

Abstract
The Notch signaling pathway is fundamental to proper cardiovascular development and is now recognized as an important player in tumor angiogenesis. Two key Notch ligands have been implicated in tumor angiogenesis, Delta-like 4 and Jagged1. We introduce the proteins and how they work in normal developing vasculature and then discuss differing models describing the action of these Notch ligands in tumor angiogenesis. Endothelial Dll4 expression activates Notch resulting in restriction of new sprout development; for instance, in growing retinal vessels. In agreement with this activity, inhibition of Dll4-mediated Notch signaling in tumors results in hypersprouting of nonfunctional vasculature. This Dll4 inhibition may paradoxically lead to increased angiogenesis but poor tumor growth because the newly growing vessels are not functional. In contrast, Jagged1 has been described as a Notch ligand expressed in tumor cells that can have a positive influence on tumor angiogenesis, possibly by activating Notch on tumor endothelium. A novel Notch inhibitor, the Notch1 decoy, which blocks both Dll4 and Jagged1 has been recently shown to restrict tumor vessel growth. We discuss these models and speculate on therapeutic approaches.
Keywords: angiogenesis, Notch, Jagged1, Dll4, sprout, VEGFR-2
Introduction
The Notch pathway is a conserved ligand–receptor signaling mechanism that modulates cell fate and differentiation (Greenwald, 1998; Artavanis-Tsakonas et al., 1999). The Notch receptor is a single-pass transmembrane protein consisting of a signal peptide, an extracellular domain that is responsible for ligand interaction, a transmembrane domain that is involved in receptor activation and an intracellular signaling domain. Notch signaling is initiated when the extracellular domain of the receptor engages ligand found on neighboring cells that are in close proximity to one another. Thus, Notch signaling depends on cell-contact-dependent interactions. This leads to a cascade of enzymatic cleavages and release of the extracellular domain, whereas the intracellular domain is released and then translocated to the nucleus where it interacts with CSL (CBF1, Su(H) and Lag-2) transcriptional repressors and converts them to transcriptional activators. In many cases, the cell that presents the ligand is a cell that does not have Notch signaling present, thus distinguishing two neighboring cells into one with ligand with little Notch signaling and one with receptor and high Notch signaling.
In mammals there are four Notch receptors, Notch1, Notch2, Notch3 and Notch4. There are five ligands named Jagged1, Jagged2 (homologues to Serrate), Delta-like (Dll) 1, Dll3 and Dll4. Of note for this review are two ligands in particular; Dll4 and Jagged1. The Dll4 ligand is a Notch ligand with a pattern of strong expression in the endothelium of tumor blood vessels (Mailhos et al., 2001). In addition, Dll4 expression is found in arterial endothelium during mouse embryogenesis, such as the endothelium of the axial dorsal aorta and umbilical artery, as well as in the endothelium of blood vessels (Mailhos et al., 2001). Jagged1 is a ligand thought to be involved in early cardiovascular development by regulation of endothelial and vascular smooth muscle cells.
It is clear that the Notch family is critically important for the proper construction of the vascular system. Global knockouts of Notch1 alone or Notch1/4 together are embryonic lethal with severe vascular defects (Krebs et al., 2000). Haploinsufficiency of the Notch ligand gene Dll4 results in embryonic death due to vascular defects in mice (Gale et al., 2004; Krebs et al., 2004). Global as well as endothelium-specific knockouts of Notch ligand gene Jagged1 induce embryonic death with vascular defects (Xue et al., 1999; High et al., 2008). Endothelium-specific ectopic expression of activated Notch4 in mice results in embryonic death with vascular defects in mice (Uyttendaele et al., 2001). These results show that proper regulation of the Notch pathway is indispensable for vascular development during embryogenesis in mammals. Notch pathway components have also been shown to be required for postnatal angiogenesis (Limbourg et al., 2005; Takeshita et al., 2007).
These signaling events and Notch family members are discussed in greater detail in other reviews presented in this issue. However, the pertinence to angiogenesis and the mechanisms by which Notch signaling influences tumor angiogenesis will be discussed here. We highlight the concept that ligand–receptor interactions in Notch signaling depend on contact between two cells and these may be two different cell types. Notch ligands involved in tumor angiogenesis may be presented by tumor endothelium, tumor cells or by inflammatory cells. We will discuss the consequence of such diverse presentation modes and speculate on outcomes of these different types of signaling.
Notch in arterial differentiation
A role for Notch family members in arterial differentiation is well established (Lawson and Weinstein, 2002). Expression of Notch ligands and Notch family members sets the stage for this process. At approximately e9.5–e11.5 of murine embryogenesis the primary vascular plexus begins remodeling leading to the establishment of arteries and veins. During initiation of this remodeling, Notch1, Notch4 and Dll4 are expressed within and around the developing vasculature (Alva and Iruela-Arispe, 2004), however by e13.5, expression of Dll4, Notch1 and Notch4 become restricted to the arterial endothelium (Villa et al., 2001). At the same time, members of the Eph signaling pathway, ephrinB2 and EphB4, are expressed and serve as markers of arterial and venous identity, respectively (Wang et al., 1998). Loss of Notch signaling results in reduced expression of the arterial marker ephrinB2. Definitive studies using zebrafish as a model for cardiovascular development conclude that Notch functions upstream of eprhinB2 vessel identity, promoting arterial specification (Zhong et al., 2000, 2001; Lawson et al., 2001). Studies in endothelial cells (Shawber et al., 2003) and in mice (Carlson et al., 2005; Iso et al., 2006) also point to this signaling axis as specifying arterial cell fate.
Sprouting angiogenesis and the influence of Dll4/Notch
Angiogenesis is the process of generating a new blood vessel from a pre-existing vessel. The process consists of a series of endothelial cell responses to angiogenic stimulation; including degradation of extracellular matrix (ECM), budding, proliferation, migration, tube formation, maturation and maintenance of quiescent endothelium. Figure 1 schematizes the steps in angiogenesis as a cycle; going from a preexisting vessel to a nascent sprout to development of a new tube to maturation resulting in a new vessel. The preexisting vessel consists of a tubular structure made up of quiescent endothelium with blood flowing through it and in association with mural cells, which can be pericytes or vascular smooth muscle cells. A new sprout will form at a distinct location in the preexisting vessel and will be initiated by degradation of the ECM followed by development of a new bud from the endothelial cell. This bud will elongate and the endothelial cell will then migrate toward the source of angiogenic factor. During this phase, endothelial cell proliferation is activated leading to increased cell numbers in the growing sprout. The process culminates with tube formation and maturation. Tube formation can occur through a variety of mechanisms that are as yet poorly defined. Maturation consists of recruitment of perivascular cells, referred to as mural cells, which can be pericytes or vascular smooth muscle cells. Pericytes are thought to stabilize capillaries, whereas vascular smooth muscle cells are critical for arterial function. All of these events occur during the process of tumor angiogenesis. Tumor vessels can exist either in an immature state (lack of mural cells) or in association with tumor pericytes/vascular smooth muscle cells.
Figure 1.
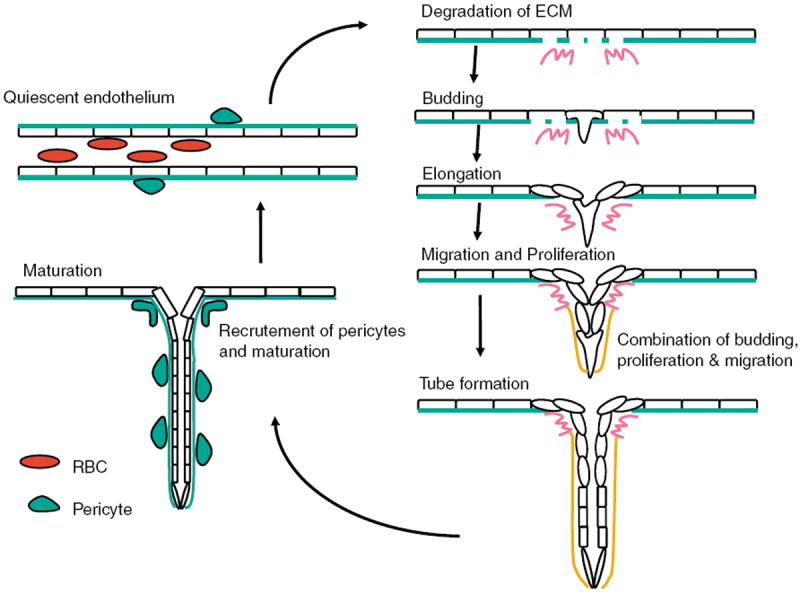
This figure explains angiogenic progression. Progression starts with degradation of extracellular matrix (ECM), and followed by survival and budding of activated endothelial cell, migration and proliferation. By recruiting smooth muscle cell (SMC), such as pericytes, maturation proceeds.
A further discussion of the distinct cell types found in a newly sprouting vessel is merited before a discussion of the role of Notch in this process. During angiogenesis, equipotent endothelial cells must adopt different roles to ensure the creation of a stable vasculature. This fact is typified in sprouts formed during the initiation of angiogenesis. At the angiogenic front endothelial ‘sprouts’ form from preexisting vessels. In these sprouts endothelial cells ‘differentiate’ into three distinct phenotypes. One is a tip cell that resides at the very end of the growing sprout (Gerhardt et al., 2003). The endothelial tip cell takes on a nonproliferative, highly motile, tubeless phenotype. This phenotype is restricted to the tip cell. There is only one endothelial tip cell per sprout. Directly behind the endothelial tip cell are endothelial cells that adopt another phenotype. These cells are highly proliferative, motile and vacuolated. Further from the tip cell these proliferating endothelial cells develop into endothelial tube cells. Endothelial tube cells are the lumen-containing, nonproliferating, immobile cells that make up the blood-carrying vasculature. The tube cell phenotype is stable and eventually all the endothelial cells in a persistent sprout take on this phenotype.
We know that the Notch pathway is important in determining how endothelial cells decide both spatially and temporally when to adopt tip or tube phenotypes (Hellstrom et al., 2007; Lobov et al., 2007; Siekmann and Lawson, 2007; Suchting et al., 2007). It has been shown that the endothelial tip cell expresses high levels of the Notch ligand Dll4 (Claxton and Fruttiger, 2004). The Dll4-expressing endothelial tip cell activates Notch activity on the adjacent proliferating endothelial cell. In the context of the tip-cell/tube-cell model presented in Figure 2, Dll4 is involved in the endothelial ‘tip cell’, where it is highly expressed. The role of Dll4 in the tip cell is well understood in the retina where Notch signaling functions in modeling of the retinal vascular plexus. Disruption of Dll4 or endothelium-specific loss of Notch1 increases the superficial plexus vascular density and causes an excess of angiogenic sprouts (Hellstrom et al., 2007; Lobov et al., 2007; Suchting et al., 2007). This loss of Notch signaling is associated with an increase in endothelial vascular endothelial growth factor receptor (VEGFR)-2 expression (Suchting et al., 2007). The current model for Dll4/Notch role in angiogenesis proposes that the leading ‘tip cell’ expresses Dll4 and has little Notch activity (Figure 2). Dll4 signals through Notch1 in the adjoining stalk cell to initiate a vascular endothelial growth factor (VEGF) feedback loop, limiting sprouting by downregulation of VEGFR-2. Inhibition or mutation of Notch activity causes excess tip cells and possibly alters stalk cell proliferation. Thus, Dll4 is functioning in an antiangiogenic capacity to restrict new sprouts. It is not fully determined if Notch signaling, downstream of Dll4 has select proangiogenic functions, but this concept will be considered later.
Figure 2.

This figure schematizes where Dll4 and Notch are expressed during steps of sprouting angiogenesis.
Dll4 and Notch in tumor angiogenesis
The roles of Dll4 defined by analysis of retinal vasculature have been validated through the analysis of Dll4 in tumor angiogenesis. Notch signaling components are expressed in tumor endothelial cells but the most notable in this class is Dll4. The Notch ligand Dll4 is robustly expressed in tumor endothelial cells of both murine and human tumors, especially in comparison to the expression of Dll4 in neighboring, normal tissue vessels (Mailhos et al., 2001). In this setting, Dll4 expression is regulated by VEGF. For instance, VEGF blockade in experimental tumors in mice results in a large reduction in tumor endothelial Dll4 expression (Thurston et al., 2007). In contrast, Dll4 expression in tumors is positively correlated with VEGF expression levels (Patel et al., 2005, 2006). One theory proposes that the regulation of Dll4 expression is found in settings where VEGF levels are very high; the endothelial tip cell or the endothelium growing into the center of a hypoxic tumor.
Experimental intervention of Dll4 activity in tumor models has been reported by several different groups seeking to test the hypothesis that Dll4 is involved in tumor angiogenesis. When mice were treated with Dll4 or Notch inhibitors the results were somewhat paradoxical but, on further examination, could be explained when one considers the role of Dll4 described in retinal angiogenesis. Dll4 inhibition caused overgrowth of the tumor vasculature. Despite this observed overgrowth, blockade of Dll4 or Notch inhibited the growth of tumors in a variety of tumor models (Noguera-Troise et al., 2006; Ridgway et al., 2006; Scehnet et al., 2007). The explanation for this growth inhibition comes from analysis of the resultant tumor vasculature. The reduced growth was associated with an increase in tumor vessel density and increased vessel sprouts with numerous interconnecting branches but the tumor vessels were nonfunctional, when tested for the ability to perfuse compounds. That is, although Dll4 blockade stimulated overgrowth of a disorganized tumor vasculature, the resulting tumor vessels could not efficiently deliver blood thus adversely affecting tumor growth. Another piece of evidence to support this concept was the observation that, although tumors shrunk due to Dll4 blockade, tumors were more hypoxic. Thus came the new concept that one could reduce tumor growth by ‘abnormalization’ of tumor vasculature (Thurston et al., 2007). To put it another way, a therapy that results in formation nonfunctional tumor vessels can adversely affect tumor growth, even if this therapy leads to increased tumor endothelium.
Other roles for Notch in tumor angiogenesis: a role for Jagged1?
Mice lacking Jagged1 are embryonic lethal and have severe vascular defects (Xue et al., 1999). In humans, Jagged1 has been linked to congenital heart disease (High and Epstein, 2008). Jagged1 is expressed in the vasculature, as well as in many other tissues. A recent report finds that endothelium-specific deletion of Jagged1 results in embryonic lethality and cardiovascular defects (High et al., 2008). Embryos deficient in endothelial Jagged1 show defects in vascular smooth muscle. In contrast, arterial–venous differentiation appears normal in these embryos. Endothelial Jagged1 mutant embryos are phenotypically distinct from embryos in which Notch signaling is inhibited in endothelium. These data suggest that endothelial Jagged1 may signal to neighboring smooth muscle cells. In this setting, Jagged1 is expressed in one cell type, the endothelial cell, and acts on Notch in a different cell type, vascular smooth muscle cells.
As with the example given above, where endothelial Jagged1 effects vascular smooth muscle cells, another paradigm has been uncovered whereby Jagged1 may signal from cancer cell to tumor endothelium. An extensive analysis of head and neck squamous cell carcinoma (SCC) suggests that upregulation of Jagged1 expression in SCC cells may provide a unique mechanism to control tumor angiogenesis (Zeng et al., 2005). The Jagged1-expressing tumor cells were proposed to promote angiogenesis. The Jagged1 expressed by cancer cells was dependent on activation of the mitogen-activated protein kinase (MAPK) signaling pathway. Thus, MAPK activation led to expression of Jagged1 which in turn could influence tumor neovascularization. Evidence in support of this model includes the fact that knockdown of Jagged1 expression significantly inhibited the proangiogenic effects of the SCC, when assessed in vitro. These results point to the fact that contact-dependent Notch ligand signals provided by tumor cells may be important in endothelial cell differentiation. In support, analysis of Jagged1 expression in human head and neck squamous cell carcinoma (HNSCC) samples suggested that Jagged1 was associated with the level of vasculature in tumors.
Another setting where tumor cell expression of Jagged1 may influence the tumor microenvironment is that of breast cancer. Notch signaling may be activated in greater than 50% of human breast cancers (Pece et al., 2004), implicating its role in tumor development (Stylianou et al., 2006). Moreover, expression of Notch1 and its ligand, Jagged-1, on breast tumor cells has been shown to correlate with poor prognosis in terms of survival (Reedijk et al., 2005, 2007). The role of Jagged1 expressed in breast tumor cells can be diverse, influencing tumor cell growth, tumor angiogenesis and/or the inflammatory response.
A recent report suggests that Jagged1 expressed in breast tumor cells can influence tumor angiogenesis (Funahashi et al., 2008). This report utilized a novel, soluble form of the Notch1 receptor (Notch1 decoy; Funahashi et al., 2008), which functions as a ligand-dependent Notch antagonist, and assessed its effect on angiogenesis. The Notch1 decoy can reduce Notch1 signaling stimulated by the action of at least three distinct Notch ligands; Dll1, Dll4 and Jagged1. Notch1 decoy was also shown to block the activity of Notch4 expressed by endothelial cells. Thus, Notch1 decoy functions as an antagonist of ligand-dependent Notch signaling and may be a ‘pan’ Notch ligand inhibitor. In mice, Notch1 decoy inhibits VEGF-induced angiogenesis in skin, establishing a role for Notch receptor function in this process.
The effects of the Notch1 decoy on tumor angiogenesis was tested using a mouse mammary Mm5MT cells overexpressing fibroblast growth factor (FGF) 4 (Mm5MT-FGF4; Funahashi et al., 2008). This study reports that FGF-expressing Mm5MT cells show a significant increase in Jagged-1 expression by tumor cells when xenografted into syngeneic C3H mice. This pattern resembled the case described above for SCC, where Jagged1 expression was robust on tumor cells. In this case, FGF4 via FGF receptor signaling significantly elevated the expression of tumor cell Jagged1. Treatment of mice bearing Mm5MT-FGF4 xenografts with Notch1 decoy, via expression of the decoy in the tumor cells, resulted in a significant reduction in tumor growth. This establishes that the FGF-driven mammary tumorigenesis is dependent on Notch signal activation, presumably through Jagged1. The Notch1 decoy may thus either block tumor epithelial cells, tumor angiogenesis or both. Interestingly, little effect was seen on Mm5MT-FGF4 tumor growth in vitro as a result of Notch1 decoy expression. The study concludes that the likely scenario entails Jagged1 expression by tumor cells promoting the growth or stability of tumor vessels.
Comparing and contrasting Dll4 and Jagged1 in tumor angiogenesis
Tumor angiogenesis is regulated by multiple signaling pathways; including those responding to VEGF, FGFs, angiopoietins and Notch ligands. Among these, VEGF represents an angiogenic factor involved in virtually all steps of normal and pathological angiogenesis. As such, VEGF represents the prime target for therapeutic intervention in tumors and in fact VEGF inhibitors reduce angiogenesis in preclinical models, and have been clinically validated as cancer therapy (Jain et al., 2006). However, despite the excitement engendered by the success of VEGF inhibitors in preclinical models and in the clinic, different tumor types exhibit widely varying susceptibility to VEGF blockade. Notch ligands have come to represent an alternate or complementary target to VEGF, based on the preclinical studies carried out with Dll4 inhibitors and the Notch1 decoy.
The effects documented for the Notch1 decoy on dermal and tumor angiogenesis differ from those reported for Dll4 blockade in models of tumor angiogenesis. One major difference is the lack of overgrowth of blood vessels in response to Notch1 decoy, in contrast to Dll4 blockade. The activity of the Notch1 decoy was tested in a model of dermal angiogenesis and two models of tumor neovascularization and no major overgrowth of blood vessels was observed. In contrast, in multiple tumor models and in retinas, Dll4 blockade or loss of Dll4 function resulted in overgrowth of endothelium. Thus, the action of the Notch1 decoy appears unique from that of Dll4 blockade.
The differences in angiogenic phenotype observed when Dll4 activity is specifically blocked as compared to the activity of the Notch1 decoy suggest that tumor-specific patterns of Notch ligand–receptor interaction may fine-tune vessel assembly. It is worth considering how the different ligands may act in this context and we will focus on difference between Dll4 and Jagged1.
Dll4 is an endothelial ligand that is thought to act on endothelially expressed Notch. In current models, Dll4-expressing endothelial cells have little apparent Notch signaling. In contrast, the neighboring endothelial cell expresses Notch and the cell has Notch signal activation. The net result being a restriction of new sprouts in the Notch activated endothelial cell, via downregulation of VEGFR-2. In tumors, Dll4 activity is thought to function similarly as described above. Dll4 activates Notch in neighboring endothelial cells and restricts new tumor vessel sprout formation. This may be a vestige of the feedback loop that Notch engages in during normal sprouting but which is also relevant to pathological vessel sprouting.
In contrast, endothelium-specific Jagged1 ablation does not appear to function in a similar capacity, at least not during early embryonic development. Endothelially expressed Jagged1 signals to neighboring vascular smooth muscle cells to promote differentiation. In tumors, a similar distinction of ligand-receptor cell types may occur. Tumor endothelial cells that express Jagged1 may be involved in recruitment, maintenance or differentiation of tumor-associated vascular smooth muscle cells or pericytes. As noted, tumor cells themselves can also express Jagged1; for instance in HNSCC and breast cancer. Jagged1 expressed by tumor cells may engage Notch in neighboring tumor cells, tumor endothelium, stroma or inflammatory cells of the tumor. Two published reports suggest that tumor cells expressing Jagged1 can act in a proangiogenic manner. It is not clear what specific cellular events in angiogenesis are stimulated by Jagged1 activity in these models. Interference with Jagged1 via the Notch1 decoy may disrupt this proangiogenic activity thus blocking tumor growth by blocking tumor angiogenesis. The Notch1 decoy also has the potential to interfere with Dll1 and Dll4 activities (Funahashi et al., 2008). Dll1 is less studied in the vasculature but has been reported to function in pathological angiogenesis (Takeshita et al., 2007). It will be interesting to determine the consequences of select inhibition of ligands such as Jagged1 and Dll1 and to compare their roles in tumor neovascularization, with that documented for Dll4. In addition, tumor vessels likely express multiple Notch ligands and the ability to intervene therapeutically may depend on understanding which Notch ligands are expressed in tumor vessels. With this knowledge in hand and further delineation of ligand-specific effects on angiogenesis, one may better be able to predict the effects of diverse Notch signaling inhibitors on tumor angiogenesis and tumor growth. Clearly, this is an important area for investigation for researchers interested in the complexities of the tumor microenvironment or for those interested in the potential for treating tumors with anti- or proangiogenic factors.
References
- Alva JA, Iruela-Arispe ML. Notch signaling in vascular morphogenesis. Curr Opin Hematol. 2004;11:278–283. doi: 10.1097/01.moh.0000130309.44976.ad. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, et al. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci USA. 2005;102:9884–9889. doi: 10.1073/pnas.0504391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S, Fruttiger M. Periodic Delta-like 4 expression in developing retinal arteries. Gene Expr Patterns. 2004;5:123–127. doi: 10.1016/j.modgep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68:4727–4735. doi: 10.1158/0008-5472.CAN-07-6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I. LIN-12/Notch signaling: lessons from worms and flies. Genes Dev. 1998;12:1751–1762. doi: 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Maeno T, Oike Y, Yamazaki M, Doi H, Arai M, et al. Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun. 2006;341:708–714. doi: 10.1016/j.bbrc.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM. Arteries and veins: making a difference with zebrafish. Nat Rev Genet. 2002;3:674–682. doi: 10.1038/nrg888. [DOI] [PubMed] [Google Scholar]
- Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailhos C, Modlich U, Lewis J, Harris A, Bicknell R, Ish-Horowicz D. Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation. 2001;69:135–144. doi: 10.1046/j.1432-0436.2001.690207.x. [DOI] [PubMed] [Google Scholar]
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- Patel NS, Dobbie MS, Rochester M, Steers G, Poulsom R, Le Monnier K, et al. Up-regulation of endothelial delta-like 4 expression correlates with vessel maturation in bladder cancer. Clin Cancer Res. 2006;12:4836–4844. doi: 10.1158/1078-0432.CCR-06-0285. [DOI] [PubMed] [Google Scholar]
- Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005;65:8690–8697. doi: 10.1158/0008-5472.CAN-05-1208. [DOI] [PubMed] [Google Scholar]
- Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, et al. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9805-3. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–1087. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood. 2007;109:4753–4760. doi: 10.1182/blood-2006-12-063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawber CJ, Das I, Francisco E, Kitajewski J. Notch signaling in primary endothelial cells. Ann NY Acad Sci. 2003;995:162–170. doi: 10.1111/j.1749-6632.2003.tb03219.x. [DOI] [PubMed] [Google Scholar]
- Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445:781–784. doi: 10.1038/nature05577. [DOI] [PubMed] [Google Scholar]
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- Suchting S, Freitas C, Ie Noble F, Benedito R, Breant C, Duarte A, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita K, Satoh M, Li M, Silver M, Limbourg FP, Mukai Y, et al. Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res. 2007;100:70–78. doi: 10.1161/01.RES.0000254788.47304.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci USA. 2001;98:5643–5648. doi: 10.1073/pnas.091584598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108:161–164. doi: 10.1016/s0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8:13–23. doi: 10.1016/j.ccr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zhong TP, Childs S, Leu JP, Fishman MC. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414:216–220. doi: 10.1038/35102599. [DOI] [PubMed] [Google Scholar]
- Zhong TP, Rosenberg M, Mohideen MA, Weinstein B, Fishman MC. gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science. 2000;287:1820–1824. doi: 10.1126/science.287.5459.1820. [DOI] [PubMed] [Google Scholar]