

Abstract
The transient nucleolus plays a central role in the upregulated synthesis of ribosomal RNA (rRNA) to sustain ribosome biogenesis, a hallmark of aberrant cell growth. This function, in conjunction with its unique pathohistological features in malignant cells and its ability to mediate apoptosis, renders this subnuclear structure a potential target for chemotherapeutic agents. In this Minireview, structurally and functionally diverse small molecules are discussed that have been reported to either interact with the nucleolus directly or perturb its function indirectly by acting on its dynamic components. These molecules include all major classes of nucleic acid-targeted agents, antimetabolites, kinase inhibitors, anti-inflammatory drugs, natural product antibiotics, oligopeptides, as well asnano-sized particles. Together, these molecules are invaluable probes of structure and function of the nucleolus. They also provide a unique opportunity to develop novel strategies for more selective and therefore better tolerated chemotherapeutic intervention. In this regard, inhibition of RNA polymerase I-mediated rRNA synthesis appears to be a promising mechanism of cancer cell kill. The recent development of molecules targeted at G-quadruplex forming rRNA gene sequences, which are currently undergoing clinical trials, seems to attest to the success of this approach.
Keywords: antitumor agents, apoptosis, DNA-targeted, p53, ribosome biogenesis
Introduction
Multifactorial drug resistance and systemic toxicity continue to be the major limitations in the treatment of malignant disease. The management of many aggressive cancers, such as non-small-cell lung cancer (NSCLC), faces an urgent need for novel strategies that provide longer-lasting responses at all stages of the disease and improve overall survival.[1,2] Classical chemotherapy often provides no significant survival benefit but relieves symptoms only for a short duration.[2] Molecularly targeted therapies designed to selectively intercept tumor angiogenesis and growth signaling usually exhibit more favorable toxicity profiles. Unfortunately, such treatments often show a rapid onset of resistance and provide a modest improvement only if administered in combination with common cytotoxics.[3] Most importantly, several aggressive cancers, such as triple-negative breast cancer (TNBC) and squamous cell carcinoma (SQCC) of the lung, a form of non-small-cell lung cancer (NSCLC), do not respond to common targeted therapies because of their genetic make-up.[4,5] Thus, in order to overcome the above shortcomings, it is important to constantly evaluate and validate new cancer targets. At the core of such strategies are targets that improve the performance and safety of cytotoxic agents by combining their classical antiproliferative properties with features directed at cancer cell-specific abnormalities.[1] One such target, the cancer cell’s nucleolus, has begun to show great promise in cancer chemotherapy. By developing new technology capable of exploiting the functional and cytomorphological differences between the nucleoli in cancerous versus normal cells, a wide range of cancers could potentially be targeted with manageable safety risks to the patient.
Structure and function of the nucleolus
The nucleolus can be described as a dynamic, non-membrane-bound subnuclear structure, whose most widely studied function involves ribosome biogenesis. This process can be studied indirectly by monitoring ribosomal RNA (rRNA) transcription by RNA polymerase I (Pol I), which correlates with the transient nature of the nucleolus.[6] In microscopic images, the nucleolus first becomes visible at the end of mitosis during telophase, as does the first detection of Pol I transcription. As cells enter into interphase (the cell growth phase between cell divisions), the formation of pre-nucleolar bodies (PNBs) and translocation of the transcription and processing machinery to the assembled nucleolar organizer regions (NORs) are observed (Figure 1).[7] This activity, which increases during early G1, leads to multiple nucleoli which fuse into a single nucleolus as the cell cycle progresses. The nucleolus disassembles during the beginning of cell division, in prophase, and is not detected again until the end of mitosis in telophase (Figure 1).[7,8]
Figure 1.

Assembly and disassembly of the nucleolus (no) in the nucleus (nu) during cell-cycle progression. Times are based on data reported for HeLa human cervix adenocarcinoma cells (see ref. 7).
The nucleolus is composed of three distinct components: the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC), which is believed to contain inactive rDNA and non-transcribing Pol I.[9] The rDNA found in the nucleolus accounts for around 50% of the total RNA production in the cell. Early accumulation and processing of rRNA occurs within the DFC, while late rRNA processing and assembly of the ribosomal components occurs in the GC.[10]
NORs describe the head-to-tail tandem repeats of the ribosomal genes (rDNA).[11] Approximately 400 copies of the 13,357 base-pair 45S pre-mRNA gene (NCBI reference: NR 046235)are found in the NORs of 5 acrocentric chromosome pairs, which are processed in the nucleoli.[6] A silver-staining method, AgNOR,[12] has proven useful to distinguish actively transcribed from non-transcribed rDNA, as silver ions only stain those NORs that contain transcriptionally active protein complexes. Important observations have been made regarding AgNOR levels and Pol I activity, indicating that increased AgNOR staining results from an increase in rRNA transcription. This observation has been significant in the histological and pathological assessment of tumor aggression, carcinogenic staging, and neoplastic potential.[13,14]
Proteomic studies suggest that the nucleolus is involved in a multitude of cellular processes independent of ribosome biogenesis. A recent study has identified 271 distinct proteins found in nucleolar extracts of HeLa cells. Aside from the expected transcription and processing factors, another large subgroup of proteins localized to the nucleolus were DEAD-box helicases, chaperones, and other nucleotide-binding proteins, such as topoisomerases I and II.[15,16] Tubulin, a heterodimer of two alpha and two beta chains, is the major subunit of the microtubules.[17] This globular protein was previously thought to exist solely in the cytoplasm of cells, where it is incorporated into the cytoskeleton. Immunofluorescence studies indicate that the βII-subunit of tubulin localizes to the nuclei, with a strong aggregation in the nucleoli of many different types of carcinoma cells (C6, T98G, MCF-7, MDA-MB-435, and HeLa).[18] Upon addition of the drug taxol, an inhibitor of mitosis targeted at the microtubules of dividing cells,[19] βII-tubulin underwent rearrangement in the nuclei and displayed the formation of micronuclei, which are markers of apoptotic cell death.[18] Nucleolar involvement has also been implicated in the biogenesis of certain ribonucleoproteins, such as telomerase[20] and the signal recognition particle.[21] Nucleolar localization sequences (NOLSs), which contain a conserved (R/K)(R/K)(R/K)X(R/K) motif, have been demonstrated to aid in the direction of proteins to the nucleolus versus the nucleus.[22] To this end, nucleolar hub proteins may also bind certain proteins leading to their localization in the nucleolus.[22] Knowledge of the location, function, and time of residency[23] of proteins in the nucleolus is critical in elucidating additional drug targets in the nucleolus.
The nucleolus as a drug target
An important aspect regarding nucleolus structure and function are the morphological differences that exist between normal somatic cells and neoplastic/malignant cells. It has been well established that an increase in rRNA transcription, as is found in rapidly replicating cells, results in a prominent increase in nucleolus size.[24] This relationship is even more pronounced when the tumor suppressor gene p53 is either mutated or absent, leading to large, hypertrophic nucleoli.[25] Notably, p53 mutations are found in greater than 50% of all human cancers.[26] Enlarged nucleoli are required to support the rapid, aberrant growth of tumor cells. Because the nucleoli persist throughout all growth phases of the cell cycle, downmodulation of rRNA synthesis at this locus can be considered a direct way of inhibiting the proliferative potential of tumor cells (“ribosome starvation”). Disruption of rRNA transcription and processing, as well as other forms of nucleolar stress, can lead to cell cycle arrest and programmed cell death in G1 or G2. This includes p53-mediated pathways in cells with functional p53.[27–29] Another important pathway involving the retinoblastoma protein (pRb) has been shown to affect RNA transcription and nucleolar hypertrophy. Together, cancers with inactive p53 and pRb have been characterized with increased aggressiveness and a more negative prognosis.[30] In addition, the Myc and PI3K-AKT-mTOR pathways have demonstrated a cooperative mechanism for promoting ribosome biogenesis. Consequentially, when these pathways are hyperactive, they are more likely to lead to more aggressive and resistant forms of cancer.[31] From the distinct change in nucleolus size seen in carcinogenesis, drugs targeting the nucleolus have the potential to produce an immediate and detrimental impact on cancer cells, while sparing a larger portion of normal cells.
Molecules interacting with the nucleolus or modulating nucleolar activity
1. Actinomycin D
Actinomycin D, a polypeptide antibiotic, has been shown to bind duplex DNA efficiently and decrease RNA synthesis by stalling Pol I. This inhibition is considered a reversible process due to the dissociation of actinomycin D (a reversible intercalating agent) from duplex DNA, which restarts normal transcription.[32] Actinomycin D is unique in that it inhibits rRNA and cytoplasmic RNA synthesis, but does not inhibit the synthesis of chromosomal RNA or DNA at low concentrations.[33] This suggests that the antibiotic selectively acts on the rDNA template in the nucleolus by inhibiting rRNA transcription, which quenches ribosome biogenesis and cytoplasmic RNA levels.[32] This evidence for a nucleolus-mediated mechanism is supported by studies demonstrating that a redistribution of nucleolar components occurs during actinomycin treatment.[16,24,34]
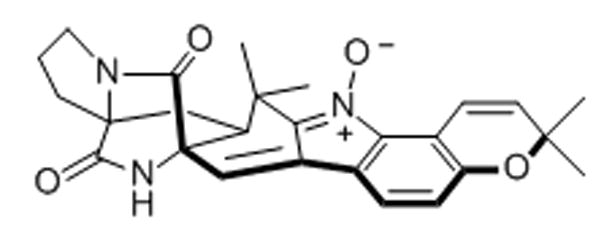
2. Avrainvillamide
A naturally occurring alkaloid, (+)-avrainvillamide, has been demonstrated to trigger anti-proliferative and apoptotic events in cells.[35] The alkaloid was fluorescently labeled with a dansyl moiety for use in fluorescence microscopy.[35] The conjugate was found to accumulate in the nucleoli and cytosol of HeLa-S3 cervical cancer and T-47D human ductal breast epithelial tumor cells. Moreover, co-localization is observed with fluorescently-tagged nucleophosmin, a nucleolar protein expressed in many cancers, which has been correlated with tumorigenesis.[36] Nucleophosmin, which is mainly located in the GC of the nucleolus, has demonstrated functions involving the stimulation of rDNA transcription, the processing of pre-rRNA, and as both a positive and negative modulator of p53-mediated apoptosis depending on the stimulus and cell type.[37] More recent studies have shown that the compound is able to bind to sulfur of Cys-275 in nucleophosmin, which could explain its co-localization and anti-proliferative effects.[38]
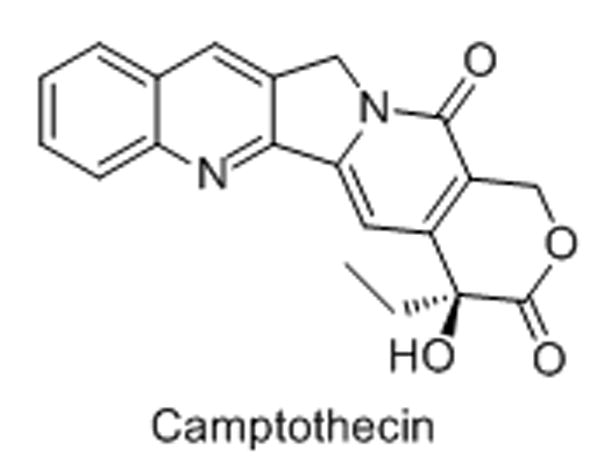
3. Camptothecin and analogues
Camptothecin and its clinically active derivatives, irinotecan (pro-drug) and topotecan (water-soluble analogue), are quinolone alkaloids used as chemotherapeutic agents.[39,40] The drugs are potent inhibitors of topoisomerase I (Topo I), an enzyme responsible for relieving strain in super-coiled DNA that occurs during transcription and replication.[41] Topo I has been shown to predominantly reside in the nucleolus of cells, aiding in rRNA transcription elongation.[41,42] In cells treated with this class of drugs, Topo I is redistributed to the nucleoplasm and a build-up of covalent Topo I–DNA cleavable complex, which is stabilized by camptothecin and its analogues, is observed, ultimately leading to cell death.[41,43]
4. CDK9 inhibitors
Cyclin-dependent kinase 9 (CDK9) inhibitors such as DRB, flavopiridol, and roscotivine have been shown to inhibit early rRNA processing and transcription.[44,45] Transcription inhibition occurs by inhibiting the transcription elongation step catalyzed by Pol I. The blocking of transcription and early rRNA processing leads to nucleolar disintegration and relocation of nucleolar proteins, such as nucleophosmin.[45]
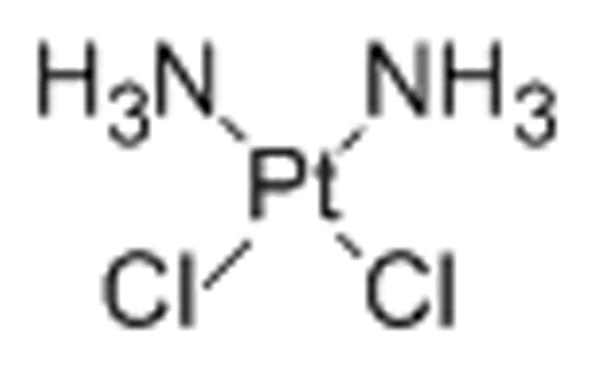
5. Cisplatin
Cisplatin, cis-diamminedichloridoplatinum(II), is a platinum chemotherapeutic agent widely used in the clinic. It forms 1,2-intrastrand cross-links and covalently platinates the N7 position of adjacent purine bases in double-stranded, Watson–Crick base-paired DNA.[46] Cisplatin was shown to inhibit rRNA synthesis indirectly in cell culture by causing a redistribution of the rRNA transcription machinery in the nucleolus.[46] These include the upstream binding factor (UBF), TATA-binding protein (TBP), TBP-associated factors for Pol I (TAFIs), and Pol I itself.[46] Along with the redistribution of nucleolar proteins, a segregation of nucleolar RNA occurred (micronucleoli) and a cascade of caspases was activated ultimately leading to apoptosis.[47]
6. CX-3543 (Quarfloxin)
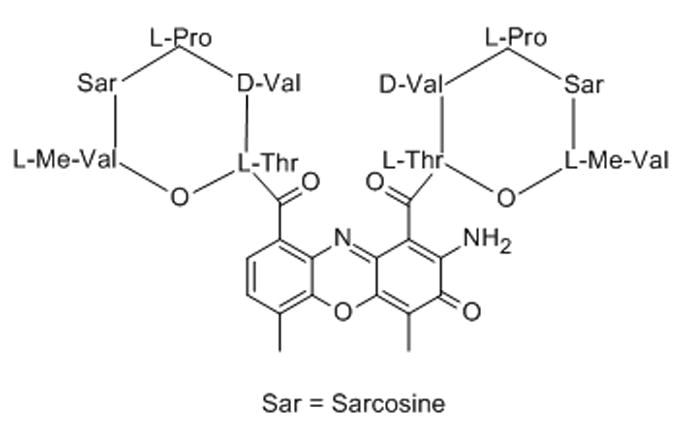
Quarfloxin, a fluoroquinoline derivative, developed after a long structure–activity relationship study evolving from fluoroquinoline antibiotics, was shown to localize to the nucleolus of A549 lung adenocarcinoma cells by fluorescence microscopy.[48] Quarfloxin selectively inibits rRNA synthesis by interfering with Pol I transcription elongation, and was shown to induce apoptosis in cancer cells.[48] Quarfloxin’s ability to release nucleolin, a G-quadruplex binding nucleolar phosphoprotein, into the nucleoplasm, has been interpreted as evidence that the drug’s mechanism involves competitive binding to this DNA secondary structure in rDNA.[49] Although tolerated well by patients in Phase I clinical trials, with no major organ toxicities, quarfloxindid not proceed past Phase II clinical trials due to its limited bioavailability.[49] Quarfloxin was the first G-quadruplex binding agent to enter human clinical trials.
7. CX-5461
CX-5461 is an orally available anti-cancer agent capable of selectively inhibiting Pol I, whose development was inspired by chemically related quarfloxin.[50–51] In melanoma and pancreatic carcinomas, the primary pathway to cell death was found to be autophagy,[51] in contrast to leukemia and lymphoma models where the primary pathway was p53-mediated apoptosis.[50] CX-5461 induces selective inhibition of Pol I-mediated rRNA synthesis resulting in cancer cell-specific toxicity, but has no effect on DNA, mRNA, or protein synthesis.[51]
8. Distamycin–ellipticine hybrid agent (“Distel”)
Cationic conjugates were synthesized containing the classical DNA minor groove binder distamycin and the intercalator ellipticine with the goal of generating molecules able to recognize specific DNA sequences. Biophysical and biochemical experiments were performed to confirm the proposed hybrid binding mode of Distel, which distorts the structure of B-form DNA significantly.[52] Fluorescence microscopy was used to detect Distel and ellipticine in treated cells.[53] Both the conjugate and the intercalator itself preferentially accumulated in the nuclei of HeLa cells. Unlike ellipticine fluorescence, which showed a high intensity across the entire nuclei, Distel fluorescence was less pronounced and was localized to the nucleoli.
9. Ethidium bromide
Ethidium bromide (EtdBr), the classical DNA-intercalating agent, was studied for cellular localization and metabolism in isolated rat hepatocytes.[54] It was determined by confocal laser scanning microscopy (CLSM) that ethidium bromide is rapidly taken up into the nucleoli, nuclear membrane, and endoplasmic reticulum of the cultured cells. Likewise, the related analogue, propidium iodide (PI), a classical red-fluorescent non-cell-permeable DNA stain, showed time-dependent accumulation in live HeLa cells.[55] The agent had no major effect on cell cycle progression and did not appear to affect the response of cells to other cytotoxic agents in co-treatment experiments.
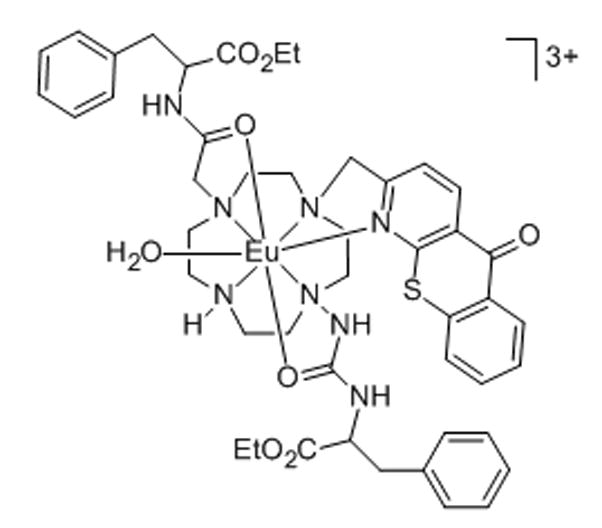
10. Europium–azathiaxanthone complex
Due to their favorable photophysical properties, lanthanide(III) complexes are well-suited as molecular probes. When equipped with a sensitizing moiety to overcome their extremely low molar absorptivities, these metal complexes are able to produce narrow spectral bands and high signal/noise ratios in optical spectroscopies.[56,57] One such complex, a Eu(III) complex with an azathiaxanthone sensitizing moiety, was shown to localize to the nucleoli of cells.[57] This complex was shown to be non-toxic to cells at 100μM, to enter cells by means other than endocytosis, and to co-localize with SYTO-RNA-Select dye (see below), making it an ideal probe for nucleolar localization.[57] The complex was tested in NIH 3T3 (standard fibroblast) cells, HeLa (human cervix endothelial carcinoma cells), and HDF (non-transformed human dermal fibroblast) cells. In NIH 3T3 cells, the fluorescence intensity in the nucleolus was found to be three times higher than that in the cytoplasm.[57]
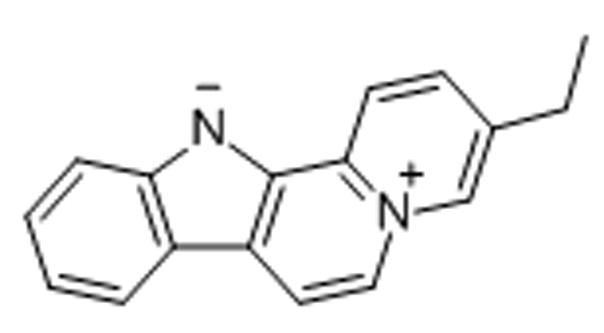
11. Flavopereirine
Flavopereirine (PB-100), a plant-derived alkaloid, has demonstrated excellent anticancer activity in several malignant cell lines but did not inhibit the growth of normal cells.[58] The blue-fluorescent zwitterionic agent was reported to rapidly accumulate in the nucleoli of U251 glioblastoma cells, but did not enter normal astrocyte control cells.[59] PB-100 appears to have a distinct binding preference for purine-rich nucleic acids.[59]
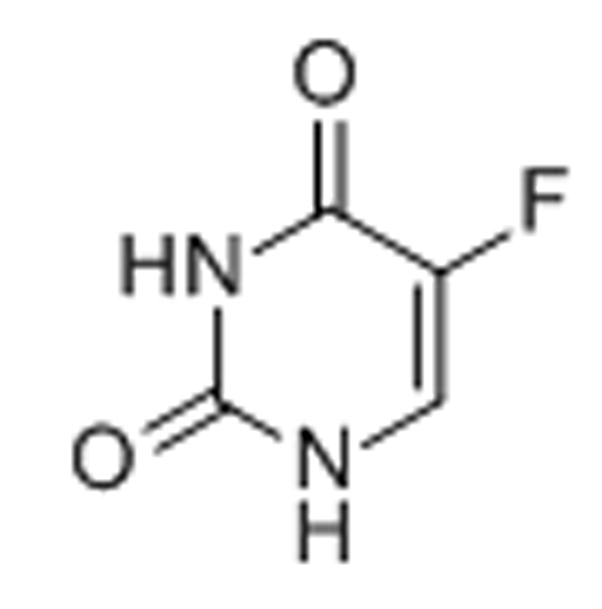
12. 5-Fluorouracil
The anticancer drug 5-fluorouracil, a pyrimidine analogue, is actively metabolized in the cell to fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP). The deoxy-metabolites result in DNA damage primarily via inhibition of thymidylate synthase by FdUMP.[60,61] In addition, major RNA damage occurs due to incorporation of FUTP into various forms of RNA, which leads to a disruption of its processing and vital functions.[60,61] In particular, the processing of late rRNA in the nucleolus is inhibited, leading to nucleolar stress, an alteration of nucleolar structure, and activation of p53-mediated apoptosis.[61]
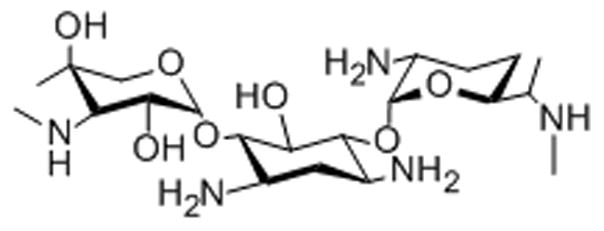
13. Gentamicin
Gentamicin, an aminoglycoside antibiotic used to treat tuberculosis and Gram-negative bacterial infections, was conjugated to the fluorophore Texas Red to form a drug-fluorophore conjugate GTTR.[62] Two kidney cell lines, OK and MDCK, were incubated with GTTR for two hours and observed by fluorescence microscopy to determine the cellular localization of the drug.[62] Along with endosomal accumulation, intra-nuclear accumulation occurred in the nucleoli and trans-nuclear tubules.[62] Localization to the nucleoli can potentially be explained by previous studies illustrating the propensity for gentamicin to bind ribosomes and rRNA.[63,64]
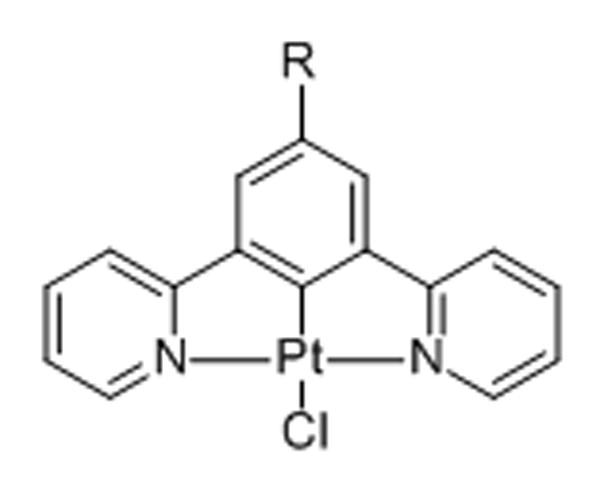
14. Monofunctional planar complexes [PtLCl][65]
Cyclometalated, planar monochloroplatinum(II)(“pincer”) complexes were studied using time-resolved emission imaging microscopy (TREM) in HDF, C8161, and CHO cells. The metallointercalators showed minimal cytotoxicity in colorimetric cell proliferation assays. When determining cellular distribution, the neutral complexes displayed accumulation in the nuclei (as confirmed by co-imaging with the nuclear stain DAPI) with the highest emission intensity localized to the nucleoli.
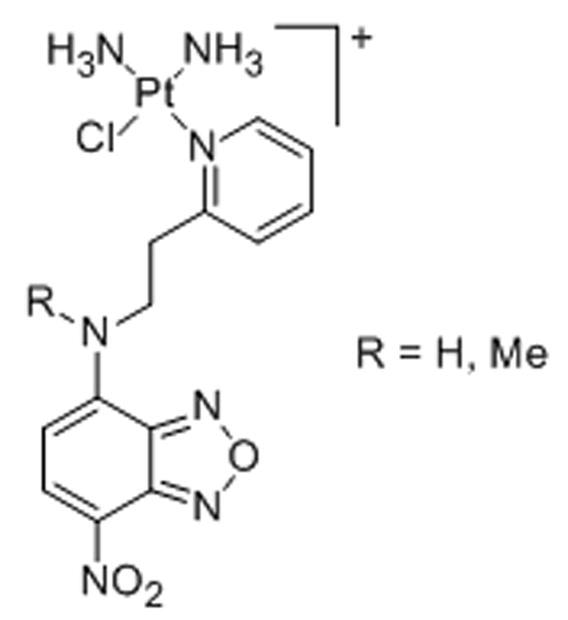
15. Monofunctional nonplanar complexes [PtClL]+
Nucleolar accumulation was also reported for platinum complexes derived from the antitumor-active monofunctional cationic complex cis-diamminechloridopyridine chloride (pyriplatin).[66] The complexes, which were labeled with green-fluorescent dye 7-nitrobenzafurazan, were detected in the nucleoli of HeLa cells and appear to induce cell death by a mechanism different from that of cisplatin.[67]
16. Mitomycin C
Mitomycin C is a potent antibiotic, originally isolated from the Gram-negative bacteria Streptomyces caespitosus. It is used as a chemotherapeutic agent, typically in bladder, colon, and breast cancers.[68] It is a bio-reductive molecule, which, after enzymatic activation in the cytosol, induces site-specific DNA cross-links.[68,69] While generally thought of as an alkylating agent damaging the heterochromatin, mitomycin C has recently been demonstrated to cause irreparable cross-links in rDNA leading to inhibition of rRNA synthesis.[68] In MCF-7 breast carcinoma cells, treatment with mitomycin C led to a 25% decrease in the 18S rRNA levels, most likely due to the binding and degradation of the rDNA transcript by the drug.[68] Early electron microscopy data suggests that mitomycin C causes a segregation and redistribution of nucleolar constituents upon treatment with the drug, further corroborating a nucleolus-mediated mechanism of action.[70]
17. Nano-sized particles
Nanoparticles (NPs) are currently being developed for a variety of different applications including those in the biomedical field. Recent literature suggests that some NPs can translocate to the nucleus, occasionally to the nucleolus.[71] Polyethyleneglycol-coated (“pegylated”) single-walled carbon nanotubes (SWCNTs) have been labeled with fluorescein dye and their cellular uptake and distribution monitored by time-lapse fluorescence microscopy.[72] The nanotubes showed cell line-specific accumulation in the nucleoli of various cancer cells but did not perturb their morphology.[72] Interestingly, nucleolar localization of these particles was reversible, and redistribution to the nucleoplasm and cytoplasm was observed when the nanoparticles were removed from the cell culture media.[72] Similarly, NP beads conjugated to the Tat peptide, a portion of the HIV-1 Tat protein, displayed significant accumulation in the nucleoli of permeablized HeLa cells.[73] Nanoconjugates consisting of TiO2 nanoparticles and oligonucleotides containing matching sequences to rDNA were found to localize in the nucleolus with similar uptake to the oligonucleotides alone.[74] Contrary to the previous examples, fluorescent SiO2 NPs were not shown to localize to the nucleolus, however they did effect a time-dependent redistribution of the nucleolar protein Topo I.[71]
18. Naphthalene diimides[75]
Naphthalene diimide derivatives were synthesized and tested for G-quadruplex selectivity, telomerase inhibition, senescence, and intracellular localization. Tetra-substituted ligands displayed remarkable G-quadruplex stabilization and selectivity over duplex DNA when compared to the tri- and di-substituted derivatives. Since the tetra- and tri-substituted derivatives were inherently fluorescent, localization experiments with confocal microscopy were completed in MCF-7 breast carcinoma cells. Of the derivatives that displayed localization exclusively in the nucleus, accumulation was observed in the nucleolus. The naphthalene diimides localizing to the nucleolus displayed IC50 values in the nanomolar range in A549 lung carcinoma cells.
19. Nucleolar targeting peptides[76]
Nucleolar targeting peptides are a family of cell-penetrating peptides (CPPs) that are capable of targeting and localizing to the nucleolus of cells. One such peptide sequence, NrTP1 (YKQCHKKGGKKGSG), was fluorescently labeled at the N-terminus and incubated with HeLa cells, pancreatic adenocarcinoma (BxPC-3), ductal mammary gland carcinoma (BT-474) and murine neuroblastoma (N2A) cells. In all cases except for N2A cells, NrTP1 was shown by confocal microscopy to accumulate in the nucleolus of cells, regardless of cell cycle phase. It was observed that the polypeptide was taken up by cells in a dose-dependent manner by clathryn-mediated endocytosis.
Another CPP, fluorescein-labeled β3-octaarginine,[77] was shown by confocal microscopy to localize to the nucleoli of erythrocytes infected with Plasmodium falciparum, however, was unable to enter intact, healthy erythrocytes. These properties could lead to the development of novel drug-conjugated CPPs that selectively deliver drugs to infected (or malignant) cells.
20. Nonsteroidal anti-inflammatory drugs
The efficacy of non-steroidal anti-inflammatory drugs (NSAIDS) as anti-tumor agents in colorectal cancers has stimulated several studies into their cellular mechanism in recent years. In the case of aspirin, sulindac, sulindac sulfone, and indomethacin, the NF-κB signaling pathway is activated.[78] NF-κB, a heterodimer of the p50 and RelA (p65) polypeptides, has been shown to regulate gene expression, affecting both cell growth and apoptotic pathways.[79] Aspirin, sulindac, sulindac sulfone, and indomethacin cause translocation of the RelA (p65) subunit into the nucleolus, inducing an apoptotic response in colorectal cancers.[78] These responses were demonstrated by the co-localization of RelA with fibrillin or nucleolin using confocal microscopy in conjunction with immunocytochemical techniques.[78]
21. Platinum–acridine hybrid agent
Platinum–acridines derived from the prototypical agent PT-ACRAMTU ([PtCl(en)(ACRAMTU)](NO3)2; en = ethane-1,2-diamine, ACRAMTU = 1-[2-(acridin-9-ylamino)ethyl]-1,3-dimethylthiourea) show promising anticancer activity in various solid tumors, in particular non-small-cell lung cancer (NSCLC).[80] The agents produce a unique form of DNA damage involving a covalent–intercalative binding mode and cellular effects unlike that of cisplatin.[80] To gain insight into the subcellular distribution of this pharmacophore, an analogue compatible with copper-catalyzed azide–alkyne cycloaddition chemistry was developed.[81] The azide-modified conjugate[82] was incubated with NCI-H460 NSCLC cells and post-labeled with the fluorophore Alexa Fluor 488-alkyne.[81] Cells in interphase exposed to the complex showed significant accumulation of hybrid agent in the nucleus and a 50% increased fluorescence in the nucleoli relative to the heterochromatin (Figure 2).[81]
Figure 2.

Intracellular distribution of “clickable” azide-modified platinum–acridine in an NCI-H460 cell in interphase co-stained with nuclear dye (Hoechst 33342). Single confocal image planes of (A) bright-field image (red scale bar = 5μm), (B) Hoechst (blue) channel, (C) Alexa Fluor 488 (green) channel, and (D) green and blue channels merged. The nucleolus (no), which is readily identified as a nuclear region unstained by Hoechst dye, appears as an area of high fluorescence intensity in the Alexa Fluor 488 and merged-channel images. Images were generated from data acquired in ref. 81.
22. Polynuclear platinum complexes[83]
Nano-scale secondary ion mass spectrometry (NanoSIMS) was employed to study the localization of a trinuclear platinum complex, TriplatinNC. A 15N-labeled analogue of TriplatinNC was employed to monitor the 195Pt and 15N isotope localization in whole cells. Resulting ion maps were indicative of nucleolar and cytoplasmic accumulation of the 15N-TriplatinNC and suggest that nucleolar targets may be involved in the mechanism of these noncovalent, purely electrostatic DNA binders.
23. Rapamycin and analogues
Rapamycin derivatives (“rapalogues”), including temsirolimus, everolimus, and deformolimus, are macrolides that are typically used as immunosuppressants or chemotherapeutic agents in the clinic.[84] They are potent inhibitors of the PI3K-AKT-mTOR signaling pathway that is mutated in a high percentage of cancers. Specifically, this class of drugs inhibits the mTORC1 complex which plays a role in rRNA transcription and ribosome biogenesis.[84,85] The rapalogues inactivate an important component of the RNA polymerase I machinery, TIF-1A, by a change in phosphorylation.[85] These drugs may also play a role in inhibiting rRNA processing, observed by a build-up of downstream rRNA intermediates.[85]
24. SYTO-RNA Select[86]
SYTO-RNA Select is a cell-permeable proprietary cyanine dye with maximal staining of nucleoli. It exhibits enhanced green fluorescence when bound to RNA versus DNA and proteins. The relative fluorescence intensity of the nucleolus in bovine pulmonary artery endothelial cells is approximately 2.5 times that of the nucleus and approximately 10 times that of the cytoplasm.
25. UNBS1450[87]
UNBS1450, a hemi-synthetic cardenolide, was discovered to modify the shape and protein localization of the nucleolus in human cancer cells. When comparing the nucleolar organization of cells treated with 100nM UNBS1450 using immunofluorescence, normal human lung and fibroblast cells displayed no change, whereas DU145 and PC-3 pancreatic carcinomas displayed a marked change in nucleolus organization. In agreement with this observation, the expression levels of the upstream binding factor (UBF) and fibrillan were shown to vastly decrease over a 24h period in PC-3 cells. UNBS1450 displayed a low-nanomolar IC50 value in PC-3 cells, which was around a 100-fold enhancement over oxaliplatin, irinotecan and etoposide. In vivo, PC-3 tumor bearing mice displayed an increased survival of 36% and 77% when injected with 10mg/kg i.p. and 20mg/kg i.p., respectively, with five injections per week for six weeks.
Outlook
The role of the nucleolus in supporting cell growth provides an attractive opportunity for chemotherapeutic intervention. A diverse array of molecules has been identified that target this subnuclear structure directly or affect its functions in some indirect ways. These include DNA intercalators, minor groove binders, metalating and alkylating electrophiles, as well as hybrid agents that combine multiple binding modes. However, the range of nucleolus-targeted molecules is by no means limited to DNA-interacting agents. Antimetabolites, kinase inhibitors, anti-inflammatory drugs, natural product antibiotics, oligopeptides, as well as nano-sized particles, also show nucleolus-specific properties.
Among the nucleic acid-directed agents, compounds that selectively inhibit transcription of rRNA genes (rDNA), are particularly promising and have demonstrated initial clinical success.[48,50,88] The tandem repeats of rDNA (45S gene) are G-rich (~75%; see NCBI database entry: NR 046235) and, thus, area “natural” high-affinity target for many electrophilic agents (platinums, mitomycin, etc.). A major challenge in designing nucleolus-directed DNA binders is to equip these molecules with properties that would allow more selective targeting of this structure while sparing sequences of similar base content within the surrounding heterochromatin. One particularly promising approach in this regard is to design molecules that recognize and bind to non-duplex nucleic acid structures (CX-3543, CX-5461, naphthalene diimides; see discussion above), such as G-quadruplex forming sequences, which are abundant in the 45S gene copies.[48] In addition to the de novo design of such pharmacophores, an opportunity might exist for repurposing existing clinical and experimental cytotoxics as nucleolus-directed therapies using vector-based delivery (peptides, nanoparticles; see discussion above). These include chemically promiscuous electrophiles and non-specific DNA binders that have either demonstrated unfavorable side effects or have been dismissed entirely as too toxic for clinical use. Finally, a strong case could be made for targeting the nucleolus with agents that produce permanent adducts in DNA. Unlike adducts in genomic DNA transcribed by RNA polymerase II (Pol II), damage in nucleolar rDNA is not efficiently removed by transcription-coupled nucleotide excision repair (NER).[89–91] NER, a major form of repair of clinically relevant drug-induced DNA adducts and cross-links, is a mediator and biomarker of tumor resistance.[92] In conclusion, the diverse nature of molecules acting on the cell’s nucleolus provides a powerful platform for designing urgently needed therapies that overcome several of the drawbacks associated with current chemotherapies.
Scheme 1.
Scheme 2.
Scheme 3.
Scheme 4.

Scheme 5.
Scheme 6.
Scheme 7.
Scheme 8.

Scheme 9.

Scheme 10.
Scheme 11.
Scheme 12.
Scheme 13.
Scheme 14.
Scheme 15.
Scheme 16.
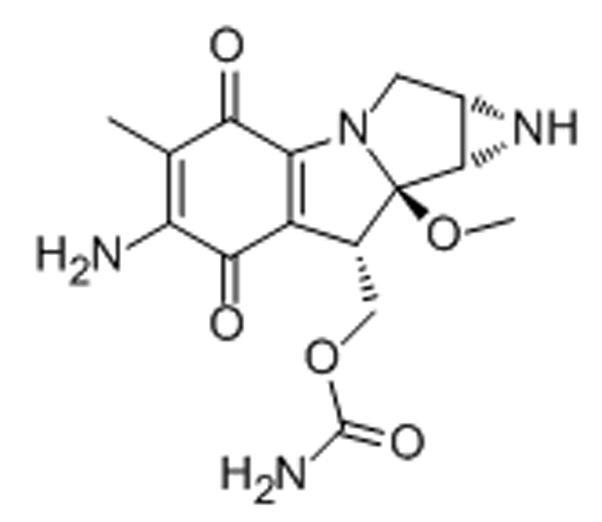
Scheme 17.
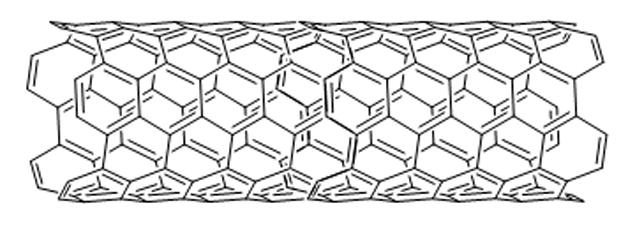
Scheme 18.
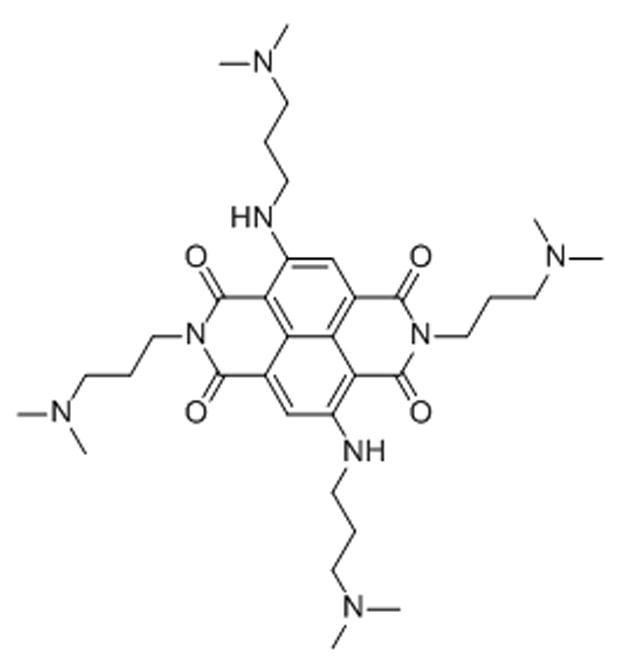
Scheme 20.

Scheme 21.
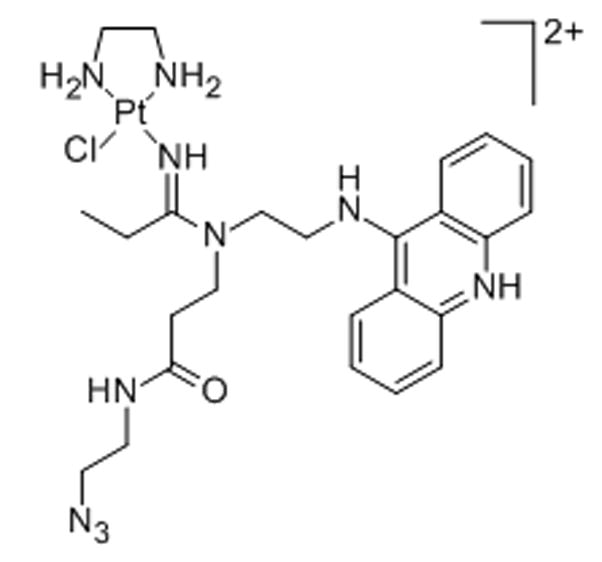
Scheme 22.
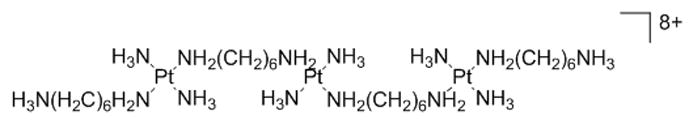
Scheme 23.
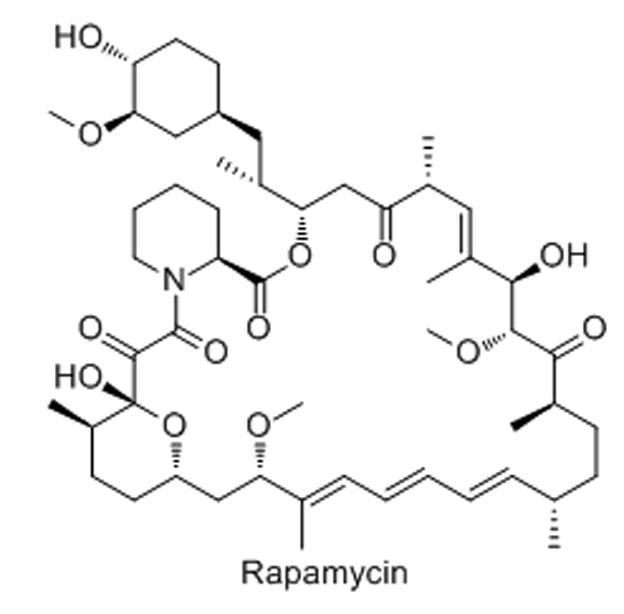
Scheme 25.

Acknowledgments
The confocal images in this contribution were acquired by Dr. Xin Qiao (Tianjin Medical University, PR China) during a research stay at Wake Forest University sponsored by the China Scholarship Council (CSC). This study was supported by the US National Institutes of Health (CA 101880).
References
- 1.Hambley TW, Hait WN. Cancer Res. 2009;69:1259–1262. doi: 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA., III Oncogene. 2009;28(Suppl 1):S4–13. doi: 10.1038/onc.2009.196. [DOI] [PubMed] [Google Scholar]
- 3.Dempke WC, Suto T, Reck M. Lung Cancer. 2010;67:257–274. doi: 10.1016/j.lungcan.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Nature. 2012;489:519–525. [Google Scholar]
- 5.The Cancer Genome Atlas Research Network. Nature. 2012;490:61–70. [Google Scholar]
- 6.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. Nat Rev Mol Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez-Verdun D. Nucleus. 2011;2:189–194. doi: 10.4161/nucl.2.3.16246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roussel P, Andre C, Comai L, Hernandez-Verdun D. J Cell Biol. 1996;133:235–246. doi: 10.1083/jcb.133.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheer U, Hock R. Curr Opin Cell Biol. 1999;11:385–390. doi: 10.1016/S0955-0674(99)80054-4. [DOI] [PubMed] [Google Scholar]
- 10.Scheer U, Thiry M, Goessens G. Trends Cell Biol. 1993;3:236–241. doi: 10.1016/0962-8924(93)90123-i. [DOI] [PubMed] [Google Scholar]
- 11.Raska I, Shaw PJ, Cmarko D. Curr Opin Cell Biol. 2006;18:325–334. doi: 10.1016/j.ceb.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Trere D. Micron. 2000;31:127–131. doi: 10.1016/s0968-4328(99)00069-4. [DOI] [PubMed] [Google Scholar]
- 13.Derenzini M. Micron. 2000;31:117–120. doi: 10.1016/s0968-4328(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 14.Ceccarelli C, Trere D, Santini D, Taffurelli M, Chieco P, Derenzini M. Micron. 2000;31:143–149. doi: 10.1016/s0968-4328(99)00071-2. [DOI] [PubMed] [Google Scholar]
- 15.Andersen JS, Lyon CE, Fox AH, Leung AKL, Lam YW, Steen H, Mann M, Lamond AI. Curr Biol. 2002;12:1–11. doi: 10.1016/s0960-9822(01)00650-9. [DOI] [PubMed] [Google Scholar]
- 16.Christensen MO, Krokowski RM, Barthelmes HU, Hock R, Boege F, Mielke C. J Biol Chem. 2004;279:21873–21882. doi: 10.1074/jbc.M400498200. [DOI] [PubMed] [Google Scholar]
- 17.Howard J, Hyman AA. Nature. 2003;422:753–758. doi: 10.1038/nature01600. [DOI] [PubMed] [Google Scholar]
- 18.Xu K, Luduena RF. Cell Motil Cytoskeleton. 2002;53:39–52. doi: 10.1002/cm.10060. [DOI] [PubMed] [Google Scholar]
- 19.Exposito O, Bonfill M, Moyano E, Onrubia M, Mirjalili MH, Cusido RM, Palazon J. Anticancer Agents Med Chem. 2009;9:109–121. doi: 10.2174/187152009787047761. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell JR, Wood E, Collins K. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 21.Politz JC, Yarovoi S, Kilroy SM, Gowda K, Zwieb C, Pederson T. Proc Natl Acad Sci U S A. 2000;97:55–60. doi: 10.1073/pnas.97.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossalam M, Dixon AS, Lim CS. Ther Deliv. 2010;1:169–193. doi: 10.4155/tde.10.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coute Y, Burgess JA, Diaz JJ, Chichester C, Lisacek F, Greco A, Sanchez JC. Mass Spectrom Rev. 2006;25:215–234. doi: 10.1002/mas.20067. [DOI] [PubMed] [Google Scholar]
- 24.Phillips DM, Phillips SG. J Cell Biol. 1971;49:803–815. doi: 10.1083/jcb.49.3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montanaro L, Trere D, Derenzini M. Am J Pathol. 2008;173:301–310. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sigal A, Rotter V. Cancer Res. 2000;60:6788–6793. [PubMed] [Google Scholar]
- 27.Rubbi CP, Milner J. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma H, Pederson T. Mol Biol Cell. 2013;24:1334–1342. doi: 10.1091/mbc.E12-12-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma H, Pederson T. Mol Biol Cell. 2007;18:2630–2635. doi: 10.1091/mbc.E07-03-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trere D, Ceccarelli C, Montanaro L, Tosti E, Derenzini M. J Histochem Cytochem. 2004;52:1601–1607. doi: 10.1369/jhc.4A6454.2004. [DOI] [PubMed] [Google Scholar]
- 31.Ruggero D. Sci Signal. 2012;5:pe38. doi: 10.1126/scisignal.2003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry RP, Kelley DE. J Cell Physiol. 1970;76:127–139. doi: 10.1002/jcp.1040760202. [DOI] [PubMed] [Google Scholar]
- 33.Perry RP. Exp Cell Res. 1963;29:400–406. [Google Scholar]
- 34.Dousset T, Wang C, Verheggen C, Chen DY, Hernandez-Verdun D, Huang S. Mol Biol Cell. 2000;11:2705–2717. doi: 10.1091/mbc.11.8.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wulff JE, Siegrist R, Myers AG. J Am Chem Soc. 2007;129:14444–14451. doi: 10.1021/ja075327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindstrom MS. Biochem Res Int. 2011;2011:195209. doi: 10.1155/2011/195209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colombo E, Alcalay M, Pelicci PG. Oncogene. 2011;30:2595–2609. doi: 10.1038/onc.2010.646. [DOI] [PubMed] [Google Scholar]
- 38.Grummitt CG, Townsley FM, Johnson CM, Warren AJ, Bycroft M. J Biol Chem. 2008;283:23326–23332. doi: 10.1074/jbc.M801706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas CJ, Rahier NJ, Hecht SM. Bioorg Med Chem. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 40.Pondarre C, Strumberg D, Fujimori A, Torres-Leon R, Pommier Y. Nucleic Acids Res. 1997;25:4111–4116. doi: 10.1093/nar/25.20.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mo YY, Yu Y, Shen Z, Beck WT. J Biol Chem. 2002;277:2958–2964. doi: 10.1074/jbc.M108263200. [DOI] [PubMed] [Google Scholar]
- 42.Christensen MO, Barthelmes HU, Feineis S, Knudsen BR, Andersen AH, Boege F, Mielke C. J Biol Chem. 2002;277:15661–15665. doi: 10.1074/jbc.C200066200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang H, Wang JC, Liu LF. Proc Natl Acad Sci U S A. 1988;85:1060–1064. doi: 10.1073/pnas.85.4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bensaude O. Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burger K, Mühl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, Kellner M, Gruber-Eber A, Kremmer E, Hölzel M, Eick D. J Biol Chem. 2010;285:12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jordan P, Carmo-Fonseca M. Nucleic Acids Res. 1998;26:2831–2836. doi: 10.1093/nar/26.12.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horky M, Wurzer G, Kotala V, Anton M, Vojtesek B, Vacha J, Wesierska-Gadek J. J Cell Sci. 2001;114:663–670. doi: 10.1242/jcs.114.4.663. [DOI] [PubMed] [Google Scholar]
- 48.Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, Lim JK, Von Hoff D, Anderes K, Rice WG. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 49.Balasubramanian S, Hurley LH, Neidle S. Nat Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, Huser N, Proffitt C, Bliesath J, Haddach M, Schwaebe MK, Ryckman DM, Rice WG, Schmitt C, Lowe SW, Johnstone RW, Pearson RB, McArthur GA, Hannan RD. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, Streiner N, Quin JE, Sanij E, Bywater MJ, Hannan RD, Ryckman D, Anderes K, Rice WG. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 52.Bailly C, Michaux C, Colson P, Houssier C, Sun JS, Garestier T, Helene C, Henichart JP, Rivalle C, Bisagni E, Waring MJ. Biochemistry. 1994;33:15348–15364. doi: 10.1021/bi00255a016. [DOI] [PubMed] [Google Scholar]
- 53.Bailly C, Leclere V, Pommery N, Colson P, Houssier C, Rivalle C, Bisagni E, Henichart JP. Anticancer Drug Des. 1993;8:145–164. [PubMed] [Google Scholar]
- 54.Tettey JNA, Skellern GG, Midgley JM, Grant MH, Wilkinson R, Pitt AR. Xenobiotica. 1999;29:349–360. doi: 10.1080/004982599238551. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H, Oczos J, Janowski P, Trembecka D, Dobrucki J, Darzynkiewicz Z, Wlodkowic D. Cytometry A. 2010;77:399–405. doi: 10.1002/cyto.a.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montgomery CP, Murray BS, New EJ, Pal R, Parker D. Acc Chem Res. 2009;42:925–937. doi: 10.1021/ar800174z. [DOI] [PubMed] [Google Scholar]
- 57.Yu JH, Parker D, Pal R, Poole RA, Cann MJ. J Am Chem Soc. 2006;128:2294–2299. doi: 10.1021/ja056303g. [DOI] [PubMed] [Google Scholar]
- 58.Beljanski M. Gen Mol Biol. 2000;23:29–33. [Google Scholar]
- 59.Beljanski M, Crochet S. Int J Oncol. 1995;7:81–85. doi: 10.3892/ijo.7.1.81. [DOI] [PubMed] [Google Scholar]
- 60.Longley DB, Harkin DP, Johnston PG. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 61.Sun XX, Dai MS, Lu H. J Biol Chem. 2007;282:8052–8059. doi: 10.1074/jbc.M610621200. [DOI] [PubMed] [Google Scholar]
- 62.Myrdal SE, Johnson KC, Steyger PS. Hear Res. 2005;204:156–169. doi: 10.1016/j.heares.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lynch SR, Puglisi JD. J Mol Biol. 2001;306:1023–1035. doi: 10.1006/jmbi.2000.4419. [DOI] [PubMed] [Google Scholar]
- 64.Yoshizawa S, Fourmy D, Puglisi JD. EMBO J. 1998;17:6437–6448. doi: 10.1093/emboj/17.22.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Botchway SW, Charnley M, Haycock JW, Parker AW, Rochester DL, Weinstein JA, Williams JA. Proc Natl Acad Sci U S A. 2008;105:16071–16076. doi: 10.1073/pnas.0804071105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lovejoy KS, Serova M, Bieche I, Emami S, D’Incalci M, Broggini M, Erba E, Gespach C, Cvitkovic E, Faivre S, Raymond E, Lippard SJ. Mol Cancer Ther. 2011;10:1709–1719. doi: 10.1158/1535-7163.MCT-11-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu S, Wang X, Zhu C, Song Y, Wang J, Li Y, Guo Z. Dalton Trans. 2011;40:10376–10382. doi: 10.1039/c1dt10555h. [DOI] [PubMed] [Google Scholar]
- 68.Snodgrass RG, Collier AC, Coon AE, Pritsos CA. J Biol Chem. 2010;285:19068–19075. doi: 10.1074/jbc.M109.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rey JP, Scott R, Muller H. Mutat Res. 1993;289:171–180. doi: 10.1016/0027-5107(93)90067-p. [DOI] [PubMed] [Google Scholar]
- 70.Lapis K, Bernhard W. Cancer Res. 1965;25:628–645. [PubMed] [Google Scholar]
- 71.Chen M, von Mikecz A. Exp Cell Res. 2005;305:51–62. doi: 10.1016/j.yexcr.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 72.Cheng J, Fernando KA, Veca LM, Sun YP, Lamond AI, Lam YW, Cheng SH. ACS Nano. 2008;2:2085–2094. doi: 10.1021/nn800461u. [DOI] [PubMed] [Google Scholar]
- 73.Nitin N, LaConte L, Rhee WJ, Bao G. Ann Biomed Eng. 2009;37:2018–2027. doi: 10.1007/s10439-009-9768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paunesku T, Vogt S, Lai B, Maser J, Stojicevic N, Thurn KT, Osipo C, Liu H, Legnini D, Wang Z, Lee C, Woloschak GE. Nano Lett. 2007;7:596–601. doi: 10.1021/nl0624723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cuenca F, Greciano O, Gunaratnam M, Haider S, Munnur D, Nanjunda R, Wilson WD, Neidle S. Bioorg Med Chem Lett. 2008;18:1668–1673. doi: 10.1016/j.bmcl.2008.01.050. [DOI] [PubMed] [Google Scholar]
- 76.Radis-Baptista G, de la Torre BG, Andreu D. Chem Biol Drug Des. 2012;79:907–915. doi: 10.1111/j.1747-0285.2012.01377.x. [DOI] [PubMed] [Google Scholar]
- 77.Kamena F, Monnanda B, Makou D, Capone S, Patora-Komisarska K, Seebach D. Chem Biodivers. 2011;8:1–12. doi: 10.1002/cbdv.201000318. [DOI] [PubMed] [Google Scholar]
- 78.Loveridge CJ, MacDonald ADH, Thoms HC, Dunlop MG, Stark LA. Oncogene. 2008;27:2648–2655. doi: 10.1038/sj.onc.1210891. [DOI] [PubMed] [Google Scholar]
- 79.Stark LA, Dunlop MG. Mol Cell Biol. 2005;25:5985–6004. doi: 10.1128/MCB.25.14.5985-6004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suryadi J, Bierbach U. Chem Eur J. 2012;18:12926–12934. doi: 10.1002/chem.201202050. [DOI] [PubMed] [Google Scholar]
- 81.Ding S, Qiao X, Suryadi J, Marrs GS, Kucera GL, Bierbach U. Angew Chem. 2013;125:3434–3438. doi: 10.1002/anie.201210079. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:3350–3354. doi: 10.1002/anie.201210079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ding S, Qiao X, Kucera GL, Bierbach U. J Med Chem. 2012;55:10198–10203. doi: 10.1021/jm301278c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wedlock LE, Kilburn MR, Liu R, Shaw JA, Berners-Price SJ, Farrell NP. Chem Commun (Camb) 2013 doi: 10.1039/C3CC42098A. Advance Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Le Tourneau C, Faivre S, Serova M, Raymond E. Br J Cancer. 2008;99:1197–1203. doi: 10.1038/sj.bjc.6604636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iadevaia V, Huo Y, Zhang Z, Foster LJ, Proud CG. Biochem Soc Trans. 2012;40:168–172. doi: 10.1042/BST20110682. [DOI] [PubMed] [Google Scholar]
- 86.Johnson I, Spence MTZ, editors. The Molecular Probes Handbook - A Guide to Fluorescent Probes and Labeling Technologies. 11. Life Technologies Corporation; Carlsbad, CA: 2010. [Google Scholar]
- 87.Mijatovic T, De Neve N, Gailly P, Mathieu V, Haibe-Kains B, Bontempi G, Lapeira J, Decaestecker C, Facchini V, Kiss R. Mol Cancer Ther. 2008;7:1285–1296. doi: 10.1158/1535-7163.MCT-07-2241. [DOI] [PubMed] [Google Scholar]
- 88.Drygin D, Rice WG, Grummt I. Annu Rev Pharmacol Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 89.Conconi A. DNA Repair (Amst) 2005;4:897–908. doi: 10.1016/j.dnarep.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 90.Christians FC, Hanawalt PC. Biochemistry. 1993;32:10512–10518. doi: 10.1021/bi00090a030. [DOI] [PubMed] [Google Scholar]
- 91.Balajee AS, May A, Bohr VA. Nucleic Acids Res. 1999;27:2511–2520. doi: 10.1093/nar/27.12.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stewart DJ. Crit Rev Oncol Hematol. 2010;75:173–234. doi: 10.1016/j.critrevonc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]