4 M), or the cyclooxygenase inhibitor indomethacin (10−5 M).
Response to Phenylephrine (PE)
After washing and resting for 20 min, MRA segments were exposed to a cumulative concentration-response curve of PE by exposing arteries to fourteen (10−8–10−4.5M) increasing concentrations in fourthlog steps, with each subsequent dose being introduced only after a steady response had been reached.
Response to Endothelin-1 (ET-1)
Under resting tension, MRA segments were exposed to a cumulative concentration-response curve of ET-1 by exposing arteries to nine (10−11–10−7 M) increasing concentrations in half-log steps, with each subsequent dose being introduced after a steady response had been reached (every 4 min). At the end of the ET-1 curve, arteries were washed and allowed to recover.
Response to Acetylcholine and Sodium Nitroprusside
Arteries were washed and stimulated with a sub-maximal dose of PE, between 10−6 M-3×10−5M, in order to attain an equivalent level of contraction. A dose response curve to acetylcholine (10−9 -10−5M) was performed. Data of all the vessels studied were included in the analysis, denuded arteries showed less than 10% relaxation to acetylcholine. After washing in KHB for at least 30 min, PE stimulation was repeated and after a stable contraction was reached, increasing concentrations of sodium nitroprusside (10−9-10−3 M) were added at 3 min intervals.
Functional Vascular Na+-K+ ATPase Activity Assay
Functional vascular Na+-K+ ATPase activity was determined as previously described (40). At resting, the media in the myograph chamber was changed to KHB without K+ (all K+ was replaced by Na+). After a 15 min incubation in KHB-K+ free media, arteries were stimulated with PE (3×10−6 M) and when a stable contraction was reached, K+ concentration was increased in steps at 2 min intervals by adding small volumes from a concentrated KCl solution. After full relaxation was reached, arteries were washed with normal KHB. After 15 min of resting, arteries were incubated again in KHB-K+ free media and 10−4 M ouabain was added to the chamber. After 15 min of pre-incubation, arteries were stimulated with PE and the dose response to K+ was repeated.
Drugs
Nω-nitro-L-arginine methyl ester (L-NAME), phenylephrine (PE), acetylcholine (ACh), sodium nitroprusside (SNP), and ouabain were from Sigma (St Louis, MO) and stock solutions were prepared in distilled water. Indomethacin (Sigma) was dissolved in 50 mM NaCO3 in KHB. Endothelin-1 (ET-1) (California Peptide Research Inc., Napa, CA) was dissolved in KHB with 1% BSA. All other chemical reagents were from Sigma.
Data Analysis
Maximal contractile responses to KCl were expressed in absolute values, whereas maximal responses to PE and ET-1 were expressed as a percent of the maximal response induced by KCl (%KMAX). Vasodilatory responses to ACh and SNP were expressed as % of pre-constricted tone. Na+-K+ ATPase activity was expressed as the difference in area under the curve (%dAUC) in the absence and presence of 10−4 M ouabain in the myograph chamber. Concentration-response curves for KCl, PE, ACh, SNP and ET-1 were analyzed by fitting individual experimental data to a logistic curve to determine the maximal response and sensitivity. The curve was of the form Y = bottom + (top − bottom) / (1 +10(LogEC50 − X)* Hill Slope)) where X is the logarithm of the concentration and Y is the response; the sensitivity values reported are derived from these fits. The contractile response to KCl was expressed in mN/mm as units of arterial wall tension (AWT) (AWT = force / 2 × length of vessel). For each arterial segment, the maximal response to KCl was calculated, and the response to contractile agonists was expressed as percent of the corresponding maximal response to KCl. Sensitivity was expressed as pD2 (pD2 = −log [EC50]) with EC50 being the concentration of agonist producing 50% of the maximal response. Data are expressed as mean ± SEM. One-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons was used to determine significant differences. A p < 0.05 was accepted as an indication of statistical significance.
RESULTS
Blood pressure measurements
Treatment with ouabain lead to an increase in systolic blood pressure (SBP) compared to control animals. The baseline SBP for the total of animals studied was 130.7±8 mmHg (mean±SEM; n=11). At the end of the ouabain-treatment, SBP values were 170±10 mmHg (n=6) for ouabain-treated and 127±10 mmHg (n=5) for the control group. The increase in SBP reached stable values after 7 weeks of ouabain treatment (130±12 vs 103.1±2.5% of baseline, p<0.05) (Figure 1).
Figure 1. Systolic blood pressure during Ouabain treatment.

SBP was measured daily via tail-cuff plethysmography as described in Materials & Methods in control (엯, n=5) and Oua-treated rats (●, n=6). Data are expressed as mean±SEM. * p<0.05 vs control.
Vascular measurements
Contractile responses
Potassium chloride
Similar maximal responses to KCl were observed between Oua and control MRA, with a greater sensitivity in MRA from Oua-treated animals (p<0.05, Figure 2, Table 1). Endothelial denudation increased sensitivity to KCl in control and Oua-treated arteries with the greater sensitivity being maintained in Oua-treated arteries (Figure 2B, Table 1). Pre-incubation with indomethacin decreased maximal responses in both groups with no effect on sensitivity (Figure 2C), whereas pre-incubation with L-NAME increased both maximal response and sensitivity in control, such that there is no longer a difference with Oua-treated arteries (Figure 2D). The higher sensitivity to KCl in untreated Oua MRA compared to control MRA was abolished by pre-incubation with indomethacin or L-NAME (Figure 2, Table 1),
Figure 2. Contractile responses to potassium chloride (KCl) in MRA from Control and Ouabain-treated rats.

A. Intact arteries (control 엯, n=13; Oua ●, n=9). B. Denuded arteries (control 엯, n=4; Oua ●, n=4). C. Arteries pre-incubated with indomethacin 10−5M (control 엯, n=10; Oua ●, n=8). D. Arteries pre-incubated with L-NAME 10−4M (control 엯, n=10; Oua ●, n=9). Data are expressed as mean±SEM. * p<0.05 in sensitivity vs control arteries.
Table 1.
Maximal responses (RMAX) and sensitivity (pD2) to contractile agonists (potassium chloride (KCl), phenylephrine (PE), and endothelin-1 (ET-1)) in mesenteric arteries from Control and Ouabain-treated (OUA) rats. Results are shown for intact, and denuded arteries and arteries pre-incubated with indomethacin or L-NAME as indicated.
| Intact | Denuded | |||
|---|---|---|---|---|
|
|
||||
| CONTROL | OUA | CONTROL | OUA | |
|
|
||||
| KMAX, mN/mm | 3.69±0.18 | 3.51±0.27 | 3.11±0.6 | 3.2±0.3 |
| pD2 | 1.38±0.01 | 1.43±0.03* | 1.43±0.03† | 1.58±0.05†* |
|
| ||||
| PEMAX, % KMAX | 107.6±9 | 116.8±11 | 127.4±5† | 129.4±4† |
| pD2 | 5.96±0.1 | 6.02±0.16 | 6.0±0.07 | 6.11±0.11 |
|
| ||||
| ET-1MAX, % KMAX | 137±8 | 126±11 | 146±10 | 129±15 |
| pD2 | 7.98±0.1 | 8.48±0.17* | 9.3±0.2† | 9.6±0.4† |
|
| ||||
| + Indomethacin | + L-NAME | |||
|
|
||||
| CONTROL | OUA | CONTROL | OUA | |
|
|
||||
| KMAX, mN/mm | 2.51±0.29† | 2.3±0.44† | 4.23±0.33† | 4.1±0.31† |
| pD2 | 1.36±0.03 | 1.41±0.03 | 1.44±0.02† | 1.44±0.03 |
|
| ||||
| PEMAX, % KMAX | 78.5±10.3† | 68.5±8.5† | 117±12.5 | 119±13.7 |
| pD2 | 6.1±0.15 | 6.02±0.2 | 6.25±0.06† | 6.18±0.17 |
|
| ||||
| ET-1MAX, % KMAX | 98±9† | 100±6† | 135±9 | 136±18 |
| pD2 | 8.22±0.06† | 8.54±0.2* | 8.23±0.08† | 8.76±0.2* |
p<0.05 vs control;
p<0.05 vs intact arteries.
Phenylephrine
Responses to PE showed no differences between Oua-treated and control MRA (Figure 3A). Endothelial denudation increased maximal response to PE in both groups (Figure 3B, Table 1). Indomethacin reduces maximal PE response in both groups (Figure 3C), whereas L-NAME treatment increased sensitivity in control arteries (Figure 3D and D, Table 1).
Figure 3. Contractile responses to phenylephrine (PE) in MRA from Control and Ouabain-treated rats.
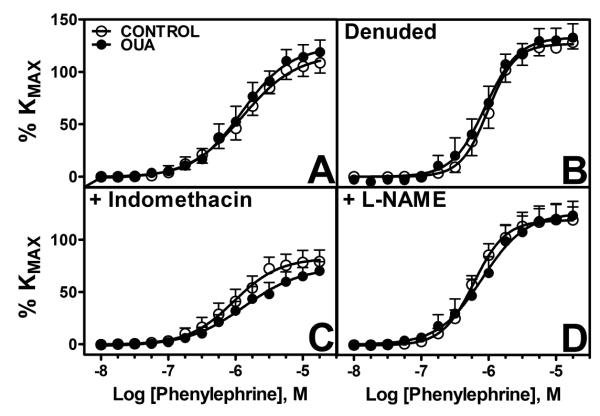
A. Untreated arteries (control 엯, n=9; Oua ●, n=8). B. Denuded arteries (control 엯, n=4; Oua ●, n=4). C. Arteries pre-incubated with indomethacin 10−5M (control 엯, n=9; Oua ●, n=8). D. Arteries pre-incubated with L-NAME 10−4M (control 엯, n=8; Oua ●, n=8). Data are expressed as mean±SEM.
Endothelin-1
A greater sensitivity to ET-1 (p<0.05) was observed in arterial segments from Oua-treated animals while no differences in maximal responses were seen (Figure 4A). Endothelium denudation induced an increased sensitivity in control and Oua-treated arteries and tended to alter the maximal response to ET-1; however, this did not reach statistical significance and no differences between both groups were found (Figure 4B, Table 1). Pre-incubation with indomethacin reduced maximal responses to ET-1 (Figure 4C), whereas L-NAME treatment increased sensitivity to ET-1 in control arteries. The greater sensitivity to ET-1 was maintained after pre-incubation with Indomethacin or L-NAME (Table 1).
Figure 4. Contractile response to endothelin-1 (ET-1) in mesenteric arteries from control and ouabain-treated rats.

A. Untreated arteries (control 엯, n=9; Oua ●, n=8). B. Denuded arteries (control 엯, n=4; Oua ●, n=4). C. Arteries pre-incubated with indomethacin 10−5M (control 엯, n=9; Oua ●, n=8). C. Arteries pre-incubated with L-NAME 10−4M (control 엯, n=8; Oua ●, n=8). Data are expressed as mean±SEM. *, p<0.05 in sensitivity vs control arteries.
Vasodilatory responses
Acetylcholine
Acetylcholine-dependent relaxation showed no differences between control and Oua-treated animals (Figure 5A, Table 2). Endothelial denudation abolished ACh relaxation in both groups (Figure 5B), whereas pre-incubation with indomethacin had no effect (Figure 5C). Pre-treatment with L-NAME greatly blocked ACh relaxation in control arteries, however the inhibitory effect was lower in Oua-treated arteries compared to control arteries (Figure 5D, Table 2).
Figure 5. Endothelium-dependent dilatation to acetylcholine (ACh) in MRA from Control and Ouabain-treated rats.

A. Untreated arteries (control 엯, n=4; Oua ●, n=4). B. Denuded arteries (control 엯, n=4; Oua ●, n=4). C. Arteries pre-incubated with indomethacin 10−5M (control 엯, n=4; Oua ●, n=4). D. Arteries pre-incubated with L-NAME 10−4M (control 엯, n=4; Oua ●, n=4). Data are expressed as mean±SEM.
Table 2.
Maximal responses (RMAX) and sensitivity (pD2) to vasodilatory agonists (acetylcholine (Ach) and sodium nitroprusside (SNP)) in mesenteric arteries from Control and Ouabain-treated (OUA) rats. Results are shown for intact and denuded arteries, and arteries pre-incubated with indomethacin or L-NAME as indicated.
| Intact | Denuded | |||
|---|---|---|---|---|
|
|
||||
| CONTROL | OUA | CONTROL | OUA | |
|
|
||||
| AChMAX, % | 87±3 | 84±3 | 5.4±2.4† | 8.7±2.3† |
| pD2 | 7.3±0.1 | 7.1±0.1 | nd | nd |
|
| ||||
| SNPMAX, % | 91.4±2 | 74.8±7* | 97.3±2.2 | 73.6±8* |
| pD2 | 5.5±0.1 | 5.93±0.4 | 6.51±0.3† | 6.13±0.4 |
|
| ||||
| + Indomethacin | + L-NAME | |||
|
|
||||
| CONTROL | OUA | CONTROL | OUA | |
|
|
||||
| AChMAX, % | 88±2 | 92±8 | 26.3±0.1† | 55.6±6.9†* |
| pD2 | 6.93±0.2 | 6.81±0.1 | 6.85±0.5 | 6.87±0.1 |
|
| ||||
| SNPMAX, % | 93.6±2 | 69.1±7* | 96.2±3 | 88.2±7 |
| pD2 | 6±0.4 | 6.2±0.2 | 6.8±0.5† | 6.14±0.4 |
p<0.05 vs control;
p<0.05 vs intact arteries.
nd: not determined.
Sodium nitroprusside
Response to sodium nitroprusside (SNP) in PE pre-constricted arteries showed lower maximal relaxation in Oua-treated arteries (Figure 6A, Table 2). Endothelial denudation increased SNP sensitivity in control arteries, and had no effect in Oua-treated arteries (Figure 6B). Indomethacin had no effect on SNP responses (Figure 6C), whereas L-NAME pre-incubation induced a greater sensitivity in control arteries (Figure 6D, Table 2).
Figure 6. Endothelium-independent dilatation to sodium nitroprusside (SNP in MRA from Control and Ouabain-treated rats.

A. Untreated arteries (control 엯, n=4; Oua ●, n=4). B. Denuded arteries (control 엯, n=4; Oua ●, n=4). C. Arteries pre-incubated with indomethacin 10−5M (control 엯, n=4; Oua ●, n=4). D. Arteries pre-incubated with L-NAME 10−4M (control 엯, n=4; Oua ●, n=4). Data are expressed as mean±SEM.
Functional Na+-K+ ATPase activity
Na+-K+ ATPase activity was lower in MRA from Oua-treated animals than from control animals (65±2.2 vs 73.4±2.5 %dAUC, p<0.05, Figure 7C). Endothelial denudation had no effect on both groups, while denuded Oua-treated arteries still displayed lower Na+-K+ ATPase activity compared to controls. Pre-incubation with indomethacin (69.4±5.1 vs 77.2±3.6 %dAUC, p>0.05) or L-NAME (72.2±4.2 vs 75.6±2.3 %dAUC, p>0.05) abolished this difference (Figure 7C).
Figure 7. Functional Na+-K+ ATPase activity in mesenteric arteries.

Functional Na+-K+ ATPase activity was measured in MRA from control and ouabain-treated animals as the relaxation to low concentrations of KCl in arteries pre-constricted with 10−6M PE. The specificity of the relaxation was tested by pre-incubation with ouabain 10−4M (−Oua/+Oua). A. Arteries from control animals (n=13, 엯 − Oua/● + Oua). B. Arteries from ouabain-treated animals (n=10, 엯 −Oua/● + Oua). C. Functional Na+-K+ ATPase activity expressed as the difference in area under the curve (dAUC, %) in arteries from control (◻) and ouabain-treated animals (∎), denuded, and pre-incubated with indomethacin (Indo, 10−5M) or L-NAME (10−4M) as indicated. Data are expressed as mean±SEM. *, p<0.05 vs control arteries
Expression of Na+-K+ ATPase α2 isoform in heart and mesenteric vessels
Protein expression of Na+-K+ ATPase α2 isoform showed greater values in heart tissue from Oua-treated animals compared to control animals (Figure 8A, p<0.05). In main mesenteric artery, Na+-K+ ATPase α2 isoform expression showed no difference between control and Oua-treated animals (Figure 8B).
Figure 8. Na+-K+ α2 isoform expression in heart tissue and mesenteric artery.

A. Na+-K+ α2 isoform expression was measured by western blot in microsomes from heart tissue obtained from control (◻, n=5) and ouabain-treated (∎, n=5) rats. B. Na+-K+ α2 isoform expression was measured by western blot in total protein extracts of main mesenteric artery obtained from control (◻, n=4) and ouabain-treated (∎, n=4) rats B: Rat brain. Inset: A representative gel is shown. Results from the densitometric analysis are expressed in arbitrary units (a.u.) as described in Materials & Methods. Data are expressed as mean±SEM. *, p<0.05 vs control.
DISCUSSION
We observed an increased vasoconstrictor tone in rat small resistance mesenteric arteries induced by prolonged in vivo treatment with ouabain. This increased vasoconstrictor tone was expressed as an increased sensitivity to a depolarizing solution of KCl, increased sensitivity to ET-1, lower relaxation to SNP, and diminished Na+-K+ ATPase vascular activity. Consistent with previous reports (21;23-25;27), we found that long–term ouabain treatment increased blood pressure, reaching approximately a 30% increase over baseline values after 8 weeks of treatment. The changes we observed in the resistance vasculature suggest an increased reactivity of the smooth muscle cell that may contribute to the increased blood pressure observed in this model of hypertension.
The contractile response to a dose response curve to KCl showed no differences in maximal response between control and treated MRA. This contraction appears to be mainly dependent on smooth muscle cells and is modulated by endothelial factors since endothelial denudation increased KCl sensitivity in both groups, with Oua-treated arteries still displaying a greater sensitivity compared to control arteries. Prostaglandins are one of those endothelial factors involved in vascular control, and we observed a diminished contraction to KCl in both groups pretreated with indomethacin. The effects of blockade of prostaglandins on MRA contractile responses appears to be dependent on the protocol used to test the responses; whereas an increased effect of indomethacin after a single dose of KCl was reported (41), a detailed study of the dependence of contractile responses of rat MRA on cyclooxygenase products indicated that indomethacin decreased contraction to a dose response curve of KCl (42). These effects appear to be mediated by cyclooxygenase as well as lipooxygenase vasoconstrictor products released during contraction to KCl (42). Our results suggest involvement of vasoconstrictor prostanoids in KCl contraction, in agreement with previous findings on the effects of prostanoids on adrenergic contraction in rat arteries (43;44). Treatment with L-NAME increased sensitivity to KCl in control arteries and maximal contraction in both control and Oua-treated arteries, as expected after inhibition of nitric oxide (NO) release. The increased sensitivity in Oua-treated arteries to KCl compared to control arteries was abolished when MRA were pre-incubated with indomethacin or L-NAME. Our results suggest that in Oua-treated MRA arteries enhanced sensitivity to KCl contraction is due to increased smooth muscle cell activation and this contraction is modulated by endothelial factors. An increased endothelium-dependent modulation of contraction responses after ouabain treatment has previously been suggested in the aorta (24). Depolarization-dependent contraction is mostly dependent on Ca+2 influx via voltage dependent calcium channels (VDCC) (45), and plays an important role in blood pressure regulation (46) and hypertension (47). A greater response to KCl has been reported in the renal artery in ouabain-treated animals (22) and both VDCC blockers, verapamil and nifedipine, partially inhibit in vitro (48) and in vivo (49) vascular effects of ouabain respectively. Thus, our results suggest that VDCC are also modulating contractile responses in the resistance circulation of ouabain-treated animals.
We observed no differences in responses to the α1-adrenergic agonist PE in MRA of Oua-treated animals compared to control animals. PE response was increased after endothelial denudation, whereas indomethacin lowered maximal responses to PE as described for adrenergic contraction in the same vascular bed (42). The inhibitory effect of indomethacin on α-adrenergic contractions has been described in aorta and caudal artery in control and Oua-treated rats (24), in agreement with the inhibitory effect of indomethacin reported on adrenergic contraction (43;44). Both endothelial denudation and L-NAME treatment increased maximal responses in both groups, indicating the modulatory role that NO has in adrenergic responses. It was previously shown that ouabain treatment for five weeks alters the adrenergic responses in conductance vessels mainly as a result of increased modulation by endothelial factors (21;23;24). It appears that a longer period of ouabain treatment is required to detect effects on adrenergic responses on MRA since recent data showed greater noradrenaline contraction in MRA only after 20 weeks of ouabain treatment (27).
In conductance vessels, such as the aorta, an increased contribution of endothelial derived factors to contraction has been observed in ouabain-treated animals. Our results of similar responses to ACh in ouabain-treated and control MRA are in agreement with the absence of changes reported in ACh responses in MRA at 5 or 10 weeks of ouabain treatment (27). ACh relaxation was abolished in endothelium-denuded arteries in both groups supporting a role of endothelium-derived NO, prostacyclin and/or EDHF (50-52). In agreement with previous reports (42), indomethacin did not modify the response to ACh, indicating that prostaglandins are not involved in endothelium-mediated dilatation in these vessels. Treatment with L-NAME blocked ACh dilatation in both groups; however the remaining dilatation was greater in Oua-treated arteries compared to control arteries. Relaxation of MRA in the presence of L-NAME has been shown to be inhibited by K+ channel blockers, such as tetraethylamonium, and ascribed to an endothelium-derived hyperpolarization factor (53). Thus, one possible explanation for the differences observed on ACh vasodilatation is that Oua-treated arteries possess an enhancement of EDHF-mediated hyperpolarization, which in turn reduces intracellular smooth muscle cell Ca2+ concentration causing relaxation (54) and would act as a compensatory mechanism to the increased constriction observed. EDHF activation has been showed to also modulate contraction responses; however in our experiments PE-mediated sensitivity remained unaltered, suggesting that some additional factors compensate adrenergic contractile responses in Oua-treated MRA.
Endothelium-independent vasodilatation, expressed as the response to SNP, was lower in Oua-treated arteries, suggesting that the smooth muscle portion of the nitric oxide–dependent vasodilatory pathway is affected by ouabain treatment in MRA. In control arteries, endothelial denudation and L-NAME treatment both increased sensitivity to SNP in agreement with numerous reports showing that activation of endothelium-independent relaxations may compensate for the loss of endothelial derived dilatation (55-58). This compensation appears to be dependent on an enhanced sensitivity of the soluble form of the enzyme guanylyl cyclase (sGC) in the absence of endogenous NO (55). The effects of ouabain treatment in sGC function and expression in MRA remain to be investigated.
Since earlier studies showed that ouabain stimulates expression and release of ET-1 from endothelial cells (59), and changes in central and peripheral endothelin system have been reported in models of ouabain-induced hypertension (25;30), we analyzed the contractile responses to the main agonist of this system, ET-1. Our results showed similar maximal responses and greater sensitivity for ET-1 in intact untreated MRA from ouabain-treated animals compared to control animals. Endothelial modulation of ET-1 contraction is evident in MRA since endothelial denudation, prostaglandins or NO inhibition all increased ET-1 sensitivity in control arteries. We observed decreased maximal contraction in the presence of indomethacin in both groups, similarly to what we observed for the other contractile agents, KCl and phenylephrine, and this is in agreement with previous evidences that mesenteric (60), as well as, aortic (61) reactivity to ET-1 involves the release of constrictor prostanoids that contribute to the contractile response. The increased sensitivity observed in Oua-treated arteries was not affected by indomethacin or L-NAME, indicating that prostanoids or NO are not responsible for the increased ET-1 sensitivity in Oua-treated MRA. ET-1 contraction is dependent on the action on its receptors ETA and ETB; ETB is present in muscle and endothelial cells, whereas ETA has been found only in smooth muscle cells (28). ETA activation mediates contraction, whereas vascular ETB receptors may mediate relaxation via NO-cGMP pathways, vasodilator prostanoids or activation of voltage-activated K+ channels (62). Evidence also indicates that ETB may mediate contraction (63). In MRA from hypertensive rats, an imbalance in the ET-1 contractile and dilatory responses has been described with an increase in ETA receptors and a decrease in ETB-mediated vasodilatation contributing to an enhanced ET-1 contraction (64). The greater ET-1 response we observed in Oua-treated MRA is in agreement with the fact that both endothelin blockers lower blood pressure (25;30) and endothelin receptor (ETA) expression in the aorta is increased in ouabain-treated animals (25). This suggests that in the MRA, ouabain-induced changes in the endothelin system, probably involving increased ETA-mediated signaling, are expressed. Whether these changes we observed in MRA are accompanied by changes at the level of receptor expression and/or signal transduction remains to be determined.
A key observation in the investigations about the role of the catalytic isoform of Na+-K+ ATPase in ouabain –induced hypertension was obtained using mice carrying a Na+-K+ ATPase enzyme containing an ouabain insensitive α2 subunit (65). These animals were refractory to ouabain treatment, did not develop hypertension, and displayed an increase in vascular contractility. Since ouabain and other glycosides inhibit Na+-K+ ATPase (7), we sought to measure the activity of the vascular Na+-K+ ATPase in small MRA from ouabain-treated animals. We used the validated protocol described by Webb & Bohr (1978), in which arteries pre-constricted with phenylephrine in a K+-free media are exposed to increasing K+ concentrations (40). We observed a reduced vasodilatation to K+ in MRA from ouabain-treated animals, whereas the effect of ouabain added to the assay was similar between groups. Our result of decreased Na+-K+ ATPase activity in ouabain-treated animals is similar to those found in vessels from spontaneously hypertensive rats (SHR) (32), and hypertensive dogs (33). As with the KCl contractile responses, the differences in Na+-K+ ATPase activity were maintained after endothelial denudation and abolished after pre-incubation with indomethacin or L-NAME, suggesting that ouabain-dependent effects on vascular Na+-K+ ATPase activity are affecting smooth muscle mechanisms and are modulated by prostanoids and NO. This is in agreement with an enhanced role for endothelium-derived factors in regulating Na+-K+ ATPase activity in hypertensive models (66). Previous reports in ouabain-treated animals indicate regional differences in the vascular functional Na+-K+ ATPase activity with increased activity in the aorta, decreased activity in tail arteries, and no changes in superior mesenteric artery (21). The reduced Na+-K+ ATPase activity we observed in MRA and the associated alterations in intracellular Ca2+ homeostasis may help explain the increased sensitivity in vasoconstrictor responses we observed.
In mice carrying the ouabain-insensitive α2 subunit, the ouabain-induced increase in cardiac contractility is abolished (67), providing direct evidence for the mechanism used by glycosides to elevate cardiac inotropy. Additionally, previous reports show that expression of Na+-K+ ATPase α2 subunit is increased in hearts of hypertensive patients (35). We observed a similar increase of the α2 subunit in hearts from ouabain-treated animals. In the mesenteric artery, the decreased functional Na+-K+ ATPase activity is not a result of diminished protein levels, as we did not observe differences in expression of the Na+-K+ ATPase α2 subunit; however, these determinations were performed in the main mesenteric artery and a direct comparison with small resistance vessels is needed. Elevated intracellular Na+ and Ca+2 concentrations have also been reported in hypertensive patients and increased intracellular Ca+2 concentrations have been observed in the vasculature of ouabain-treated rats (68). The upregulation of this enzyme in heart tissue contrasts with the decreased functional activity we observed in vascular tissue where no changes in expression were found. One possible explanation is that the presence of elevated ouabain levels and the consequent inhibition of Na+-K+ ATPase activity, induce a greater expression of α2 Na+-K+ ATPase as a compensatory reaction to increased intracellular Na+ concentrations. This compensatory reaction would be more evident in cardiomyocytes than in smooth muscle cells. Further research is needed to understand the relationships between Na+-K+ ATPase expression and activity in vascular smooth muscle cells.
CONCLUSION
We have demonstrated an increased response to constrictor agonists and decreased endothelium-independent relaxation in the resistance vasculature of animals treated with the glycoside ouabain. This was accompanied with an increased expression of the α2 Na+-K+ ATPase subunit in hearts and decreased Na+-K+ ATPase activity in MRA from treated rats. Our findings are consistent with enhanced vascular smooth cell reactivity to constrictor agents and may help explain the vascular changes that contribute to the increased blood pressure reported in this hypertension model.
ACKNOWLEDGEMENTS
This research was supported by grants NIMHD MD 002303, NIGMS 5T34GM070416 and HL51952.
Footnotes
Conflicts of Interest None conflicts of interest declared.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Mendelsohn ME. In hypertension, the kidney is not always the heart of the matter. J Clin Invest. 2005;115(4):840–844. doi: 10.1172/JCI24806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61(1):9–38. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A. 1991;88(14):6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamlyn JM, Ringel R, Schaeffer J, Levinson PD, Hamilton BP, Kowarski AA, Blaustein MP. A circulating inhibitor of (Na+ + K+)ATPase associated with essential hypertension. Nature. 1982;300(5893):650. doi: 10.1038/300650a0. [DOI] [PubMed] [Google Scholar]
- 5.Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation. 1992;86(2):420–425. doi: 10.1161/01.cir.86.2.420. [DOI] [PubMed] [Google Scholar]
- 6.Lingrel JB, Kuntzweiler T. Na+,K(+)-ATPase. J Biol Chem. 1994;269(31):19659–19662. [PubMed] [Google Scholar]
- 7.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol. 2007;293(2):C509–C536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 8.Leenen FHH. The central role of the brain aldosterone-“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta. 2010;1802(12):1132–1139. doi: 10.1016/j.bbadis.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Blaustein MP. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. Am J Physiol Cell Physiol. 1993;264(6):C1367–C1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- 10.Blaustein MP, Zhang J, Chen L, Song H, Raina H, Kinsey SP, Izuka M, Iwamoto T, Kotlikoff MI, Lingrel JB, Philipson KD, Wier WG, Hamlyn JM. The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension. 2009;53(2):291–298. doi: 10.1161/HYPERTENSIONAHA.108.119974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaustein MP, Hamlyn JM. Signaling mechanisms that link salt retention to hypertension: Endogenous ouabain, the Na+ pump, the Na+/Ca2+ exchanger and TRPC proteins. Biochim Biophys Acta. 2010;1802(12):1219–1229. doi: 10.1016/j.bbadis.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M, Martin A, Wen C, Turner SW, Lewis LK, Whitworth JA. Long-term ouabain administration does not alter blood pressure in conscious Sprague-Dawley rats. Clin Exp Pharmacol Physiol. 1995;22:919–923. doi: 10.1111/j.1440-1681.1995.tb02327.x. [DOI] [PubMed] [Google Scholar]
- 13.Pidgeon GB, Richards AM, Nicholls MG, Charles CJ, Rademaker MT, Lynn KL, Bailey RR, Lewis LK, Yandle TG. Chronic ouabain infusion does not cause hypertension in sheep. Am J Physiol Endocrinol Metab. 1996;270(3):E386–E392. doi: 10.1152/ajpendo.1996.270.3.E386. [DOI] [PubMed] [Google Scholar]
- 14.Cargnelli G, Trevisi L, Debetto P, Luciani S, Bova S. Effect of long-term ouabain treatment on contractile responses of rat aortae. J Cardiovasc Pharmacol. 2000;35(4):538–542. doi: 10.1097/00005344-200004000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ, Pamnani MB. Long-term ouabain administration produces hypertension in rats. Hypertension. 1993;22(2):178–187. doi: 10.1161/01.hyp.22.2.178. [DOI] [PubMed] [Google Scholar]
- 16.Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens. 1994;12(5):549–560. [PubMed] [Google Scholar]
- 17.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res. 2000;23(Suppl):S77–S85. doi: 10.1291/hypres.23.supplement_s77. [DOI] [PubMed] [Google Scholar]
- 18.Huang BS, Huang X, Harmsen E, Leenen FH. Chronic central versus peripheral ouabain, blood pressure, and sympathetic activity in rats. Hypertension. 1994;23(6 Pt 2):1087–1090. doi: 10.1161/01.hyp.23.6.1087. [DOI] [PubMed] [Google Scholar]
- 19.Veerasingham SJ, Leenen FHH. Ouabain- and central sodium-induced hypertension depend on the ventral anteroventral third ventricle region. Am J Physiol Heart Circ Physiol. 1999;276(1):H63–H70. doi: 10.1152/ajpheart.1999.276.1.H63. [DOI] [PubMed] [Google Scholar]
- 20.Aileru AA, De Albuquerque A, Hamlyn JM, Manunta P, Shah JR, Hamilton MJ, Weinreich D. Synaptic plasticity in sympathetic ganglia from acquired and inherited forms of ouabain-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R635–R644. doi: 10.1152/ajpregu.2001.281.2.R635. [DOI] [PubMed] [Google Scholar]
- 21.Rossoni LV, Salaices M, Marín J, Vassallo DV, Alonso MJ. Alterations in phenylephrine-induced contractions and the vascular expression of Na+,K+-ATPase in ouabain-induced hypertension. Br J Pharmacol. 2002;135(3):771–781. doi: 10.1038/sj.bjp.0704501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura K, Manunta P, Hamilton BP, Hamlyn JM. Different effects of in vivo ouabain and digoxin on renal artery function and blood pressure in the rat. Hypertens Res. 2000;23:67–76. doi: 10.1291/hypres.23.supplement_s67. [DOI] [PubMed] [Google Scholar]
- 23.Xavier FE, Rossoni LV, Alonso MJ, Balfagón G, Vassallo DV, Salaices M. Ouabain-induced hypertension alters the participation of endothelial factors in α-adrenergic responses differently in rat resistance and conductance mesenteric arteries. Br J Pharmacol. 2004;143(1):215–225. doi: 10.1038/sj.bjp.0705919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossoni LV, Salaices M, Miguel M, Briones AM, Barker LA, Vassallo DV, Alonso MJ. Ouabain-induced hypertension is accompanied by increases in endothelial vasodilator factors. Am J Physiol Heart Circ Physiol. 2002;283(5):H2110–H2118. doi: 10.1152/ajpheart.00454.2002. [DOI] [PubMed] [Google Scholar]
- 25.Xavier FE, Yogi Á , Callera GE, Tostes RC, Alvarez Y, Salaices M, Alonso MJ, Rossoni LV. Contribution of the endothelin and renin-angiotensin systems to the vascular changes in rats chronically treated with ouabain. Br J Pharmacol. 2004;143(6):794–802. doi: 10.1038/sj.bjp.0705994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padilha AS, Peçanha FM, Vassallo DV, Alonso MJ, Salaices M. Ouabain treatment changes the role of endothelial factors in rat resistance arteries. Eur J Pharmacol. 2008;600(1-3):110–116. doi: 10.1016/j.ejphar.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Wenceslau CF, Davel AP, Xavier FE, Rossoni LV. Long-term ouabain treatment impairs vascular function in resistance arteries. Journal of Vascular Research. 2011;48(4):316–326. doi: 10.1159/000322576. [DOI] [PubMed] [Google Scholar]
- 28.Rodríguez-Pascual F, Busnadiego O, Lagares D, Lamas S. Role of endothelin in the cardiovascular system. Pharmacol Res. 2011;63(6):463–472. doi: 10.1016/j.phrs.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 29.Jandeleit-Dahm KAM, Watson A. The endothelin system and endothelin receptor antagonists. Curr Opin Nephrol Hypertens. 2012;21(1):66–71. doi: 10.1097/MNH.0b013e32834dde48. [DOI] [PubMed] [Google Scholar]
- 30.Di Filippo C, Filippelli A, Rinaldi B, Piegari E, Esposito F, Rossi F, D’Amico M. Chronic peripheral ouabain treatment affects the brain endothelin system of rats. J Hypertens. 2003;21(4):747–753. doi: 10.1097/00004872-200304000-00018. [DOI] [PubMed] [Google Scholar]
- 31.Hauck C, Frishman WH. Systemic hypertension: the roles of salt, vascular Na+/K+ ATPase and the endogenous glycosides, ouabain and marinobufagenin. Cardiol Rev. 2012;20(3):130–138. doi: 10.1097/CRD.0b013e31823c835c. [DOI] [PubMed] [Google Scholar]
- 32.Ponte A, Marín J, Arribas S, González R, Barrús MT, Salaices M, Sánchez-Ferrer CF. Endothelial modulation of ouabain-induced contraction and sodium pump activity in aortas of normotensive Wistar-Kyoto and spontaneously hypertensive rats. Journal of Vascular Research. 1996;33(2):164–174. doi: 10.1159/000159145. [DOI] [PubMed] [Google Scholar]
- 33.Overbeck HW, Pamnani MB, Akera T, Brody TM, Haddy FJ. Depressed function of a ouabain-sensitive sodium-potassium pump in blood vessels from renal hypertensive dogs. Circ Res. 1976;38(6 Suppl 2):48–52. doi: 10.1161/01.res.38.6.48. [DOI] [PubMed] [Google Scholar]
- 34.Herrera VL, Chobanian AV, Ruiz-Opazo N. Isoform-specific modulation of Na+, K+-ATPase alpha-subunit gene expression in hypertension. Science. 1988;241(4862):221–223. doi: 10.1126/science.2838907. [DOI] [PubMed] [Google Scholar]
- 35.Jäger H, Wozniak G, Akintürk IH, Hehrlein FW, Scheiner-Bobis G. Expression of sodium pump isoforms and other sodium or calcium ion transporters in the heart of hypertensive patients. Biochim Biophys Acta. 2001;1513(2):149–159. doi: 10.1016/s0005-2736(01)00347-9. [DOI] [PubMed] [Google Scholar]
- 36.Buñag RD. Validation in awake rats of a tail-cuff method for measuring systolic pressure. J Appl Physiol. 1973;34(2):279–282. doi: 10.1152/jappl.1973.34.2.279. [DOI] [PubMed] [Google Scholar]
- 37.Pulgar VM, Figueroa JP. Antenatal betamethasone administration has a dual effect on adult sheep vascular reactivity. Pediatr Res. 2006;60:705–710. doi: 10.1203/01.pdr.0000246481.05231.17. [DOI] [PubMed] [Google Scholar]
- 38.Pulgar VM, Yamashiro H, Rose JC, Moore LG. Role of the AT2 receptor in modulating the angiotensin II contractile response of the uterine artery at mid-gestation. J Renin Angiotensin Aldosterone Syst. 2011;12(3):176–183. doi: 10.1177/1470320310397406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulvany MJ, Halpern W. Mechanical properties of vascular smooth muscle cells in situ. Nature. 1977;260(617):619. doi: 10.1038/260617a0. [DOI] [PubMed] [Google Scholar]
- 40.Webb RC, Bohr DF. Potassium-induced relaxation as an indicator of Na+-K+ ATPase activity in the vascular smooth muscle. Blood Vessels. 1978;15:198–207. doi: 10.1159/000158166. [DOI] [PubMed] [Google Scholar]
- 41.Sogni P, Sabry S, Moreau R, Gadano A, Lebrec D, Dinh-Xuan AT. Hyporeactivity of mesenteric resistance arteries in portal hypertensive rats. J Hepatol. 1996;24(4):487–490. doi: 10.1016/s0168-8278(96)80170-x. [DOI] [PubMed] [Google Scholar]
- 42.Wu XC, Johns E, Michael J, Richards NT. Interdependence of contractile responses of rat small mesenteric arteries on nitric oxide and cyclo-oxygenase and lipoxygenase products of arachidonic acid. Br J Pharmacol. 1994;112(2):360–368. doi: 10.1111/j.1476-5381.1994.tb13080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin L, Nasjletti A. Prostanoid-mediated vascular contraction in normotensive and hypertensive rats. Eur J Pharmacol. 1992;220(1):49–53. doi: 10.1016/0014-2999(92)90010-2. [DOI] [PubMed] [Google Scholar]
- 44.Tabernero A, Giraldo J, Vila E. Effect of NG-nitro-L-arginine methylester (L-NAME) on functional and biochemical alpha 1-adrenoceptor-mediated responses in rat blood vessels. Br J Pharmacol. 1996;117(4):757–763. doi: 10.1111/j.1476-5381.1996.tb15255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolton TB, MacKenzie I, Aaronson PI. Voltage-dependent calcium channels in smooth muscle cells. J Cardiovasc Pharmacol. 1988;12(Suppl 6):S3–S7. doi: 10.1097/00005344-198812006-00003. [DOI] [PubMed] [Google Scholar]
- 46.Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22(22):6027–6034. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: Pathophysiology and therapeutic implications. Vascul Pharmacol. 2006;44(3):131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marín J, Sánchez-Ferrer CF, Salaices M. Effects of ouabain on isolated cerebral and femoral arteries of the cat: a functional and biochemical study. Br J Pharmacol. 1988;93(1):43–52. doi: 10.1111/j.1476-5381.1988.tb11403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manunta P, Tyzack J, Hamilton BP, Hamlyn JM. Impact of antihypertensive therapy on ouabain-induced hypertension. J Hypertens. 1994;12:72. (Abstract) [PubMed] [Google Scholar]
- 50.Fleming I, Busse R. NO: the Primary EDRF. J Mol Cell Cardiol. 1999;31(1):5–14. doi: 10.1006/jmcc.1998.0839. [DOI] [PubMed] [Google Scholar]
- 51.Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond) 2009;117(4):139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- 52.Parkington HC, Coleman HA, Tare M. Prostacyclin and endothelium-dependent hyperpolarization. Pharmacol Res. 2004;49(6):509–514. doi: 10.1016/j.phrs.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 53.Mcculloch AI, Bottrill FE, Randall MD, Robin Hiley C. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br J Pharmacol. 1997;120(8):1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Félétou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Tromb Vasc Biol. 2006;26(6):1215–1225. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 55.Brandes RP, Kim Dy, Schmitz-Winnenthal FH, Amidi M, Gödecke A, Mülsch A, Busse R. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35(1):231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- 56.Ortiz PA, Garvin JL. Cardiovascular and renal control in NOS-deficient mouse models. Am J Physiol Regul Integr Comp Physiol. 2003;284(3):R628–R638. doi: 10.1152/ajpregu.00401.2002. [DOI] [PubMed] [Google Scholar]
- 57.Moncada S, Rees DD, Schulz R, Palmer RM. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc Natl Acad Sci U S A. 1991;88(6):2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linder AE, Weber DS, Whitesall SE, D’Alecy LG, Webb RC. Altered vascular reactivity in mice made hypertensive by nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46(4):438–444. doi: 10.1097/01.fjc.0000175879.14994.63. [DOI] [PubMed] [Google Scholar]
- 59.Saunders R, Scheiner-Bobis G. Ouabain stimulates endothelin release and expression in human endothelial cells without inhibiting the sodium pump. Eur J Biochem. 2004;271(5):1054–1062. doi: 10.1111/j.1432-1033.2004.04012.x. [DOI] [PubMed] [Google Scholar]
- 60.Rakugi H, Nakamaru M, Tabuchi Y, Nagano M, Mikami H, Ogihara T. Endothelin stimulates the release of prostacyclin from rat mesenteric arteries. Biochem Biophys Res Commun. 1989;160(2):924–928. doi: 10.1016/0006-291x(89)92523-0. [DOI] [PubMed] [Google Scholar]
- 61.Asano H, Shimizu K, Muramatsu M, Iwama Y, Toki Y, Miyazaki Y, Okumura K, Hashimoto H, Ito T. Prostaglandin H2 as an endothelium-derived contracting factor modulates endothelin-1-induced contraction. J Hypertens. 1994;12(4):383–390. [PubMed] [Google Scholar]
- 62.Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, D’Orléans-Juste P, De Oliveira AM. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+ channels in ETB-induced relaxation. Br J Pharmacol. 2005;146(6):903–912. doi: 10.1038/sj.bjp.0706388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mickley EJ, Gray GA, Webb DJ. Activation of endothelin ETA receptors masks the constrictor role of endothelin ETB receptors in rat isolated small mesenteric arteries. Br J Pharmacol. 1997;120(7):1376–1382. doi: 10.1038/sj.bjp.0701036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montagnani M, Potenza MA, Rinaldi R, Mansi G, Nacci C, Serio M, Vulpis V, Pirrelli A, Mitolo-Chieppa D. Functional characterization of endothelin receptors in hypertensive resistance vessels. J Hypertens. 1999;17(1):45–52. doi: 10.1097/00004872-199917010-00008. [DOI] [PubMed] [Google Scholar]
- 65.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol. 2005;288(2):H477–H485. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 66.Marín J, Redondo J. Vascular sodium pump: endothelial modulation and alterations in some pathological processes and aging. Pharmacol Ther. 1999;84(3):249–271. doi: 10.1016/s0163-7258(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 67.Dostanic I, Lorenz JN, Schultz JE, Grupp IL, Neumann JC, Wani MA, Lingrel JB. The α2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J Biol Chem. 2003;278(52):53026–53034. doi: 10.1074/jbc.M308547200. [DOI] [PubMed] [Google Scholar]
- 68.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;298(1):H263–H274. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]