

Abstract
Some forms of idiopathic epilepsy in animals and humans are associated with deficiency of synapsin, a phosphoprotein that reversibly associates with synaptic vesicles. We have previously shown that the epileptic phenotype seen in synapsin II knock-out mice (SynII(−)) can be rescued by the genetic deletion of the Rab3a protein. Here we have examined the cellular basis for this rescue using whole-cell recordings from CA1 hippocampal pyramidal cells in brain slices. We find that SynII(−) neurons have increased spontaneous activity and a reduced threshold for the induction of epileptiform activity by 4-aminopyridine (4-AP). Using selective recordings of glutamatergic and GABAergic activity we show that in wild-type neurons low concentrations of 4-AP facilitate glutamatergic and GABAergic transmission in a balanced way, whereas in SynII(−) neurons this balance is shifted toward excitation. This imbalance reflects a deficit in inhibitory synaptic transmission that appears to be secondary to reduced Ca2+ sensitivity in SynII(−) neurons. This suggestion is supported by our finding that synaptic and epileptiform activity at SynII(−) and wild-type synapses is similar when GABAergic transmission is blocked. Deletion of Rab3a results in glutamatergic synapses that have a compromised responsiveness to either low 4-AP concentrations or elevated extracellular Ca2+. These changes mitigate the overexcitable phenotype observed in SynII(−) neurons. Thus, Rab3a deletion appears to restore the excitatory/inhibitory imbalance observed in SynII(−) hippocampal slices indirectly, not by correcting the deficit in GABAergic synaptic transmission but rather by impairing excitatory glutamatergic synaptic transmission.
Introduction
Synapsin I (SynI) and synapsin II (SynII) are widely expressed in the CNS where they associate with synaptic vesicles, both glutamate-containing vesicles at excitatory synapses and GABA-containing vesicles at inhibitory synapses. Synapsins tether synaptic vesicles to the cytomatrix (for review, see Shupliakov et al., 2011), and synaptic activity triggers the phosphorylation of the synapsins, allowing them to dissociate from synaptic vesicles (Chi et al., 2001, 2003). Recent studies suggest that the synapsins, in addition, may also directly regulate the releasable pool of vesicles (Humeau et al, 2001; Cousin et al., 2003), control vesicle trafficking (Messa et al., 2010), and participate in the docking of synaptic vesicles at the active zone (Coleman et al., 2008).
Parallel basic and clinical work has shown that SYN1 or SYN2, the genes coding for the proteins Syn I and Syn II, are important epilepsy susceptibility genes. At the basic science level, experiments in genetically modified mice have shown that deletion of either the Syn1 or Syn2 genes (or both) results in a strong epileptic phenotype (Li et al., 1995; Rosahl et al., 1995; Etholm and Heggelund, 2009). At the clinical level, genetic epidemiology studies have identified a human nonsense mutation in SYN1 associated with syndromic epilepsy (Garcia et al., 2004). More recent studies have discovered several other mutations in the SYN1 gene associated with epilepsy (Fassio et al., 2011a) and also a contribution of SYN2 to sporadic epilepsy predisposition (Cavalleri et al., 2007; Lakhan et al., 2010). However, exactly how synapsin deficiency contributes to the development of epilepsy is only partly understood.
Previous studies have shown that deletion of SynI and/or SynII differentially affects excitatory and inhibitory neurotransmision in the brain and more specifically that deletion of the synapsins produces a reduction in GABAergic synaptic transmission (Gitler et al., 2004; Baldelli et al., 2007; Chiappalone et al., 2009; Farisello et al., 2012) and the loss of its asynchronous component (Medrihan et al., 2013) with unchanged or increased basal synaptic transmission at glutamatergic synapses. These observations suggested that deletion of synapsins results in an excitatory/inhibitory imbalance that could explain the epileptic phenotype (Fassio et al., 2011b); however, this conjecture has been difficult to test.
We recently reported that deletion of the presynaptic protein Rab3a robustly suppressed the epileptic phenotype observed in the SynII knock-out (SynII(−)) mice (Coleman and Bykhovskaia, 2010). This observation opens an avenue for linking the changes in cellular excitability observed in vitro with the whole animal epileptic phenotype. It also suggests a novel potential target for the treatment for epilepsy. Here, we show that Rab3a gene deletion rescues the overexcitable phenotype produced by the deficit in GABAergic transmission in SynII(−) synapses and that this Rab3a action occurs via an opposite change in excitatory glutamatergic transmission.
Materials and Methods
Animal.
Mice heterozygotes for the Syn2 and Rab3a targeted mutations were purchased from The Jackson Laboratory (strains B6;129S-Syn2tm1Sud/J and B6;129S-Rab3atm1Sud/J). Homozygous lines SynII(−), Rab3a(−), and wild-type (WT) mice were derived from breeding heterozygotes. The homozygote SynII(−)/Rab3a(−) double knock-out (DKO) mice were obtained by serial breeding and genotyping, as described previously (Coleman and Bykhovskaia, 2010). Genotypes of all the breeders were independently confirmed by a commercial genotyping service (Transnetix). All the animal colonies were kept under standard conditions and on a 12 h dark/light cycle. The husbandry conditions were identical for all the strains under the study. The experiments were performed on 15- to 20-d-old animals (young) or 10-week-old animals (adult) of either sex in accordance with the guidelines of the Animal Care and Use Committee of the Universidad Central del Caribe and the National Institutes of Health of the United States Public Health Service. For the adult group, only SynII(−) animals showing strong tonic-clonic seizures when being provoked by mechanical stimulation (lifting by the tail) were selected; thus, SynII(−) adult animals were verified as being symptomatic.
Slice preparation.
Mice were anesthetized using isoflurane and decapitated as soon as reflexive reactions disappeared. Brains were removed rapidly and bathed in an ice-cold solution containing the following (in mm): 65.0 NaCl, 2.0 KCl, 5.0 MgSO4, 1.1 KH2PO4, 24.0 NaHCO3, 10 dextrose, and 100 sucrose, bubbled with a mixture of 95% O2 and 5% CO2. For adult animals, a N-methyl-d-glucamine (NMDG) based ACSF solution (Zhao et al., 2011) was used (composition in mm): 110 NMDG, 110.0 HCl, 2.5 KCl, 10.0 MgSO4, 1.2 NaH2PO4, 0.5 CaCl2, 25.0 NaHCO3, and 25 dextrose. Horizontal hippocampal slices (300 mm thickness) were cut using a Leica vibratome (model VT 1200 S). For adult animals, the slices were incubated in the NMDG based solution at 33°C for 12 min. Then the slices were washed two times in the ACSF solution (previously bubbled with the same gas mixture) containing the following (in mm): 120 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 KH2PO4, 2.5 CaCl2, 24.0 NaHCO3, 10 dextrose, 50 sucrose, with the osmolarity adjusted to 295 mOsm with sucrose, and maintained in the recording solution continuously bubbled with the same gas mixture for at least 1 h before the experiment.
Electrophysiology.
Slices were held submerged in the recording chamber continuously superfused with the ACSF solution. Whole-cell recordings were performed from hippocampal CA1 neurons. Recordings were acquired using the multiclamp 700B amplifier and PClamp 10.0 software (Molecular Devices) and digitized at 10 kHz. Recordings of spontaneous and epileptiform activity were performed using a potassium gluconate-based intracellular solution containing the following (in mm): 135 K-gluconate, 0.1 CaCl2, 2.0 MgCl2, 2.0 Na-ATP, 1.0 EGTA, 10 HEPES, pH 7.35 (adjusted with KOH), 280 mOsm (Luhmann et al., 2000). Under these recording conditions, ECl was ∼−90 mV and cells were held at −50 mV to allow a clear distinction between spontaneous EPSCs (sEPSCs) and spontaneous IPSCs (sIPSCs). To record sIPSCs, the holding potential was set to 0 mV. Recordings of evoked EPSCs were performed using pipettes filled with a cesium-based solution containing the following (in mm): 117.5 CsMeSO3, 1.0 MgCl2, 10.0 HEPES, 5 EGTA, 15.5 CsCl, 8 NaCl, 0.3 NaGTP, 5.0 QX-314-bromide, and 4.0 NaATP. Recordings of evoked IPSCs were performed using pipettes filled with a cesium-based solution containing the following (in mm): 153.3 CsCl, 1.0 MgCl2, 10.0 HEPES, 5 EGTA, 0.1 NaGTP, 5.0 QX-314-bromide, and 3.0 MgATP. Holding potentials were maintained at −65 mV in both cases. The open pipette resistance with these solutions was 3–4 MΩ, and recordings with series resistance >20MΩ were discarded.
A patch pipette filled with ACSF solution was used to evoke synaptic potentials. To evoke EPSCs, the stimulation electrode was placed in the stratum radiatum; and to evoke IPSCs, the stimulation electrode was placed in the region at the border of the stratum oriens and stratum pyramidale. Stimuli of 100 μs duration were delivered at a constant voltage at 0.2 Hz. The stimulus intensity was adjusted to achieve “nearly minimal stimulation” conditions, that is ∼50% failure rate for EPSC recordings and ∼20–40% failure rate for IPSC recordings (higher failure rates cannot be reliably obtained for inhibitory synapses).
Data analysis.
The recordings were analyzed off-line using the Clampfit software (Molecular Devices), as well as the in-house software QUANTAN (Bykhovskaia, 2008). Statistical analyses were performed using Student's unpaired two-sided t test, Student's paired one-sided t test, Kolmogorov–Smirnov test, or two-way ANOVA followed by Tukey's post hoc test, using SPSS software. Curve fitting (Hill function) was performed using Prism software (GraphPad).
Drugs.
4-Aminopyridine (4-AP, 10, 15, or 100 μm; Sigma) was applied to evoke epileptiform activity. Slices were perfused for at least 4 min in the 4-AP containing recording solution before recordings (under our perfusion conditions, the 4-AP effect reaches a plateau after 3 min). NMDA receptors were blocked in recordings of evoked activity using d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5 20 μm). For recordings of EPSCs and miniature EPSCs (mEPSCs), GABA currents were blocked by picrotoxin (Ptx, 50 μm). For recordings of IPSCs and miniature IPSCs (mIPSCs), AMPA currents were blocked using CNQX (20 μm). For recordings of mEPSCs and mIPSCs, TTX (1 μm) was applied for 10 min to block action potentials.
Results
SynII deletion enhances spontaneous synaptic and epileptiform network activity in vitro
Deletions of SynI, SynII or SynI and SynII (double knock-out) result in the development of an epileptic phenotype (Rosahl et al., 1995). In all three cases, the onset of epileptiform activity is developmentally delayed and does not become apparent until the second or third postnatal month (Ketzef et al., 2011). Previous studies have shown that deletion of SynII alters short-term synaptic plasticity in the CA1 region during the presymptomatic period (Rosahl et al., 1995; Bogen et al., 2009; Hvalby et al., 2010). However, little is known about possible changes in synaptic and network activity during this period that may contribute to the development of seizures in the adult. To address this issue and gain a general indication of network excitability, we recorded spontaneous activity from pyramidal cells of the CA1 region under conditions that allowed for the separation of sEPSCs from sIPSCs in the absence of receptor blockers (Fig. 1A,E). As illustrated in Figure 1, deletion of SynII resulted in a significant increase in sEPSC activity and a significant decrease of sIPSC activity. Overall, we detected a 2.3-fold increase in sEPSC frequency (p = 0.001, n = 7) without a significant increase in sEPSC amplitude (p = 0.35, Fig. 1B–D). The sIPSC frequency was decreased to 70% of control (p = 0.002, n = 11), whereas sIPSC amplitude remained unchanged (Fig. 1F–H, p = 0.42). In this same recording in the presence of TTX, we observed a modest but statistically significant increase in mEPSC frequency (57% increase, p = 0.04, n = 6) and no change in mEPSC amplitude (p = 0.50, n = 6). The frequency and amplitude of mIPSCs were not affected by SynII deletion (n = 6, p = 0.30 and p = 0.25, respectively). These results reveal a robust increase in hippocampal network excitability in slices derived from presymptomatic SynII(−) mice.
Figure 1.

SynII deletion increases spontaneous synaptic activity in the CA1 region. A, Sample recordings of glutamate-mediated inward (sEPSCs) and GABA-mediated outward synaptic currents at −50 mV. CNQX selectively blocks the inward synaptic currents, confirming that they are mediated by AMPA-kainate receptors. B, Histograms illustrating the distribution of sEPSC amplitudes for WT and SynII(−) synapses. There is an increase in the number of sEPSCs recorded in SynII(−) but the lack of a significant difference in sEPSC amplitude (p = 0.18 per Kolmogorov–Smirnov test). C, Cumulative plot illustrating the increase in sEPSC frequency detected at SynII(−) synapses (p = 0.0001 per Kolmogorov–Smirnov test). B, C, Histograms represent the results for all the experiments pooled together for each genotype and normalized by the number of recordings. Synaptic events were collected for 5 min each from 6 WT cells in 3 mice and 7 Syn II(−) cells in 4 mice. D, Scatter plot illustrating the sEPSC frequencies recorded in the experiments illustrated in A–D but sorted by cell and genotype. The sEPSC frequency is significantly increased at SynII(−) synapses (p = 0.001 per unpaired two sided t test). E, Sample recordings of spontaneous GABAergic sIPSCs at 0 mV. Bottom, Application of Ptx completely blocks these synaptic events. F, Histogram illustrating the distribution of sIPSC amplitudes recorded at WT and SynII(−) synapses. There is a decrease in the number of sIPSCs detected at SynII(−) synapses but no significant shift in their amplitudes (p = 0.32 per Kolmogorov–Smirnov test). G, Cumulative histogram depicting the increased interevent interval detected at SynII(−) synapses (p = 0.0008 per Kolmogorov–Smirnov test). F, G, sIPSCs were recorded for 5 min from 12 WT cells in 5 mice and 11 SynII(−) cells in 4 mice. H, Scatter plot illustrating the results of the experiments illustrated in E–G sorted by cell and genotype. The sIPSC frequency is significantly reduced at SynII(−) synapses (p = 0.002 per unpaired two sided t test).
Such an increase in network excitability during the presymptomatic period could constitute the precondition for the subsequent development of epileptic seizures. To explore this possibility, we built upon the work of Boido et al. (2010) who had previously shown the usefulness of the proconvulsant potassium channel blocker 4-AP to unmask the epileptiform phenotype of the SynI/II/III triple-knock-out (TKO) mice in vitro. Specifically, their study showed that the administration of 200 μm 4-AP to brain slices could induce interictal (I-IC) discharges in slices derived from both WT and TKO mice, but that the increase in fast I-IC discharges was much larger in TKO than in WT mice. One limitation of this work is that 200 μm 4-AP induced epileptiform activity in WT mice as well as in mice carrying the SynI/Syn II/SynIII triple deletion. We reasoned that perhaps the large effect of high micromolar 4-AP on release could be occluding an effect of genotype. To test this idea, we assessed the effect of low concentrations of 4-AP on hippocampal slices derived from both young (presymptomatic) and adult SynII(−) and WT mice. As illustrated in Figure 2, administration of 10 μm 4-AP to slices derived from young WT mice resulted in a significant increase in sEPSCs (142 ± 37%, p = 0.006, n = 6; Fig. 2A1) but failed to elicit the appearance of interictal I-IC or ictal events (Fig. 2B2, n = 9). In contrast, administration of 10 μm 4-AP to slices derived from presymptomatic SynII(−) mice (Fig. 2A2) resulted in significantly larger increase in excitatory synaptic transmission (195 ± 16%, p = 0.001, n = 5) and the appearance of epileptiform I-IC activity (Fig. 2B2). Consistent with previous findings, application of high micromolar concentrations of 4-AP induced robust I-IC activity in slices derived from both WT and SynII(−) young mice (Fig. 2B1,B3).
Figure 2.

Low concentrations of 4-AP unmask the hyperexcitable SynII(−) phenotype in slices derived from young asymptomatic mice. A, Effect of 10 μm 4-AP on sEPSC frequency recorded in slices derived from WT and SynII(−) mice. A1, A2, Administration of 10 μm 4-AP induces a much larger increase in sEPSC frequency in slices derived from young asymptomatic SynII(−) mice (195 ± 16%, p = 0.001, n = 5) than in slices derived from young WT mice (142 ± 38% increase, p = 0.006, n = 6). A3, A4, In contrast, administration of 10 μm 4-AP to slices derived from adult mice of both genotypes results in much more modest increases in sEPSC frequency (WT: 31.0 ± 6.4%, p = 0.050, n = 8; SynII(−): 55.1 ± 7.3%, p = 0.01, n = 8). Data collected from three young and four adult animals per strain (on average, two recordings per animal). B, Deletion of SynII reduces the threshold for epileptiform activity in slices derived from young presymptomatic animals. B1, Sample recordings of I-IC discharges induced by 4-AP (100 μm). B2, Administration of 10 μm 4-AP induces the appearance of I-IC discharges in slices derived from presymptomatic SynII(−) mice but not in slices derived from young WT animals. B3, Administration of 10 μm 4-AP fails to elicit I-IC discharges in slices derived from either WT or symptomatic SynII(−) adult animals. *p = 0.0003 (ANOVA followed by post hoc Tukey test). n = 9. B3, Increasing 4-AP concentration to 100 μm occludes the difference in I-IC activity between the genotypes of presymptomatic animals. However, at this higher concentration of 4-AP, slices derived from adult symptomatic SynII(−) animals exhibit significantly increased I-IC activity. *p = 0.013 (ANOVA followed by post hoc Tukey test). n = 8. Data collected from four mice per age group per strain.
In slices derived from adult WT animals, administration of 10 μm 4-AP elicited only a modest but statistically significant increase in sEPSC frequency (31 ± 6%, p = 0.05, n = 8, Fig. 2A3). Slices derived from adult mice were also more resistant to the epileptogenic effects of 4-AP such that administration of 100 μm 4-AP induced I-IC activity only approximately half as intense as that seen in slices derived from young mice (Fig. 2B3). These results are in agreement with earlier studies, which suggest that immature brain has increased excitability (Holmes et al., 1998; Ben-Ari, 2006). In adult symptomatic Syn II(−) animals, administration of 10 μm 4-AP again produced a stronger increase in sEPSC activity (55 ± 7% increase, p = 0.007, n = 8, Fig. 2A4), but this increase was less robust than that seen in young animals (Fig. 2A4 vs Fig. 2A2). Furthermore, administration of 10 μm 4-AP failed to elicit any epileptiform activity in Syn II(−) slices derived from adult animals (Fig. 2B2), whereas administration of 100 μm 4-AP elicited robust epileptiform activity surpassing that seen in control slices (0.042 ± 0.006 Hz in WT vs 0.107 ± 0.015 Hz in SynII(−), p = 0.006, Fig. 2B3). These results support the idea that the changes in excitability induced by SynII deletion are present even in asymptomatic animals and that 4-AP can be used to unmask the latent epileptiform phenotype induced by the deletion of SynII. They also support the view that the epileptic phenotype seen in synapsin-deficient mice is associated with a number of secondary adaptations that develop to compensate for the deficiencies in synaptic transmission and plasticity in young presymptomatic mice (Ketzef et al., 2011; Farisello et al., 2012; Ketzef and Gitler, 2012).
SynII deficiency creates excitatory/inhibitory imbalance through an effect on GABAergic synaptic transmission
Previous studies have suggested that the epileptic phenotype resulting from synapsin deletion results from the development of an imbalance in excitatory and inhibitory synaptic transmission. The ability of 4-AP to reveal the epileptic phenotype in slices derived from the SynII(−) mice suggested a potential avenue for identifying the basis for this imbalance. To explore this idea, we tested the effect of 4-AP on EPSCs and IPSCs evoked by electrical stimulation. As illustrated in Figure 3, administration of low concentrations of 4-AP capable of selectively inducing epileptiform activity in slices derived from SynII(−) mice elicited a significant increase in the amplitude of the EPSCs in both WT and SynII(−) mice (Fig. 3A). This increase in EPSC amplitude, however, was highly asymmetrical and much larger in slices derived from SynII(−) mice than in slices derived from control mice (42.1 ± 12.1% for WT, n = 9 vs 89.6 ± 19.9% for Syn II(−), n = 6; p = 0.05; Fig. 3A1,A2,A5). Consistent with these findings, 4-AP also induced a significantly larger decrease in failure rate in slices derived from SynII(−) mice (44.4 ± 7.0% for WT vs 76.9 ± 7.5%, p = 0.008; Fig. 3A3,A4). In contrast to these observations on EPSCs, we could not detect a significant effect of 4-AP on IPSC amplitude or failure rates in slices derived from SynII(−) mice (Fig. 3B2,B4), even though 4-AP produced a significant increase in the amplitude of IPSC recorded in slices derived from WT mice (Fig. 3B1,B3). Together, these result shows that the epileptogenic agent 4-AP produces a balanced increase in excitatory and inhibitory synaptic transmission in WT mice. In contrast, in slices derived from SynII(−) mice, administration of 4-AP results in a preferential facilitation of excitatory synaptic transmission. This differential effect of 4-AP can be expected to magnify any latent imbalance in excitatory/inhibitory synaptic transmission and could explain the transition to the epileptic state.
Figure 3.

4-AP promotes excitatory and inhibitory transmission in a balanced way at WT synapses but creates an imbalance toward excitation at SynII(−) synapses. A, EPSCs recorded in slices derived from young presymptomatic SynII(−) mice exhibit an enhanced sensitivity to 4-AP (10 μm). A1, Effect of 4-AP on EPSC amplitude in WT slices (p = 0.014, n = 9). A2, Effect of 4-AP on EPSC amplitude on SynII(−) slices (p = 0.011, n = 6). A3, Effect of 4-AP on the EPSC failure rate in WT slices (p = 0.0002, n = 9). A4, Effect of 4-AP on the EPSC failure rate in SynII(−) slices (p = 0.0001, n = 6). A5, The synaptic facilitation induced by 4-AP effect is significantly (p = 0.049) stronger at SynII(−) synapses. The scatter graph represents the ratio of EPSC amplitudes with and without 4-AP. Data collected from 4 WT mice and 3 SynII(−) mice. A1, A2 insets, Average EPSCs (50 sweeps) recorded from representative experiments. Thin gray lines (A1–A4) and circles (A5) represent individual experiments. The depicted values correspond to the average of 50 EPSCs. Asterisks indicate a statistically significant difference. One-sided paired t test was used to determine an increase in synaptic response upon 4-AP application for the experiments depicted in A1–A4. Two-sided unpaired t test was used to compare the results depicted in A5 to determine the difference between genotypes. B, IPSCs recorded in slices derived from young presymptomatic SynII(−) mice exhibit diminished sensitivity to 4-AP (10 μm). B1, Effect of 4-AP on the IPSC amplitude recorded in WT slices (p = 0.010, n = 8). B2, Effect of 4-AP on the IPSC amplitude recorded in SynII(−) slices (p = 0.67, n = 6). B3, Effect of 4-AP on the IPSC failure rate recorded in WT slices (p = 0.05, n = 8). B4, Effect of 4-AP on the IPSC failure rate recorded in SynII(−) slices (p = 0.44, n = 6). B5, The effect of 4-AP is significantly (p = 0.036) stronger at WT synapses. Data collected from 4 WT mice and 3 SynII(−) mice.
We next asked what mechanism could modify the sensitivity of SynII(−) synapses to 4-AP. Because the action of 4-AP at synapses is thought to involve an increase in Ca2+ influx secondary to the blockade of potassium channels, we considered whether the deletion of SynII could affect the Ca2+ sensitivity of release. To test the idea that SynII(−) inhibitory synapses have a reduced sensitivity to Ca2+ and that this is the underlying mechanism for the observed differential effect of 4-AP on EPSCs and IPSCs in SynII(−) slices, we repeated the above experiments using 5 mm extracellular calcium instead of 4-AP. Increasing the extracellular Ca2+ concentration mimicked the effect of 4-AP in both WT and SynII(−) slices, with a balanced increase in EPSCs and IPSCs in WT slices, and selective increase in EPSCs without a corresponding increase in IPSCs in SynII(−) slices (Fig. 4). These results are consistent with the possibility that hippocampal inhibitory SynII(−) synapses may exhibit an impaired Ca2+ sensitivity of release.
Figure 4.

Raising extracellular Ca2+ concentration mimics the effect of 4-AP application on excitatory and inhibitory transmission at WT and SynII(−) synapses. A, EPSCs recorded in slices derived from young presymptomatic SynII(−) mice exhibit an enhanced sensitivity to elevated extracellular calcium concentration (5 mm). A1, Effect of raising extracellular calcium on the amplitude of EPSCs in WT slices (p = 0.005, n = 10). A2, Effect of raising extracellular calcium on the amplitude of EPSCs in SynII(−) slices (p = 0.031, n = 8). A3, Effect of raising extracellular calcium on the EPSCs failure rate in WT slices (p = 0.047, n = 10). A4, Effect of raising extracellular calcium on the EPSCs failure rate in SynII(−) slices (p < 0.001, n = 8). A5, The effect of Ca2+ elevation is significantly stronger at SynII(−) synapses (p < 0.04). Data collected from 5 WT mice and 4 SynII(−) mice. B, Increasing extracellular Ca2+ concentration produces a robust increase in evoked inhibitory transmission in WT synapses but no detectable increase at SynII(−) synapses in slices derived from young mice. B1, Effect of raising extracellular calcium on the amplitude of IPSCs in WT slices (p = 0.033, n = 8). B2, Effect of raising extracellular calcium on the amplitude of IPSCs in SynII(−) slices (p = 0.45, n = 7). B3, Effect of raising extracellular calcium on the IPSCs failure rate in WT slices (p = 0.028, n = 8). B4, Effect of raising extracellular calcium on the IPSCs failure rate in SynII(−) slices (p = 0.5, n = 7). B5, The effect of calcium is significantly stronger at WT synapses (p = 0.026). Data collected from 5 WT mice and 4 SynII(−) mice.
The results outlined above suggest that the impaired sensitivity of SynII(−) inhibitory synapses to elevated Ca2+ levels may produce an imbalance of excitatory and inhibitory transmission, leading to the overexcitable epileptic phenotype. Alternatively, it is possible that the overexcitability observed in the SynII(−) genotype is caused by an enhanced response of glutamatergic transmission to an epileptogenic stimulus. To discriminate between these two possibilities, we tested whether blocking inhibitory synaptic transmission would occlude the difference in spontaneous sEPSC activity and epileptiform discharge between the WT and SynII(−) mice. We recorded spontaneous synaptic activity in the presence of 4-AP in slices derived from WT and SynII(−) mice and administered Ptx at a concentration that could completely block inhibitory synaptic transmission (Fig. 5A). As illustrated in Figure 5B, C, the robust difference in sEPSC activity between phenotypes observed in the presence of low concentrations of 4-AP was completely occluded by administration of Ptx such that sEPSC frequency became indistinguishable in slices derived from WT and SynII(−) mice. Similarly, administration of Ptx occluded the differences in epileptiform activity observed between WT and SynII(−) genotypes (Fig. 5D). These results are consistent with the idea that the epileptic phenotype revealed by administration of 4-AP in the SynII(−) mice reflects the unmasking of a latent imbalance between excitatory and inhibitory synaptic transmission resulting from a deficit in GABAergic synaptic transmission.
Figure 5.
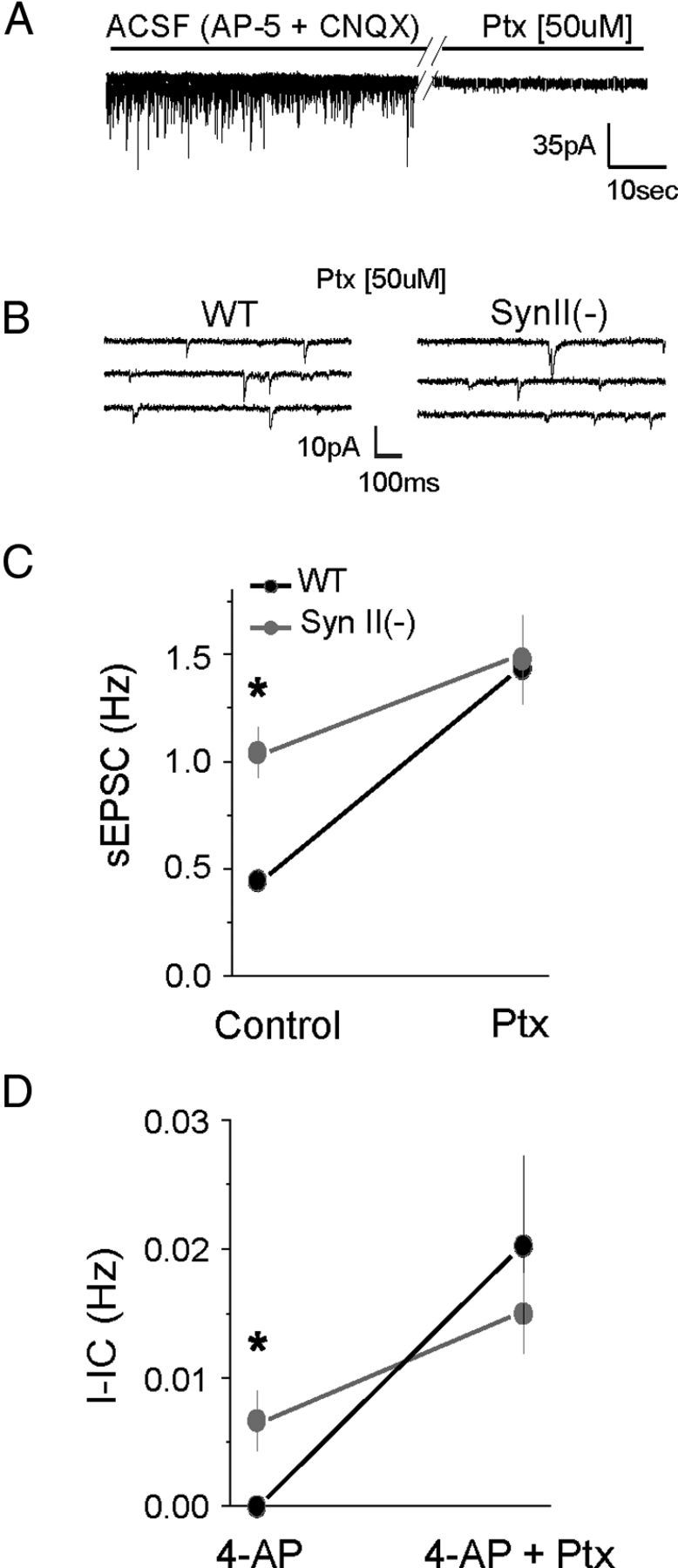
The increased synaptic and I-IC activity observed in slices derived from young presymptomatic Syn II(−) mice depends on synaptic inhibition. A, Application 50 μm Ptx eliminates inhibitory transmission. Recording were made at −65 mV holding potential in cesium-based solution in the presence of CNQX and AP-5. B, Spontaneous glutamatergic activity is comparable in WT and SynII(−) slices when inhibitory transmission is blocked by Ptx. C, Ptx application occludes the difference in sEPSC frequency between the WT and SynII(−) slices. D, Ptx application occludes the difference in I-IC activity between the WT and SynII(−) slices. *Significant difference (p < 0.05 per ANOVA followed by post hoc Tukey) between genotypes in the absence of Ptx. Data collected from 6 cells (3 animals) per genotype. Spontaneous activity was recorded for 5 min in the absence and for 5 min in the presence of Ptx.
SynII deficiency compromises Ca2+ sensitivity of inhibitory synaptic transmission
To further test the hypothesis that inhibitory SynII(−) synapses may exhibit an impaired Ca2+ sensitivity of release, we titrated GABAergic responses with increasing concentrations of extracellular Cd2+, a protocol that allows for the examination of release as a function of calcium influx. To cover a wide range of release probabilities, the extracellular Ca2+ concentration was elevated to 5 mm. We found that IPSC amplitudes at elevated Ca2+ levels (5 mm Ca2+ and 0–2 μm Cd2+) were significantly reduced in SynII(−) slices, whereas the blockade of Ca2+ channels by higher Cd2+ levels (5–50 μm) eliminated this difference (Fig. 6A,B). In addition, fitting the data by the Hill function (Fig. 6C) demonstrated that significantly higher Cd2+ concentrations were required for half-maximal inhibition of release at SynII(−) slices (K50 = 10.12 ± 0.85 in WT vs 15.09 ± 1.87 in SynII(−), p = 0.023), although Cd2+ cooperativity was not altered (Hill coefficient n = 1.51 ± 0.17 in WT vs 1.81 ± 0.3 in SynII(−), p = 0.4). This result suggests that the presence of SynII may be needed to enable GABAergic synapses to enhance their efficiency in response to Ca2+ elevation.
Figure 6.
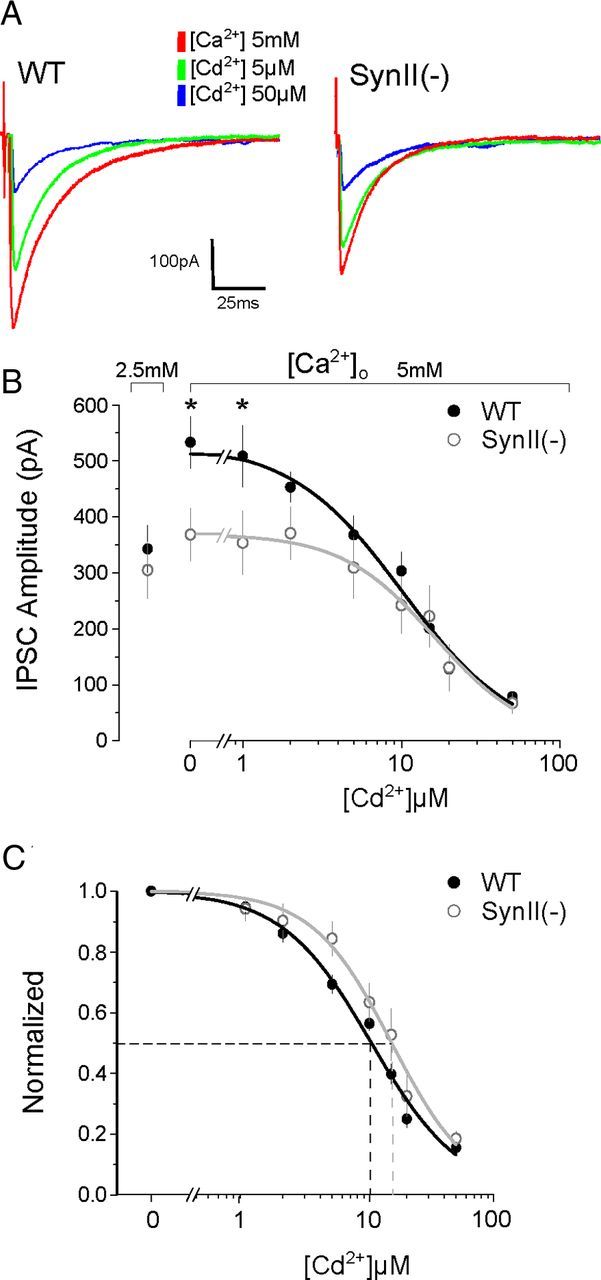
SynII(−) inhibitory synapses have reduced sensitivity to Ca2+ channel blockade by Cd2+, as well as a reduced ability to respond to Ca2+ elevation. A, Representative IPSC recordings obtained in 5 mm extracellular Ca2+ and different Cd2+ concentrations (red represents no cadmium; green represents 5 μm; blue represents 50 μm). Each trace is the average of 50 sweeps recorded in the same experiment. B, Cd2+ titration of inhibitory transmission demonstrates that WT GABAergic synapses have a stronger facilitation by elevated Ca2+ than SynII(−) synapses. In WT synapses, IPSC amplitudes are increased at elevated Ca2+ and low (<2 μm) Cd2+ concentrations (p = 0.04, n = 6 per two-way ANOVA), whereas higher Cd2+ concentrations abolish this difference. The stimulation intensity was adjusted to evoke similar responses in both lines at normal Ca2+ levels (2.5 mm Ca2+). Data collected from 3 WT and 3 SynII(−) presymptomatic mice (2 cell for each animal, 50 sweeps) were recorded at each Ca2+ and Cd2+ concentration. C, Fit to the data presented in the B by the Hill function. K50 is significantly higher in SynII(−) synapses (p = 0.035).
Rab3a balances SynII deficiency through an effect on glutamatergic synaptic transmission
Deletion of Rab3a results in a suppression of the epileptic phenotype seen in Syn II(−) mice (Coleman and Bykhovskaia, 2010). The availability of the in vitro experimental model outlined above offered the possibility of examining the cellular basis for this rescue. Therefore, we next examined the effect of deleting Rab3a on the SynII(−) cellular phenotype. Remarkably, baseline spontaneous synaptic activity recorded in slices derived from SynII(−)/Rab3a(−) DKO was indistinguishable from that recorded in slices derived from WT mice (Fig. 7A,B). Similarly, the effect of 4-AP on synaptic activity in the SynII(−)/Rab3a(−) DKO was also indistinguishable from that seen in WT mice (Fig. 7C). Deletion of Rab3a also suppressed the epileptiform activity unmasked by low concentrations of 4-AP in slices derived from young presymptomatic SynII mice (Fig. 7D). Thus, Rab3a normalized the increase in network excitability induced by the deletion of SynII(−).
Figure 7.

Rab3a deletion balances the increase in sEPSC frequency and epileptiform activity observed in presymptomatic Syn II(−) synapses. A, Recordings of sEPSCs (inward) and sIPSCs (outward) activity in the absence of blockers at −50 mV holding potential in WT and SynII(−)/Rab3a(−) slices. B, sEPSC frequency in SynII(−)/Rab3a(−) synapses is comparable with that recorded in WT (p = 0.38 per Kolmogorov–Smirnov test) but significantly different from that seen in SynII(−) synapses (p = 0.008). C, 4-AP has a modest but significant effect on spontaneous activity in SynII(−)/Rab3a(−) DKO synapses (p = 0.043), which is comparable with the effect observed in WT synapses (p = 0.6) but significantly smaller than that observed in SynII(−) synapses (p = 0.05). The thin green lines represent individual experiments. D, 4-AP-induced epileptiform activity in SynII(−)/Rab3a(−) DKO is similar to that recorded in WT slices but significantly lower than that recorded in SynII(−) slices at low 4-AP concentrations (10 and 15 μm). Asterisks indicate a significant difference between SynII(−)/Rab3a DKO and SynII(−) lines (p < 0.05 per ANOVA followed by post hoc Tukey test). Dotted lines indicate the replotted data from Figures 1 and 2. Data collected from 4 Syn II(−)/Rab3a(−) animals (9 cells).
To understand how Rab3a deletion balances SynII deficiency, we investigated synaptic activity in Rab3a(−) animals. First, we examined spontaneous activity in slices derived from Rab3a(−) mice. The basal spontaneous activity at Rab3a(−) synapses was undistinguishable from WT (Fig. 8A–C), but application of 4-AP (10 μm) to slices derived from Rab3a(−) mice resulted in a somewhat weaker increase in sEPSC frequency (70 ± 18% increase in Rab3a(−) vs 142 ± 37% in WT, Fig. 8D), although the difference between Rab3a(−) and WT phenotypes was not significant (p = 0.22). Consistent with these findings, application of 4-AP (10 μm) failed to elicit any detectable effect on EPSC amplitude (Fig. 8E1,E3), although it produced a significant increase in the IPSC amplitude (Fig. 8E2,E3). Elevating extracellular Ca2+ (to 5 mm) mimicked this effect of 4-AP, suggesting that Rab3a deletion may compromise Ca2+ sensitivity at glutamatergic but not GABAergic synapses. These results led us to suggest that deletion of Rab3a normalizes the hyperexcitability phenotype characteristic of the SynII(−) not by reversing the deficit in GABAergic synaptic transmission but instead by inducing a compensatory deficit in glutamate-mediated synaptic transmission.
Figure 8.

Rab3a deletion does not affect spontaneous synaptic activity but inhibits EPSC enhancement produced by either 4-AP application or increased extracellular Ca2+. A, Recordings of sEPSCs (inward) and sIPSCs (outward) activity in the absence of blockers at −50 mV holding potential in WT and Rab3a(−) slices derived from young mice. B, sEPSC amplitude histograms for WT and Rab3a(−) synapses. No significant shift in the sEPSC amplitude in Rab3a(−) synapses was detected (p = 0.5 per Kolmogorov–Smirnov test). C, Cumulative plot for the sEPSC interevent interval recorded in WT and Rab3a(−) slices. The interevent interval is slightly increased in Rab3a(−) synapses, but this effect is not statistically significant (p = 0.16 per Kolmogorov–Smirnov test). B, C, Histograms represent results from all the experiments pooled together for each genotype. Data were collected from 7 Rab3a(−) cells (4 mice). WT data (dotted lines) are replotted from Figures 1 and 2. D, 4-AP elicits a significant enhancement in the sEPSC frequency in Rab3a(−) synapses (p = 0.028, n = 7) This affect appears to be weaker than in WT synapses, although the difference between the phenotypes is not significant (p = 0.22). E, In Rab3a neurons, administration of 4-AP (10 μm) fails to produce any detectable change in EPSC amplitude (E1,E3, p = 0.425, n = 7), whereas it elicits a significant increase in IPSC amplitude (E2,E3, p = 0.010, n = 7). E3, The effect of 4-AP on EPSCs and IPSCs in the two genotypes. F, In Rab3a neurons, elevating extracellular calcium fails to produce a statistically significant change in EPSC amplitude (F1, p = 0.10, n = 7), whereas it elicits a significant increase in IPSC amplitude (F2, p = 0.05, n = 6). F3, The effect of elevated calcium on EPSCs and IPSCs in the two genotypes. The effect of 4-AP was quantified as the ratio between EPSC or IPSC amplitudes after and before treatment. Data were collected from 4 mice. Fifty responses at each condition were collected in each experiment.
To test this idea, we repeated these experiments in slices derived from SynII(−)/Rab3a(−) DKO mice. We found that, in the absence of both SynII and Rab3a, neither excitatory (Fig. 9A1,A3) nor inhibitory (Fig. 9A2,A3) transmission showed a detectable enhancement in response to low concentrations of 4-AP (10 μm) or elevation of extracellular Ca2+ (Fig. 9B). This suggests that the Rab3a(−) rescue of the SynII(−) phenotype reflects impaired Ca2+ sensitivity at glutamategic synapses. These results are summarized in the Figure 9C, which shows that low concentrations of 4-AP unmask an excitatory/inhibitory imbalance in slices derived from SynII(−) mice, that Rab3a deletion abolishes the effect of 4-AP on EPSC amplitude, and that the double gene deletion produces a phenotype where no detectable effect of 4-AP on either EPSC or IPSC amplitude is observed.
Figure 9.
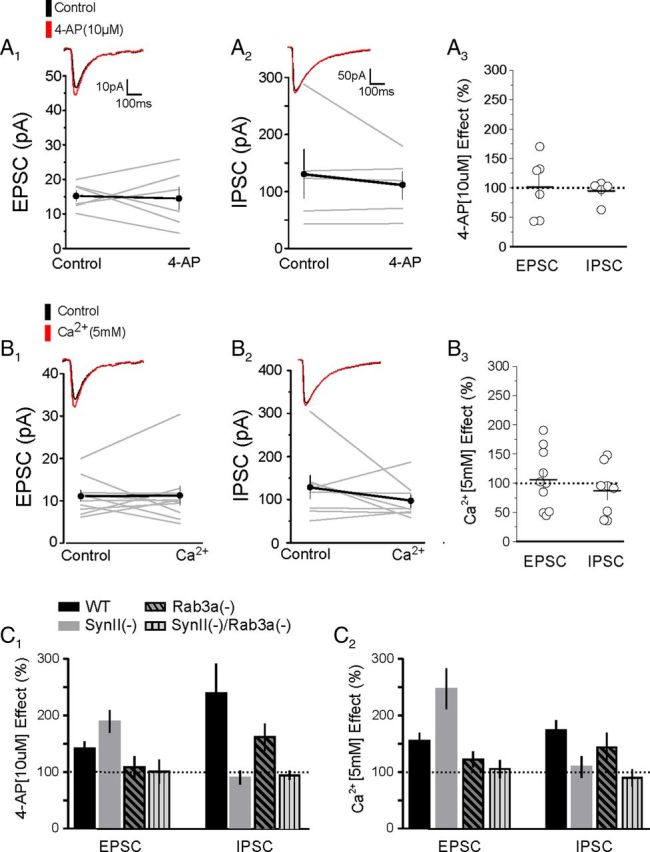
Lack of effect of 4-AP or elevated extracellular Ca2+ on EPSC/IPSC amplitudes at SynII(−)/Rab3a(−) synapses. A, In SynII(−)/Rab3a(−), DKO synapses in slices derived from young mice 4-AP (10 μm) fails to produce a detectable increase in either EPSC (A1, A3, p = 0.43, n = 6) or IPSC amplitude (A2, A3, p = 0.50, n = 5). B, In Syn II(−)/Rab3a(−), DKO synapses elevating extracellular calcium to 5 mm fails to elicit a statistically significant increase in either EPSCs (B1, B3, p = 0.48, n = 10) or IPSCs (B2, B3, p = 0.48, n = 8). C, Bar graph summarizing the results of 4-AP (C1) and elevated calcium (C2) on EPSCs and IPSCs in the different genotypes. These plots summarize the data presented in Figures 3, 4, 8, and 9A, B.
Interestingly, Rab3a deletion also negated the enhancing effect of SynII deletion on the sensitivity of excitatory transmission to low-dose 4-AP. Indeed, low-dose 4-AP produced a significantly stronger enhancement of EPSC amplitude in SynII(−) synapses than in WT synapses (89.6 ± 19.8% vs 45.1 ± 10.7% increase, p = 0.048), whereas in the absence of Rab3a, SynII deletion had no effect on the sensitivity of glutamatergic transmission to low-dose 4-AP or Ca2+, and no increase in EPSC amplitude was detected in either Rab3a(−) or SynII(−)/Rab3a(−) DKO line upon 10 μm 4-AP application or Ca2+ elevation (Fig. 9C1,C2).
Thus, our results indicate that Rab3a could normalize the hyperexcitable phenotype of the Syn II(−) mouse not by directly compensating the Syn II(−)-induced deficit in GABAergic synaptic transmission but rather by inducing a compensatory reduction in glutamate excitatory synaptic transmission, which is likely to result from impaired Ca2+ sensitivity.
Discussion
In the present study, we investigated synaptic activity in SynII(−) presymptomatic animals to advance our understanding of the seizure phenotype in synapsin-deficient brain. We found that spontaneous synaptic activity in hippocampal slices derived from SynII(−) animals is enhanced and that this enhancement reflects a deficiency in synaptic inhibition, which may be associated with impaired Ca2+ sensitivity of inhibitory synapses in SynII(−) animals. We also took advantage of SynII(−)/Rab3(−) animals to understand how Rab3a deletion modifies the synaptic effects of SynII(−) deletion. We found that the increase in synaptic and epileptiform activity observed in SynII(−) was balanced by Rab3a gene deletion through an effect on glutamatergic synaptic transmission. Thus, this study demonstrates GABA-dependent overexcitability in brain slices of presymptomatic SynII(−) animals and identifies a cellular mechanism through which Rab3a deletion could suppress synapsin-dependent epilepsy.
Excitation/inhibition imbalance in presymptomatic SynII(−) animals may trigger overexcitability and epileptic activity
Previous work has identified the synapsins as important epilepsy susceptibility genes in animals and humans (Fassio et al., 2011a), making the question of how synapsin regulates neuronal and epileptic activity especially intriguing. An earlier study (Chiappalone et al., 2009) reported an increased firing rate in synapsin-deficient cortical neurons in culture, suggesting overexcitability in synapsin-deficient neurons in vitro. Here we have extended these findings to show that spontaneous synaptic activity is increased in SynII(−) hippocampal slices and that this effect cannot be ascribed solely to the increase in mEPSCs.
To unmask the epileptic phenotype in synapsin-deficient mice in vitro, we used 4-AP, which has previously proven useful for studying epileptic activity in synapsin-deficient neurons (Boido et al., 2010; Ketzef et al., 2011). We found that administration of low concentrations of 4-AP preferentially increases spontaneous synaptic activity in SynII(−) neurons and selectively induces epileptiform activity in slices derived from presymptomatic SynII(−) animals. Interestingly, the slices of adult symptomatic animals had reduced excitability, suggesting that epileptic activity in adult animals is strongly influenced by developmental effects and secondary adaptations (Ketzef et al., 2011; Farisello et al., 2012; Ketzef and Gitler, 2012).
Previous studies suggested the likely involvement of changes in inhibitory synaptic transmission as causative agents for the seizure phenotype seen in synapsin-deficient mice. Thus, recordings from autaptic cultured hippocampal neurons demonstrated that inhibitory synapses derived from synapsin TKO (Gitler et al., 2004) and SynI(−) animals (Baldelli et al., 2007) had reduced evoked (but not spontaneous) release. A subsequent study (Farisello et al., 2012) examined the input-output relationship for excitatory and inhibitory transmission in hippocampal slices and found that high stimulation intensities evoke significantly higher excitatory and lower inhibitory currents in both adult and presymptomatic TKO synapsin mice. These studies point to a deficit in inhibitory synaptic transmission, leading to an excitatory/inhibitory imbalance in synapsin knock-out brain as a likely reason for the synapsin-dependent overexcitability.
To try to link specific changes in synaptic activity in slices derived from SynII(−) mice to the epileptic phenotype, we examined the effects of low concentrations of 4-AP on excitatory and inhibitory synaptic transmission. As previously seen by others (Buckle and Haas, 1982; Gu et al., 2004), administration of 4-AP to WT mice facilitated excitatory and inhibitory evoked synaptic transmission in a balanced way. In contrast, administration of 4-AP to slices derived from SynII(−) mice resulted in a strong selective increase in the EPSC amplitude with no detectable effect on the IPSC amplitude. These results suggested a key role for changes in inhibitory synaptic transmission. If this was the case, then the effect of genotype on spontaneous synaptic and epileptiform activity should be occluded by blockade of GABAergic synaptic transmission. Consistent with this idea, administration of a saturating concentration of Ptx abolished the differential effect of Syn II(−) on synaptic activity. We interpret these results to indicate that the hyperexcitability observed in hippocampal slices derived from presymptomatic SynII(−) animals requires the SynII(−)-induced deficit in inhibitory synaptic transmission.
Because 4-AP is thought to promote transmitter release by prolonging Ca2+ influx (Vyshedskiy et al., 2000), we asked whether we could mimic the effect of 4-AP in vitro by manipulating extracellular calcium. An effect of synapsin deletion on the Ca2+ sensitivity of release has been previously detected at the neuromuscular synapse (Coleman et al., 2008; Coleman and Bykhovskaia, 2010), and such an effect would be consistent with the inhibition of release by microinjected dephosphosynapsin (Llinás et al., 1985, 1991; Hackett et al., 1990; Coleman et al., 2008) and the regulation of synapsin phosphorylated state by several Ca2+/calmodulin and calcineurin-dependent pathways (Jovanovic et al., 2001; Yamagata et al., 2002; Cesca et al., 2010). Additional support for this idea comes from the observation that SynI(−) hippocampal inhibitory synapses have reduced sensitivity to the blockade of Ca2+ influx by Cd2+ (Baldelli et al., 2007). In the current study, we explored the effect of increasing extracellular Ca2+ concentration on glutamatergic and GABAergic transmission and discovered that it closely mimicked the effect of 4-AP treatment. Such Ca2+ elevation promoted both excitatory and inhibitory transmission in WT slices but did not produce a significant increase in GABAergic transmission in SynII(−) slices. Titration of GABAergic release by Cd2+ demonstrated that SynII(−) inhibitory synapses have reduced sensitivity to Cd2+ blockade, as well as to Ca2+ elevation. These observations are consistent with recent findings indicating that SynII interacts with presynaptic Ca2+ channels (Medrihan et al., 2013). Thus, our study suggests that impaired Ca2+ sensitivity of the GABAergic release, produced by synapsin deficiency, is responsible for the compromised response of inhibitory neurons to an epileptogenic stimulus.
Rab3a deletion restores the excitation/inhibition imbalance in SynII(−) neurons via the regulation of excitatory synaptic transmission
Given the epileptogenic effect of synapsin deletion, understanding how to balance the deleterious effect of these deletions represents an important therapeutic goal. Unfortunately, rational approaches to this problem to date have not yielded effective strategies. For example, epilepsy in synapsin-deficient animals proved to be remarkably resistant to the antiepileptic drug levetiracetam (Boido et al., 2010), which has a presynaptic target. We have previously shown that deletion of the synapsin binding partner Rab3a rescues the seizure phenotype observed in synapsin knock-out animals (Coleman and Bykhovskaia, 2010). Because this is the only manipulation discovered to date that rescues synapsin-dependent seizures at the behavioral level, it is essential to understand the functional interaction of these two proteins at the level of synapses and networks. In this study, we demonstrated in vitro that this genetic manipulation balances the overexcitability observed in SynII(−) neurons, in line with the effect of Rab3a gene deletion on synapsin-dependent epileptic activity in vivo.
Rab3a is a small vesicle-associated protein (Fischer von Mollard et al., 1991; Mizoguchi et al., 1992), which is thought to target vesicles to active zones (Nonet et al., 1997; Leenders et al., 2001; Coleman et al., 2007; Tian et al., 2012). Rab3a and synapsins interact on vesicles (Giovedì, 2004b), and this interaction changes the function of both proteins (Giovedì et al., 2004a). At the neuromuscular junction, these two proteins appear to interact in a complex hierarchical way to regulate release (Coleman and Bykhovskaia, 2010). Thus, Rab3a deletion could rescue the SynII(−) phenotype directly, by correcting the SynII(−)-induced deficit in GABAergic synaptic transmission. However, we have tested this possibility and found that this is not the case. Rab3a deletion had no effect on enhancement in GABAergic hippocampal synapses induced by 4-AP, independently of the presence or absence of SynII. Strikingly, however, Rab3a deletion significantly compromised the enhancement in glutamatergic hippocampal synapses induced by 4-AP. Although 4-AP application produced a robust increase in the EPSC amplitude in WT excitatory synapses, no significant increase in EPSCs was detected upon administration of 4-AP to Rab3a(−) synapses, independently of SynII presence. Extracellular Ca2+ elevation mimicked this effect of 4-AP, suggesting that the Ca2+ sensitivity of the release process at glutamatergic hippocampal synapses may depend on Rab3a. This observation is consistent with the proposed role of the Rab3a protein in targeting vesicles to Ca2+ channels (Schlüter et al., 2006) and with the impaired Ca2+ sensitivity observed at the neuromuscular synapse of Rab3a(−) animals (Coleman et al., 2007; Coleman and Bykhovskaia, 2009). We interpret these results to indicate that Rab3a deletion restores the excitation/inhibition imbalance produced by the SynII(−) deletion by suppressing excitatory synaptic transmission.
More broadly, our findings suggest that Rab3a deletion balances the epileptogenic effect of SynII deletion at the level of networks. Specifically, we show that both SynII and Rab3a affect the Ca2+ sensitivity of release and that this effect is synapse-specific. SynII deletion suppresses the Ca2+ sensitivity of GABA release, thus creating an excitation/inhibition imbalance. In contrast, Rab3a deletion has no apparent effect on GABA release but suppresses the Ca2+ sensitivity of glutamate release, thus compensating for the deletion of SynII. In addition, SynII deletion appears to enhance the sensitivity of glutamate release to 4-AP, and this effect is suppressed by Rab3a deletion. Thus, our study reveals functional interaction of SynII and Rab3a at the level of single synapses and networks and demonstrates that the latter is critical for the compensatory effect of Rab3a deletion on overexcitability in the SynII-deficient brain.
Footnotes
This work was supported by National Science Foundation Grant IOS 1145010 and National Institutes of Health Grant NS083924 to M.B. and National Institutes of Health Grants MH43987 and MH100850 to R.A.
The authors declare no competing financial interests.
References
- Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci. 2007;27:13520–13531. doi: 10.1523/JNEUROSCI.3151-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Basic developmental rules and their implications for epilepsy in the immature brain. Epileptic Disord. 2006;8:91–102. [PubMed] [Google Scholar]
- Bogen IL, Jensen V, Hvalby O, Walaas SI. Synapsin-dependent development of glutamatergic synaptic vesicles and presynaptic plasticity in postnatal mouse brain. Neuroscience. 2009;158:231–241. doi: 10.1016/j.neuroscience.2008.05.055. [DOI] [PubMed] [Google Scholar]
- Boido D, Farisello P, Cesca F, Ferrea E, Valtorta F, Benfenati F, Baldelli P. Cortico-hippocampal hyperexcitability in synapsin I/II/III knockout mice: age-dependency and response to the antiepileptic drug levetiracetam. Neuroscience. 2010;171:268–283. doi: 10.1016/j.neuroscience.2010.08.046. [DOI] [PubMed] [Google Scholar]
- Buckle PJ, Haas HL. Enhancement of synaptic transmission by 4-aminopyridine in hippocampal slices of the rat. J Physiol. 1982;326:109–122. doi: 10.1113/jphysiol.1982.sp014180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaia M. Making quantal analysis more convenient, fast, and accurate: user-friendly software QUANTAN. J Neurosci Methods. 2008;168:500–513. doi: 10.1016/j.jneumeth.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalleri GL, Weale ME, Shianna KV, Singh R, Lynch JM, Grinton B, Szoeke C, Murphy K, Kinirons P, O'Rourke D, Ge D, Depondt C, Claeys KG, Pandolfo M, Gumbs C, Walley N, McNamara J, Mulley JC, Linney KN, Sheffield LJ, et al. Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol. 2007;6:970–980. doi: 10.1016/S1474-4422(07)70247-8. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chiappalone M, Casagrande S, Tedesco M, Valtorta F, Baldelli P, Martinoia S, Benfenati F. Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I-deficient cortical networks. Cereb Cortex. 2009;19:1422–1439. doi: 10.1093/cercor/bhn182. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/S0896-6273(03)00151-X. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bykhovskaia M. Rab3a-mediated vesicle recruitment regulates short-term plasticity at the mouse diaphragm synapse. Mol Cell Neurosci. 2009;41:286–296. doi: 10.1016/j.mcn.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bykhovskaia M. Cooperative regulation of neurotransmitter release by Rab3a and synapsin II. Mol Cell Neurosci. 2010;44:190–200. doi: 10.1016/j.mcn.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman WL, Bill CA, Bykhovskaia M. Rab3a deletion reduces vesicle docking and transmitter release at the mouse diaphragm synapse. Neuroscience. 2007;148:1–6. doi: 10.1016/j.neuroscience.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bill CA, Simsek-Duran F, Lonart G, Samigullin D, Bykhovskaia M. Synapsin II and calcium regulate vesicle docking and the cross-talk between vesicle pools at the mouse motor terminals. J Physiol. 2008;586:4649–4673. doi: 10.1113/jphysiol.2008.154666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Malladi CS, Tan TC, Raymond CR, Smillie KJ, Robinson PJ. Synapsin I-associated phosphatidylinositol 3-kinase mediates synaptic vesicle delivery to the readily releasable pool. J Biol Chem. 2003;278:29065–29071. doi: 10.1074/jbc.M302386200. [DOI] [PubMed] [Google Scholar]
- Etholm L, Heggelund P. Seizure elements and seizure element transitions during tonic-clonic seizure activity in the synapsin I/II double knockout mouse: a neuroethological description. Epilepsy Behav. 2009;14:582–590. doi: 10.1016/j.yebeh.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Farisello P, Boido D, Nieus T, Medrihan L, Cesca F, Valtorta F, Baldelli P, Benfenati F. Synaptic and extrasynaptic origin of the excitation/inhibition imbalance in the hippocampus of synapsin I/II/III knockout mice. Cereb Cortex. 2013;23:581–593. doi: 10.1093/cercor/bhs041. [DOI] [PubMed] [Google Scholar]
- Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, Pozzi D, Messa M, Defranchi E, Fadda M, Corradi A, Baldelli P, Lapointe L, St-Onge J, Meloche C, Mottron L, Valtorta F, Khoa Nguyen D, Rouleau GA, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011a;20:2297–2307. doi: 10.1093/hmg/ddr122. [DOI] [PubMed] [Google Scholar]
- Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P. Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol. 2011b;22:408–415. doi: 10.1016/j.semcdb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G, Südhof TC, Jahn R. A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature. 1991;349:79–81. doi: 10.1038/349079a0. [DOI] [PubMed] [Google Scholar]
- Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, Curtis A, Goodship JA. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet. 2004;41:183–186. doi: 10.1136/jmg.2003.013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovedì S, Darchen F, Valtorta F, Greengard P, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles: II. Functional effects of the Rab3A-synapsin I interaction. J Biol Chem. 2004a;279:43769–43779. doi: 10.1074/jbc.M404168200. [DOI] [PubMed] [Google Scholar]
- Giovedì S, Vaccaro P, Valtorta F, Darchen F, Greengard P, Cesareni G, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles: I. Identification and characterization of the synapsin I-Rab3 interactions in vitro and in intact nerve terminals. J Biol Chem. 2004b;279:43760–43768. doi: 10.1074/jbc.M403293200. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Ge SY, Ruan DY. Effect of 4-aminopyridine on synaptic transmission in rat hippocampal slices. Brain Res. 2004;1006:225–232. doi: 10.1016/j.brainres.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Cochran SL, Greenfield LJ, Jr, Brosius DC, Ueda T. Synapsin I injected presynaptically into goldfish mauthner axons reduces quantal synaptic transmission. J Neurophysiol. 1990;63:701–706. doi: 10.1152/jn.1990.63.4.701. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Gairsa JL, Chevassus-Au-Louis N, Ben-Ari Y. Consequences of neonatal seizures in the rat: morphological and behavioral effects. Ann Neurol. 1998;44:845–857. doi: 10.1002/ana.410440602. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci. 2001;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvalby O, Jensen V, Kao HT, Walaas SI. Synapsin-dependent vesicle recruitment modulated by forskolin, phorbol ester and Ca2+ in mouse excitatory hippocampal synapses. Front Synaptic Neurosci. 2010;2:152. doi: 10.3389/fnsyn.2010.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketzef M, Gitler D. Epileptic synapsin triple knockout mice exhibit progressive long-term aberrant plasticity in the entorhinal cortex. Cereb Cortex. 2012 doi: 10.1093/cercor/bhs384. doi: 10.1093/cercor/bhs384. Advance online publication. Retrieved December 12, 2012. [DOI] [PubMed] [Google Scholar]
- Ketzef M, Kahn J, Weissberg I, Becker AJ, Friedman A, Gitler D. Compensatory network alterations upon onset of epilepsy in synapsin triple knock-out mice. Neuroscience. 2011;189:108–122. doi: 10.1016/j.neuroscience.2011.05.030. [DOI] [PubMed] [Google Scholar]
- Lakhan R, Kalita J, Misra UK, Kumari R, Mittal B. Association of intronic polymorphism rs3773364 A>G in synapsin-2 gene with idiopathic epilepsy. Synapse. 2010;64:403–408. doi: 10.1002/syn.20740. [DOI] [PubMed] [Google Scholar]
- Leenders AG, Lopes da Silva FH, Ghijsen WE, Verhage M. Rab3a is involved in transport of synaptic vesicles to the active zone in mouse brain nerve terminals. Mol Biol Cell. 2001;12:3095–3102. doi: 10.1091/mbc.12.10.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci U S A. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Dzhala VI, Ben-Ari Y. Generation and propagation of 4-AP-induced epileptiform activity in neonatal intact limbic structures in vitro. Eur J Neurosci. 2000;12:2757–2768. doi: 10.1046/j.1460-9568.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- Medrihan L, Cesca F, Raimondi A, Lignani G, Baldelli P, Benfenati F. Synapsin II desynchronizes neurotransmitter release at inhibitory synapses by interacting with presynaptic calcium channels. Nat Commun. 2013;4:1512. doi: 10.1038/ncomms2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messa M, Congia S, Defranchi E, Valtorta F, Fassio A, Onofri F, Benfenati F. Tyrosine phosphorylation of synapsin I by Src regulates synaptic-vesicle trafficking. J Cell Sci. 2010;123:2256–2265. doi: 10.1242/jcs.068445. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Arakawa M, Masutani M, Tamekane A, Yamaguchi H, Minami N, Takai Y, Ide C. Localization of smg p25A/rab3A p25, a small GTP-binding protein, at the active zone of the rat neuromuscular junction. Biochem Biophys Res Commun. 1992;186:1345–1352. doi: 10.1016/S0006-291X(05)81554-2. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Basu J, Südhof TC, Rosenmund C. Rab3 superprimes synaptic vesicles for release: implications for short-term synaptic plasticity. J Neurosci. 2006;26:1239–1246. doi: 10.1523/JNEUROSCI.3553-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Haucke V, Pechstein A. How synapsin I may cluster synaptic vesicles [Review] Semin Cell Dev Biol. 2011;4:393–399. doi: 10.1016/j.semcdb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Tian M, Xu CS, Montpetit R, Kramer RH. Rab3A mediates vesicle delivery at photoreceptor ribbon synapses. J Neurosci. 2012;32:6931–6936. doi: 10.1523/JNEUROSCI.0265-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyshedskiy A, Allana T, Lin JW. Analysis of presynaptic Ca2+ influx and transmitter release kinetics during facilitation at the inhibitor of the crayfish neuromuscular junction. J Neurosci. 2000;20:6326–6332. doi: 10.1523/JNEUROSCI.20-17-06326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Jovanovic JN, Czernik AJ, Greengard P, Obata K. Bidirectional changes in synapsin I phosphorylation at MAP kinase-dependent sites by acute neuronal excitation in vivo. J Neurochem. 2002;80:835–842. doi: 10.1046/j.0022-3042.2001.00753.x. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]