

Abstract
Rationale
Loss of insulin action on the endothelium can cause endothelial dysfunction and atherosclerosis. Hyperglycemia and elevated fatty acids induced by diabetes can activate protein kinase C (PKC) β isoforms and selectively inhibit insulin signaling via phosphatidylinositol 3-kinase (PI3K)/Akt pathway to inhibit the activation of endothelial nitric oxide synthase (eNOS) and metabolic actions.
Objective
To demonstrate that overexpressing PKCβ2 isoform in endothelial cells can cause selective insulin resistance and exacerbate atherosclerosis in the aorta.
Methods and Results
PKCβ2 isoform was overexpressed in endothelial cells using a promoter of vascular endothelial cell-cadherin (VE-Cadherin). These mice were cross-bred with ApoE-/- mice (Tg (Prkcb)ApoE-/-). On a Western diet, Tg(Prkcb)ApoE-/- and ApoE-/- mice did not differ in systemic insulin sensitivity, glucose tolerance, plasma lipid or blood pressure. Insulin action in endothelial cells and femoral artery from Tg(Prkcb)ApoE-/- mice were impaired by ∼40% with respect to Akt/eNOS activation and leukocyte-endothelial cell binding increased in cultured lung endothelial cells from Tg(Prkcb)ApoE-/-mice compared to ApoE-/- mice. Basal and angiotensin stimulated big endothelin-1 (ET-1) levels were elevated in Tg(Prkcb)ApoE-/- mice compared to ApoE-/- mice. The severity of atherosclerosis in the aorta from Tg(Prkcb)ApoE-/- mice increased by ∼70% as measured by en face fat staining and plaque content of the number of smooth muscle cells, macrophages and extracellular matrix.
Conclusions
Specific PKCβ2 activation in the endothelial cells caused dysfunction and accelerated atherosclerosis due to loss of insulin-stimulated Akt/eNOS activation and angiotensin induced increases in ET-1 expression.
Keywords: Endothelium, endothelial dysfunction, protein kinase C, insulin resistance, atherosclerosis
Introduction
Systemic metabolic abnormalities such as hyperlipemia, hyperglycemia and hypertension are important risk factors for cardiovascular disease (CVD) associated with diabetes and insulin resistant states1, 2. Clinical studies such as DCCT/EDIC showed that intensive glycemic control lowers the risk for CVD in type 1 diabetic patients but other studies such as ACCORD and ADVANCE suggested that its modifying effects are not clear for type 2 diabetic patients3-5, indicating that other factors could be as important as glycemic control. One such metabolic or hormonal factor is insulin resistance on the vascular wall, which can cause endothelial dysfunction and contribute to the development of CVD complications in type 2 diabetes1. The importance of insulin action on atherosclerosis was clearly established recently by findings that mice with double knockout of apolipoprotein E (ApoE) and insulin receptor on endothelial cells developed atherosclerosis at 2-3 times greater severity than ApoE-/- mice6. The mechanism of insulin's anti-atherogenic actions on the vascular endothelium is predominantly mediated via the IRS/PI3K/Akt pathway, which has been shown to activate endothelial nitric oxide synthase (eNOS)7, 8 and downregulate vascular cell adhesion molecule-1 (VCAM-1)6. Expression of eNOS and VCAM-1 can play an important role in the activation of endothelial cells9, 10, which is a key initial step for atherosclerosis. Nitric oxide produced by eNOS activation is the most important endothelium-derived relaxing factor, and protects the artery from atherosclerosis by multiple mechanisms11. VCAM-1 has an important role in regulating the binding of leukocytes to endothelial cells, and its reduction can attenuate the development of atherosclerosis. In insulin resistant states such as obesity and type 2 diabetes, selective inhibition of the IRS/PI3K/Akt pathway, but not MAP kinase cascade, is probably due to abnormal elevation of metabolites such as free fatty acids and glucose12. One mechanism causing the selective inhibition of insulin action is likely due to protein kinase C (PKC) activation, which has been shown to be a consistent feature in the endothelium in diabetes or insulin resistance13. PKC activation, especially the PKCβ isoform, has been reported to selectively inhibit insulin activation of Akt and eNOS in endothelial cells of diabetic animals and patients by decreasing the phosphorylation of Akt at Ser473 and eNOS at Ser117914, 15. We have characterized the effects of PKC activation on multiple steps of the insulin signaling cascade from the receptor to Akt phosphorylation in endothelial cells, and have identified a novel phosphorylation site on p85 subunit of PI3K (threonine 86), which is partially responsive to PKC activation and blunts insulin activation of Akt/eNOS in endothelial cells16. In addition, mice with whole body PKCβ knockout exhibited decreased severity of atherosclerosis further supporting its role in the atherosclerotic process17. However, whether PKCβ activation in the endothelium can inhibit insulin action and accelerate atherosclerosis has not been reported. In this study, we have characterized the effects of overexpressing PKCβ2 isoform selectively to the endothelium on insulin signaling and action in the arteries and on the development of atherosclerosis.
Methods
Animals
Mice with endothelial specific overexpression of PKCβ2 isoform were generated as previously described18. The transgene was injected into the embryos of pure C57/BL6J mice and the background of the transgenic mice is C57/BL6J. This was crossed with ApoE-/- mice which were purchased from the Jackson Lab (Bar Harbor, Maine) also on C57/BL6J background. The following primers were used for PKCβ2 isoform genotyping: 5′-ATGGATTACAAG GATGACGACGATAAG-3′ and 5′-AGTGGCTGCAGAAGGTGGGCTGCT-3′. Male mice were fed a Western diet (Harland, 42% calories from fat, containing 0.15% cholesterol) starting at 6 weeks of age and continued for 12 weeks. All protocols for animal use and euthanasia were reviewed and approved by the Animal Care Committee of the Joslin Diabetes Center. They are in accordance with NIH guidelines following the standards established by the Animal Welfare Acts and by the documents entitled “Principles for Use of Animals” and “Guide for the Care and Use of Laboratory Animals”.
Endothelial cell culture, glucose tolerance tests, insulin tolerance tests, quantification of atherosclerotic lesion size in aorta, immunohistochemistry, collagen staining, vascular reactivity studies, western blotting, real-time PCR analysis, leukocyte endothelial cell adhesion, Angiotensin II infusion, measurement of Big ET-1 and ET-1 were performed as described in supplemental methods.
Statistical analysis
Comparisons of the two groups were made using paired or unpaired t-test, as appropriate. Comparison among more than two groups was performed by one-way ANOVA followed by the post hoc analysis with paired or unpaired t test to evaluate statistical significance between the two groups. Statistical significance was defined as p<0.05. In text and graphs, data are presented as the mean ± standard deviation.
Results
Insulin signaling in the endothelium
Endothelial targeted PKCβ2 transgenic/ApoE-/- (Tg(Prkcb)ApoE-/-) mice were generated by crossbreeding endothelial specific PKCβ2 transgenic mice (Tg(Prkcb)) with ApoE-/- mice. Levels of PKCβ2 mRNA increased by 32 ± 4-fold (p<0.01, data not shown) and PKCβ2 protein increased by 2-fold (Figures 1A, 1B) in cultured lung endothelial cells from Tg(Prkcb)ApoE-/- mice compared to that from ApoE-/- mice. Expressions of PKCβ2 mRNA levels in macrophages and smooth muscle cells were not different between ApoE-/- and Tg(Prkcb)ApoE-/- mice (data not shown). We have reported that the loss of insulin receptors on endothelial cells exacerbated atherosclerosis, which correlated to decreased phosphorylation of eNOS and increased expression of VCAM-16. Insulin receptor tyrosine phosphorylation after insulin stimulation was not different between two groups of mice (Supplemental Figures IA and B). Insulin induced Akt/eNOS activation was determined in primary cultured endothelial cells from ApoE-/- and Tg(Prkcb)ApoE-/- mice. Insulin induced Akt Ser473 phosphorylation (p-Akt) by 9.4 ± 0.9-fold (Figures 1C, 1E) and eNOS Ser1176 (mouse site, equivalents of human Ser1177 and bovine Ser1179) phosphorylation (p-eNOS) by 4.3 ± 0.8-fold (Figures 1C, 1D) in ApoE-/- mice. However, insulin-stimulated p-Akt was attenuated by 42% (p<0.01) and eNOS Ser1176 phosphorylation was reduced by 39% (p<0.01) in Tg(Prkcb)ApoE-/- mice compared to ApoE-/-mice (Figures 1C, 1D, 1E). Insulin signaling was also studied in vivo by intravenous infusion of insulin and p-eNOS Ser1176 was measured in the femoral artery. Insulin increased p-eNOS Ser1176 by 1.7 ± 0.5-fold in the femoral artery of ApoE-/- mice (Figures 1F, 1G). The basal level of p-eNOS Ser1176 was 49% lower in the femoral artery of Tg(Prkcb)ApoE-/- mice than that of ApoE-/- mice (p<0.05, Figures 1F, 1G). Insulin's effect on p-eNOS Ser1176 was impaired by 25% in Tg(Prkcb)ApoE-/- mice compared to that in ApoE-/- mice (p<0.05, Figures 1F, 1G). We reported that PKC activation phosphorylated threonine 86 of P85α and blunted insulin Akt/eNOS signaling16. Activation of PKC by PMA increased P85 phosphorylation in primary cultured endothelial cells of both ApoE-/- mice and Tg(Prkcb)ApoE-/- mice. The P85 phosphorylation increased by 1.59 ± 0.07-fold in endothelial cells from Tg(Prkcb)ApoE-/- mice compared to that from ApoE-/- mice (Figures 1H, 1I, p<0.05). Insulin-induced eNOS phosphorylation also was measured in aortic endothelial cells and observed to be increased by 1.86 ± 0.80-fold in ApoE-/- mice (p<0.05). However, insulin-induced eNOS phosphorylation was inhibited in Tg(Prkcb)ApoE-/- mice (Supplemental Figures IIIA and B).
Figure 1. PKCβ2 expression and its effects on insulin induced Akt/eNOS phosphorylation and P85α phosphorylation.
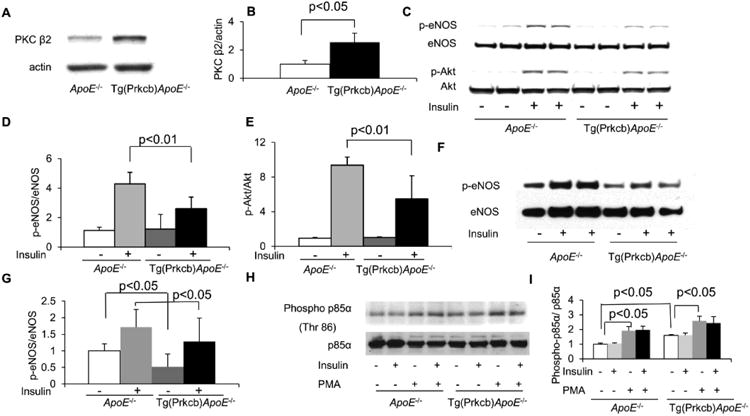
(A-B), PKCβ2 protein in cultured mouse lung endothelial cells (MLEC) from ApoE-/- or Tg(Prkcb)ApoE-/- mice. PKCβ2 protein was determined by western blotting and normalized by actin. (C-E), Phosphorylation of Akt and eNOS was stimulated by 100 nM insulin in MLECs from ApoE-/-or Tg(Prkcb)ApoE-/- mice. C. Representative western blots show Akt Ser473 and eNOS Ser1176 phosphorylation. (D-E), mean values of eNOS Ser1176 (D, ApoE-/- n=3, ApoE-/- + insulin, n=5, Tg(Prkcb)ApoE-/- n=3, Tg(Prkcb)ApoE-/- + insulin n=6) and E, Akt Ser473 phosphorylation in cultured lung endothelial cells from ApoE-/- or Tg(Prkcb)ApoE-/- mice (ApoE-/- n=4, ApoE-/- + insulin, n=7, Tg(Prkcb)ApoE-/- n=4, Tg(Prkcb)ApoE-/- + insulin n=7). (F-G), The phosphorylation of eNOS stimulated by insulin in femoral artery of ApoE-/- or Tg(Prkcb)ApoE-/- mice. Insulin (10mIU/g body weight) was injected and femoral artery was removed after 5 min. Western blotting was performed on femoral artery lysate. F, Representative western blots show eNOS Ser1176 phosphorylation. G, Quantitative analysis of eNOS Ser1176 phosphorylation in femoral artery (ApoE-/- n=6, ApoE-/- + insulin, n=6, Tg(Prkcb)ApoE-/- n=6, Tg(Prkcb)ApoE-/- + insulin n=7). (H-I), Phosphorylation of threonine 86 of P85α in MLECs. H, Representative western blots show Thr86 phosphorylation of P85 α. I, Quantitative analysis of Thr 86 phosphorylation P85α (n=3 for each group).
Insulin-induced ERK activation was also studied since it may play a role in promoting the development of atherosclerosis19. The basal ERK phosphorylation was increased by 1.82 ± 0.90-fold in endothelial cells from Tg(Prkcb)ApoE-/- mice (p<0.05) compared to ApoE-/- mice. Insulin increased ERK phosphorylation by 1.26 ± 0.2-fold in Tg(Prkcb)ApoE-/- mice, but not in ApoE-/- mice (Supplemental Figures IC and D).
Apoptosis in endothelial cells
Apoptosis induced by withdrawing growth factors was determined in endothelial cells from both groups of mice. Withdrawing growth factors increased DNA fragmentation in both groups of mice, but it was lower in endothelial cells from Tg(Prkcb)ApoE-/- mice than from ApoE-/- mice (Supplemental Figure IE).
Systemic metabolic and vascular functional parameters
All mice were fed high fat (42%) diet chow. The body weight did not differ between ApoE-/- or Tg(Prkcb)ApoE-/- mice after 12 weeks of high fat feeding (30.7 ± 5.5g vs 28.5 ± 4.6g, Supplemental Figure IIA). Blood pressure, as measured by tail vein plethysmography, was similar for systolic blood pressure (112 ± 6mmhg vs 121 ± 10mmhg) and diastolic blood pressure (94 ± 6mmhg vs 96 ± 6mmhg) between ApoE-/- and Tg(Prkcb)ApoE-/- mice (Supplemental Figure IIB). Levels of total cholesterol (462.5 ± 161.6mg/dl vs 406.8 ± 125.2mg/dl) and triglycerides (151.0 ± 82.2mg/dl vs 116.4 ± 47.4mg/dl) in plasma were not different after high fat feeding (Supplemental Figures IIC and D). The lipoprotein profile of cholesterol was also characterized by fast protein liquid chromatography (FPLC) to assess VLDL, LDL and HDL properties, which did not differ between the two groups of animals (Supplemental Figure IIE). Blood glucose levels during intraperitoneal glucose tolerance and insulin tolerance tests were similar between ApoE-/- and Tg(Prkcb)ApoE-/- mice (Supplemental Figures IIF and G). Therefore, overexpression of PKCβ2 in the endothelium did not change whole body glucose tolerance, systemic insulin sensitivity, plasma lipids, or blood pressure.
Arterial responses to acetylcholine (ACh) and sodium nitroprusside (SNP)
Functionally, pressure myograph was used to determine whether impaired eNOS phosphorylation in Tg(Prkcb)ApoE-/- mice affected arterial dilation. When arterial dilation induced by ACh was normalized by the effect of 10μM SNP, responses to multiple concentrations of ACh were impaired in Tg(Prkcb)ApoE-/- mice compared to that in ApoE-/- mice (Figure 2A). Arterial dilation induced by 0.3μM ACh was 97.4 ± 4.4% of that induced by SNP in ApoE-/- mice as compared to 84.9 ± 7.1% in Tg(Prkcb)ApoE-/- mice (p<0.05). At 1μM ACh, vasodilation was 100.6 ± 4.6% of dilation induced by SNP in ApoE-/- mice compared to 89.9 ± 6.0% in Tg(Prkcb)ApoE-/- mice (p<0.05). ACh at 3μM induced 102.3 ± 5.5% of dilation as SNP in ApoE-/- mice and was only 91.0 ± 4.8% in Tg(Prkcb)ApoE-/- mice (p<0.05). Even at 10μM ACh, the degree of vasodilation was 103.9 ± 4.1% of dilation induced by SNP in ApoE-/- mice as compared to 91.2 ± 5.2% in Tg(Prkcb)ApoE-/- mice (p<0.05). The EC50 of ACh was not different between both groups of mice (ApoE-/-vs Tg(Prkcb)ApoE-/- mice: 18.3 ± 5.7nM vs 18.3 ± 1.2nM). To study the responses of arterial smooth muscle cells directly to NO, the actions of SNP were characterized. Vasorelaxation to SNP at 10nM (73.2 ± 14.4% vs 51.3 ± 2.8%, p<0.05) and 30nM (87.2 ± 5.1% vs 73.3 ± 0.8%, p<0.01) were increased in arteries from Tg(Prkcb)ApoE-/- mice as compared to that from ApoE-/- mice, respectively (Figure 2B). However, responses to SNP at concentrations from 30nM to 10μM were not different between ApoE-/- mice and Tg(Prkcb)ApoE-/- mice.
Figure 2. Artery relaxation induced by acetylcholine (ACh) or sodium nitroprusside (SNP).
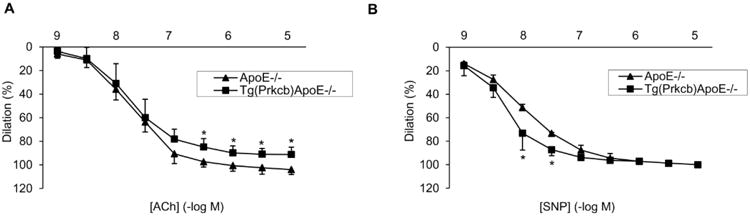
A, ACh induced common carotid artery relaxation. (ApoE-/- n=4, Tg(Prkcb)ApoE-/- n=5, * P<0.05 vs ApoE-/-). B, SNP induced common carotid artery relaxation (ApoE-/- n=4, Tg(Prkcb)ApoE-/- n=5, * P<0.05 vs ApoE-/-).
Expression of VCAM-1 and leukocyte-endothelial cell adhesion
Since insulin can inhibit VCAM-1 expression in the endothelium via the IRS/Akt pathway6, the basal levels of VCAM-1 mRNA were characterized and found to be increased by 3.5 ± 2.4-fold (p<0.01) in cultured lung endothelial cells from Tg(Prkcb)ApoE-/- mice as compared to ApoE-/- mice (Figure 3A). The addition of PMA, an activator of PKC, increased VCAM-1 mRNA by 4 ± 2.6-fold and 14 ± 4.5-fold in cultured lung endothelial cells from ApoE-/- mice and Tg(Prkcb)ApoE-/- mice, respectively (Figure 3A). However, protein expression of VCAM-1 was increased by 1.8 ± 0.6-fold in cultured lung endothelial cells from Tg(Prkcb)ApoE-/- mice vs ApoE-/- mice at basal conditions (Figure 3B and 3C). Insulin inhibited VCAM-1 protein by 44 ± 5% in cells from ApoE-/- mice (Figure 3B and 3C). In contrast, the addition of insulin did not decrease VCAM-1 expression in endothelial cells from Tg(Prkcb)ApoE-/- mice (Figure 3B). Functionally, attachments of fluorescent labeled RAW 264.7 macrophages to endothelial monolayer were determined by spectrometer. PMA increased leukocyte-endothelial cell binding by 2.1 ± 0.2-fold compared to vehicle in cultured lung endothelial cells from ApoE-/- mice (Figure 3D). Pre-incubation of cells with insulin inhibited PMA-induced leukocyte-endothelial cell binding by 36% (1.4 ± 0.1-fold vs 2.1 ± 0.2-fold, p<0.01) (Figure 3D). Leukocyte-endothelial cell binding was increased by 2.0 ± 0.1-fold (p<0.01) in cultured lung endothelial cells from Tg(Prkcb)ApoE-/- mice compared to that from ApoE-/-mice (Figure 3D). However, insulin was unable to decrease PMA-induced leukocyte-endothelial cell binding in cells from Tg(Prkcb)ApoE-/- mice (Figure 3D). The increase of VCAM-1 protein after PMA stimulation was comparable to PMA's effect on leukocyte binding to endothelium. Similarly, insulin decreased PMA-induced VCAM-1 protein, which is comparable to leukocyte binding to endothelium (Figures 3E, 3F). Basal VCAM-1 protein expression increased in Tg(Prkcb)ApoE-/- mice. PMA increased VCAM-1 protein expression by 1.48 ± 0.26-fold in ApoE-/- mice, which was inhibited by insulin in endothelial cells from ApoE-/- mice. However, the inhibitory effect of insulin on PMA-induced VCAM-1 expression was impaired in endothelial cells from Tg(Prkcb)ApoE-/- mice (Figures 3E, 3F). Basal VCAM-1 protein expressions were also measured in aortic endothelial cells and found to be 2.28 ± 0.47-fold higher in Tg(Prkcb)ApoE-/- mice than ApoE-/- mice (P<0.05, Supplemental Figures IIIC and D).
Figure 3. Expression of VCAM-1 and binding of leukocyte-endothelial cells.
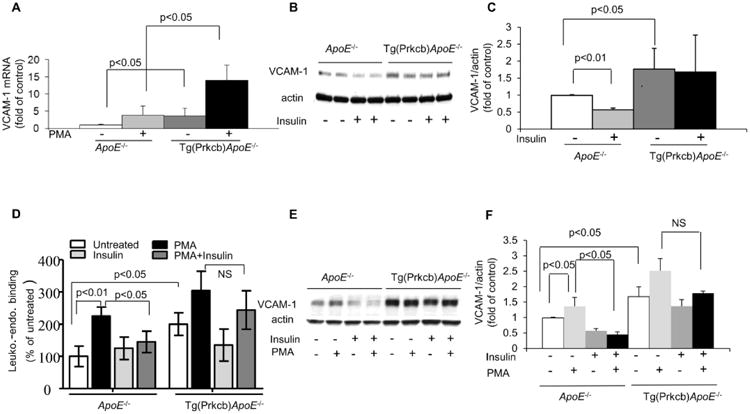
A, VCAM-1 mRNA expression in cultured lung endothelial cells. Lung endothelial cells were treated with or without 100nM PMA. VCAM-1 mRNA was determined by real time PCR and normalized by 36B4 (ApoE-/- n=8, ApoE-/- +PMA, n=3, Tg(Prkcb)ApoE-/- n=9, Tg(Prkcb)ApoE-/- + PMA n=3). (B-C), VCAM-1 protein expression in cultured lung endothelial cells. Lung endothelial cells were treated with or without 100nM insulin. VCAM-1 protein was determined by western blotting and normalized by actin. B indicates representative western blots and C shows mean value of the ratio of VCAM-1 to actin (n=4 for each group). D, Leukocyte-endothelial binding. The binding of RAW 264.7 macrophages to monolayer lung endothelial cells from ApoE-/- or Tg(Prkcb)ApoE-/- mice was measured (n=3 for each group). (E-F), VCAM-1 protein expression in endothelial cells. Endothelial cells were treated with or without 100nM insulin or 100nM PMA. VCAM-1 protein was determined by western blotting and normalized by actin. E indicates representative western blots and F shows mean value of the ratio of VCAM-1 to actin (n=3 for each group).
Angiotensin-II (AngII) actions and endothelin-1(ET-1) expression
PKCβ isoform activation can also cause insulin independent effects. We studied the actions of AngII on the expression of PDGF and ET-1 in the aorta since the actions of these cytokines have been shown to be increased in the vascular wall by diabetes or insulin resistant states20-23. Basal ET-1 mRNA expressions in the aorta of ApoE-/- mice and Tg(Prkcb)ApoE-/- mice were not different. AngII infusion increased ET-1 mRNA levels by 1.9 ± 0.4-fold compared to vehicle in the aorta of ApoE-/- mice (Figure 4A). Interestingly, AngII's actions on ET-1 mRNA expression were greater in the aorta of Tg(Prkcb)ApoE-/- mice compared to that in ApoE-/- mice by 57% (3.0 ± 1.0-fold vs 1.9 ± 0.4-fold, p<0.05, respectively (Figure 4A). PDGFα/β mRNA expressions at basal level and after AngII infusion were not different between ApoE-/- mice and Tg(Prkcb)ApoE-/- mice (data not shown). Since the half-life of ET-1 protein is only 40 seconds, it is difficult to detect changes in the aorta. Thus, we measured changes of big ET-1, a precursor of ET-1, which has a half-life of 30 minutes24. The protein levels of big ET-1 in the aorta from Tg(Prkcb)ApoE-/- mice were 2.4-fold higher than that from ApoE-/- mice at basal condition (16.0 ± 3.3ng/mg aorta vs 6.6 ± 0.6ng/mg aorta, p<0.01, respectively (Figure 4B). Compared to vehicle, AngII infusion also increased big ET-1 protein by 1.8-fold in the aorta from ApoE-/- mice (12.1 ± 3.3ng/mg aorta vs 6.6 ± 0.6ng/mg aorta, p<0.05 (Figure 4B). In Tg(Prkcb)ApoE-/- mice, AngII infusion increased the expression of big ET-1 protein in the aorta by 85% compared to that from ApoE-/- mice (22.1 ± 3.8ng/mg aorta vs 12.1 ± 3.3ng/mg aorta, p<0.05, respectively (Figure 4B). In order to determine that AngII's effect is specifically increasing ET-1 expression in the endothelial cells, cultured endothelial cells were isolated from ApoE-/- and Tg(Prkcb)ApoE-/- mice and studied. ET-1 mRNA levels were increased after AngII stimulation by 2.1-fold in Tg(Prkcb)ApoE-/-vs ApoE-/- mice (p< 0.05) (Figure 4C). Analysis of media incubated with endothelial cells showed that the levels of ET-1 were also significantly higher by 3.6-fold in endothelial cells from Tg(Prkcb)ApoE-/-vs ApoE-/- mice (Figure 4D). Insulin did not increase ET-1 in endothelial cells from either group of mice (data not shown). Since we have reported that PKCβ upregulated ET-1 expression by activation of ERK25, basal ERK phosphorylation was studied and observed to be increased by 1.82 ± 0.90-fold in cells from Tg(Prkcb)ApoE-/- mice compared to ApoE-/-mice (Figures 4E, 4F).
Figure 4. The effect of Angiotensin-II (AngII) on ET-1 expression in the aorta.
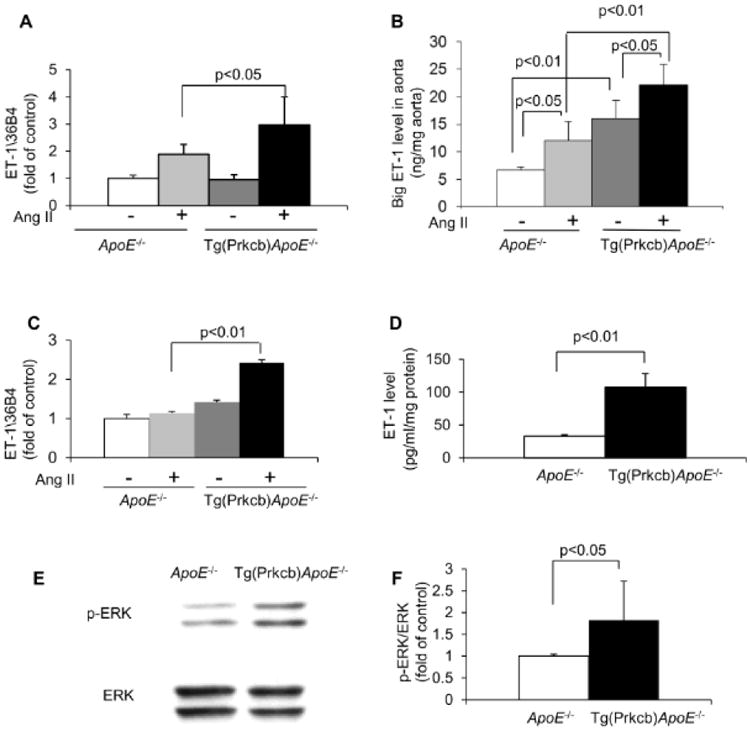
(A-B), AngII or vehicle was infused into ApoE-/- or Tg(Prkcb)ApoE-/- mice for 3 hours. A, ET-1 mRNA was determined by real time PCR (ApoE-/- infused with vehicle, n=3; ApoE-/- infused with AngII, n=5; Tg(Prkcb)ApoE-/- infused with vehicle, n=3; Tg(Prkcb)ApoE-/- infused with AngII, n=7). B. Big ET-1 in aorta was measured by ELISA. (ApoE-/- infused with vehicle, n=3; ApoE-/- mice infused with AngII, n=5; Tg(Prkcb)ApoE-/- infused with vehicle, n=4; Tg(Prkcb)ApoE-/- infused with AngII, n=5). C. Cultured mouse lung endothelial cells were treated with 100nM AngII for 16 hours and ET-1 mRNA were determined by real time PCR and normalized by 36B4. (n=3 for each group). D. ET-1 protein in medium from cultured mouse lung endothelial cells were measured by ELISA and normalized by the total protein extracted from the cells growing in the same well. (n=3 for each group). (E-F), ERK phosphorylation. Phosphorylation of ERK in MLECs. E, Representative western blots show ERK phosphorylation. F, Quantitative analysis of ERK phosphorylation (n=8 for each group).
Evaluation of atherosclerotic lesions in the aorta
In Tg(Prkcb)ApoE-/- mice, the mean area covered by aortic atherosclerotic lesions, as evaluated by en face Sudan IV staining and expressed relative to the whole aortic area, was 1.7-fold higher than that in ApoE-/- mice (p<0.05, Figures 5A, 5B). Atherosclerotic plaques increased by 2.16 ± 01.78-fold in abdominal aorta from Tg(Prkcb)ApoE-/- mice (p<0.05), but was not different in aorta root and aorta arch (Figures 5C-E). Analysis of cross sections of aortic plaque showed smooth muscle cell content in the plaque of Tg(Prkcb)ApoE-/- mice was over two times greater than that in ApoE-/- mice (10% ± 4.8% vs 4.2% ± 2.4%, p<0.05, respectively (Figures 6A, 6B). Absolute macrophage content, measured by MAC2 staining, was increased in Tg(Prkcb)ApoE-/- mice compared to ApoE-/- mice (162657.3 ± 71689.49 vs 68876.76 ± 34641.95, p<0.05 (Figures 6C, 6D). The content of the collagen as measured by aniline blue staining was increased by 2-fold in Tg(Prkcb)ApoE-/- mice compared to ApoE-/- mice (8203.5 ± 3160.9 vs 3899.7 ± 2794.5, p<0.05, respectively (Figures 6E, 6F). Area of necrosis in the plaque was denoted by a lack of staining by trichrome, which indicated the absence of cells and collagen. The necrosis area was 1.99 ± 0.55 times higher in Tg(Prkcb)ApoE-/- mice compared to ApoE-/- mice (11.0 ± 3.1% vs 5.5 ± 2.5%, p<0.05 (Figures 6G, 6H).
Figure 5. En face Sudan IV staining of aorta.
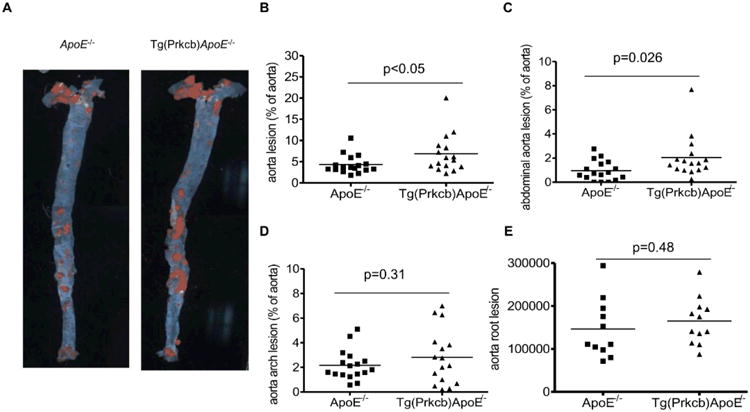
A, En face Sudan IV staining of aorta was performed in ApoE-/- (n=17) and Tg(Prkcb)ApoE-/- (n=16) mice. B, Quantitative analysis of atherosclerotic lesions in aorta (B), abdominal aorta (C), aorta arch (E) and aorta root (F) of ApoE-/- and Tg(Prkcb)ApoE-/- mice after 12 weeks of high fat feeding. Line shows the medium of the values.
Figure 6. The complexity of aortic atherosclerotic plaques.
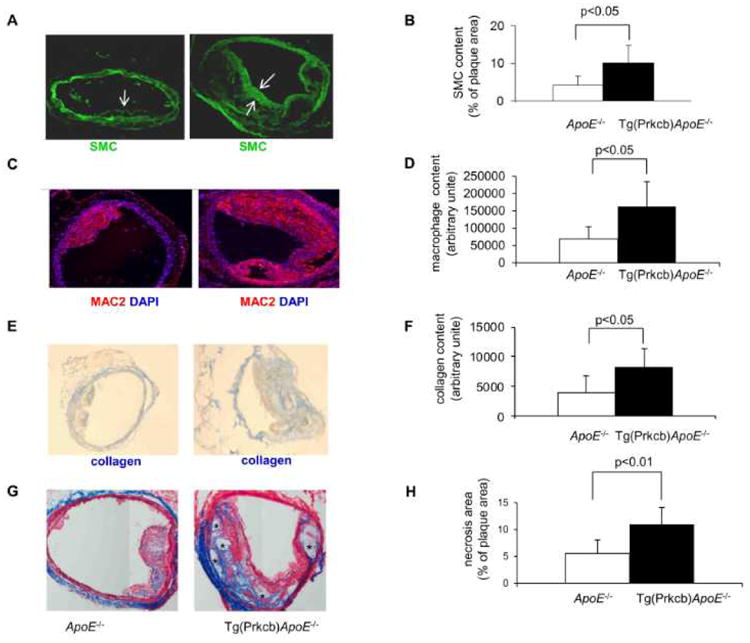
Abdominal aorta was cross-sectioned and complexity of the plaque was analyzed. Panels A–B, The slides were stained for anti-smooth muscle cells specific actin. Panel A shows representative images and Panel B, Shows the quantitative analysis of areas covered by SMC (arrows indicate smooth muscle cells, magnification 100×, n=7 for each group). Panels C-D were stained for anti-MAC2, Panel C shows representative images of macrophage stains and Panel D is the mean values of quantitative analysis (magnification 100×, n=7 for each group). Panels E-F, Collagen staining by aniline blue, Panel E, Shows representative images and Panel F is the means values of quantitative analysis (magnification 100×, n=7 for each group). Panels G-H, Trichrome staining to show necrosis area, Panel G, Shows representative images, * indicated necrosis area. Panel H is the means values of quantitative analysis (magnification 100×, n=7 for each group).
Discussion
In the present study, we have directly demonstrated that overexpression of PKCβ2 in endothelial cells inhibits insulin signaling and insulin action and increases expression of ET-1 resulting in endothelial dysfunction and accelerated atherosclerosis. These findings may help explain the elevated risk for atherosclerosis in diabetic and insulin resistant states since PKC activation, especially the β isoform, has been shown to be induced by hyperglycemia or elevated free fatty acids in many vascular tissues26. The inhibiting role of PKCβ on insulin activation of eNOS has clearly been shown in endothelial cells from diabetic patients15. At the cell signaling level, insulin's vasotropic effects such as the activation of eNOS, induction of HO-1 and VEGF, and downregulation of VCAM-1 are mediated through activation of the IRS/PI3K/Akt pathway6, 27, 28. PKC activation, especially β isoform, has been shown to selectively inhibit this pathway by serine phosphorylation of the p85 subunit of PI3K activation in the endothelial cells. Previous studies on the effects of PKCβ activation and atherosclerosis have used total body PKCβ knockout mice or systemic use of PKCβ selective inhibitor, which showed a reduction of atherosclerosis in ApoE-/- mice17. However, the systemic inhibition of PKCβ isoform cannot attribute its effects to the endothelium specifically since activation of PKCβ isoform by diabetes and its inhibition have been reported to affect many tissues and functions to regulate multiple metabolic pathways29 The present study used mice with overexpression of PKCβ2 targeted specifically to the endothelial cells by use of the VE-cadherin promoter. The Tg(Prkcb)ApoE-/- mice have allowed us to test the idea that PKCβ activation specifically to the endothelial cells can cause vascular selective insulin resistance, endothelial dysfunction, and accelerated atherosclerosis in the absence of systemic insulin resistance and its metabolic changes. Specific enhancement of PKCβ in the endothelium clearly decreased insulin signaling only in the vascular endothelium without affecting systemic insulin action indicating that decreasing insulin action on the endothelium will not contribute to systemic insulin resistance, which has been suggested. We have previously identified threonine 86 of the p85 regulatory subunit of PI3K as a possible target of PKCβ activation in endothelial cells. This phosphorylation of p85 decreased the association of IRS1 to p85/PI3K after insulin stimulation16.
A common characteristic of endothelial dysfunction of diabetes is the impairment of nitric oxide (NO) bioavailability due to either decreased production or increased degradation by oxidants. Our new findings clearly showed that the increase in PKCβ expression in endothelial cells inhibited insulin activation of p-Akt/p-eNOS resulting in decreased NO production. This is supported by the biological effect of the relaxation assay where aorta from Tg(Prkcb)ApoE-/- mice exhibited less response to acetylcholine. Another interesting finding is the arterial response to SNP, which exhibited a hyperactive response in the Tg(Prkcb)ApoE-/- mice. It is possible that this alteration is a compensatory response to lower ambient levels of NO in the Tg(Prkcb)ApoE-/- mice. One direct effect of impaired insulin-stimulated activation of the PI3K/Akt pathway is the increased expression of VCAM-1, which is normally downregulated by insulin via the IRS/Akt/PI3K pathway. Previously, we reported that VCAM-1 expression in endothelial cells or in aorta from mice with insulin receptors specifically deleted from the endothelial cells (EIRAKO mice) had upregulation of VCAM-1 and accelerated atherosclerosis6. The increase in VCAM-1 expression in the Tg(Prkcb)ApoE-/- mice also had biological significance since it is associated with elevated binding of leukocytes. The increase in leukocyte binding to the endothelium was also observed in EIRAKO mice6.
The pathology of the atherosclerotic plaque in the Tg(Prkcb)ApoE-/- mice was different from EIRAKO mice by the increased numbers of SMC cells when compared to ApoE-/- mice. The increase in SMC numbers in atherosclerotic plaques could be due to elevations of proliferation and migration as this is observed in patients with diabetes and insulin resistance30. In fact, some studies have suggested that hyperinsulinemia could be responsible for excessive SMC proliferation31, although our recent studies did not support this idea32. We also explored the possibility that PKCβ activation directly increased ET-1 expression in the endothelial cells and this effect was exacerbated by AngII, whose actions are increased in diabetes22. The results from Tg(Prkcb)ApoE-/- mice suggest that PKCβ activation in the endothelial cells increased ET-1 expression in the aorta and other arteries from Tg(Prkcb)ApoE-/- mice. Increase in PKC activity has been reported to increase ET-1 expression in several cell types possibly via the MAPK pathway23, 25. The additive effect of AngII in vivo is demonstrated by mRNA levels in the aorta of both genes. Big ET-1 was measured instead of ET-1, because the half-life of ET-1 protein is very short once secreted and protein levels of big ET-1 were also increased33. These findings are especially important for understanding the pathogenesis of accelerated atherosclerosis in diabetes and insulin resistant states since the activation of PKCβ induced both endothelial dysfunction and smooth muscle cell activation. The activation of PKC observed in diabetes or metabolic syndrome could be due to multiple mechanisms, including increased synthesis of diacylglycerol (DAG) caused by the metabolism of hyperglycemia and elevated free fatty acids. These results clearly demonstrated that PKCβ isoform activation in the endothelium can directly inhibit insulin signaling via the IRS/PI3K/Akt pathway to decrease eNOS action and reduce NO production, leading to elevation of VCAM-1 and increase monocyte attachment. Our results suggest that the increases in SMC proliferation or migration could be due to elevated expression and action of ET-1 originating from endothelial cells. This could be a direct effect of PKC activation or enhancement of AngII actions on ET-1 expression in endothelial cells. ET-1 can induce MAPK to induce migration or proliferation both of which can increase SMC numbers in atherosclerotic plaques.
The increases in atherosclerosis were observed mainly in the descending aorta and not in the root or arch in the Tg(Prkcb)ApoE-/- mice vs ApoE-/- mice. This is comparable with autopsy studies in diabetic patients which have identified that the extent of atherosclerosis is most significantly increased in the abdominal aorta compared to non-diabetic patients34. In addition, the amount of atherosclerosis in the aortic root is not so common in the clinical setting, whereas the extent of atherosclerosis in the abdominal aorta correlates well clinically with atherosclerosis in the coronary artery. In the rodent model, atherosclerosis appears earlier in the aortic root and arch and is more severe than in other parts of the aorta. This has been attributed to differences in heart rate (550bpm in mice compared to 70bpm in the average human) and flow35. The increase in atherosclerosis in the abdominal aorta in humans is thought to be the result of the turbulent flow. The induction of diabetes or overexpression of PKC in the endothelium may change the flow from laminar to turbulent in the abdominal aorta and increase atherosclerosis.
The collagen content increased in the plaque of Tg(Prkcb)ApoE-/- mice, but they were mainly located around the necrotic core and not on the cap of the lesion. Thus, the findings show an increase of the necrotic cores and the location of the extracellular matrix at the lower part of the plaque near the aventitia, which suggest that the plaques of Tg(Prkcb)ApoE-/- mice were not more stable than that of ApoE mice.
The overexpression of PKCβ in the endothelium may not be the same as activation of endogenous PKCβ in effecting the function in the endothelium and atherosclerosis. Previously, we have shown that angiotensin activation can cause inhibition of IRS/Akt/eNOS activation in bovine aortic endothelial cells and enhance MAP kinase activation16. In addition, it targeted threonine 86 phosphorylation of p85/PI3 kinase as shown in the Tg(Prkcb)ApoE-/- mice (Figures 1H, 1I). Thus, we speculate that even without overexpression of PKCβ, the activation of endogenous PKCβ will also inhibit insulin activation of phospho-Akt and eNOS and the loss of inhibition of VCAM-1.
The change of phospho-eNOS in femoral artery after insulin stimulation was not significantly different between the two groups of mice; however the maximum response to insulin was lower in Tg(Prkcb)ApoE-/- mice compared to the ApoE-/- mice. We believe this difference is important since the data shown in Figure 3 on the expression of VCAM-1 indicated that insulin's ability to inhibit VCAM-1 expression is significantly decreased and the basal level of VCAM-1 expression is increased in the Tg(Prkcb)ApoE-/- mice. Since insulin's inhibitory effect on VCAM-1 expression is due to PI3K/Akt pathway6, this increased expression of VCAM-1 suggests that the maximum effect attained by insulin through the Akt pathway will have functional significance.
Overexpression of PKCβ appears to decrease apoptosis of the endothelial cells. This is not surprising since PKCβ activation is known to enhance MAP kinase (Supplemental Figure ID) to decrease apoptosis and increase proliferation in many types of cells13.
The increase in atherosclerosis exhibited by Tg(Prkcb)ApoE-/- mice is likely due to multiple factors including the inhibition of insulin signaling to activate p-eNOS and lack of inhibition of VCAM-1 expression as shown in Figures 1C, 1 D, 3C and 3F. In addition, it is also likely that PKCβ activation can increase other atherogenic effects independent of its inhibition of insulin's activation of the IRS/Akt/eNOS pathway. This is exhibited by the enhanced basal MAP kinase phosphorylation which is known to effect PKC activation. In addition, the effect of AngII to increase ET-1 expression is also elevated independently of insulin. Multiple studies have shown that angiotensin activation can lead to PKC activation and result in actions including increased expression of ET-1. Since ET-1 expression has been shown to accelerate or exacerbate atherosclerosis, this is likely an insulin signaling independent pathway by which PKCβ activation enhanced atherosclerosis.
In summary, this study provided definitive evidence that PKCβ activation in the endothelium will accelerate atherosclerosis. In particular, an increase in migration or proliferation of SMC across the internal elastic membrane results in increased production of extracellular matrix. There could be multiple mediators of PKCβ activation that accelerate atherosclerosis including selective loss of insulin action on p-Akt/eNOS, increase in VCAM-1 expression, elevation of ET-1 expression and enhancing AngII effects. These results improve our understanding of the molecular mechanisms of atherogenesis and point to inhibition of PKCβ as a potential strategy for preventing atherosclerosis in insulin resistance, type 2 diabetes and other insulin resistant related diseases.
Supplementary Material
Novelty and Significance.
What Is Known?
Insulin has important actions in endothelial cells.
Endothelial insulin receptor and apoE double knockout mice developed more atherosclerosis than apoE-null mice.
PKCβ activated in endothelial cells in diabetes selectively inhibits Akt and eNOS activation by insulin in cultured endothelial cells and caused endothelial dysfunction.
What New Information Does This Article Contribute?
Overexpression of PKCβ2 in endothelial cells inhibits insulin's anti-atherogenic actions, including activation of Akt-eNOS and downregulation of VCAM-1.
Endothelial overexpression of PKCβ2 produced more endothelin-1 at basal level and after angiotensin-II stimulation than wild type mice.
-
Overexpression of PKCβ2 in endothelial cells accelerated the development of atherosclerosis in ApoE-/- mice.
Diabetes and states of insulin resistance accelerate atherogenesis. One risk factor for this acceleration of atherogenesis is insulin resistance in endothelial cells. PKCβ has been found to be activated in the endothelial cells of diabetic patients and has been found to selectively inhibitinsulin-induced Akt and eNOS activation. We examined the effects of overexpressing PKCβ2 isoform selectively to the endothelium on insulin signaling and action in the arteries and on the development of atherosclerosis in mice. We observed that PKCβ2 overexpression in endothelial cells accelerated the development of atherosclerosis by inhibiting insulin's antiatherogenic actions, including activation of Akt-eNOS, downregulation of VCAM-1, and elevation of ET-1 production. These findings suggest that PKCβ2 contributesto diabetic macrovascular complications and that inhibition of PKCβ r may be a promising tr treatment of macrovascular complications in diabetic patients.
Acknowledgments
We are grateful for the expert technical assistance provided by Advanced Microscopy Core, Mouse Physiology Core, Genomic Core and Special Assay Core, all of which are supported by NIH grant 5P30DK036836. Plasma lipid profiles were measured by Dr. Vladimir Babaev at the Lipid, Lipoprotein, and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers with support from NIH grant DK59637.
Sources of Funding: Q. L. was supported by ADA mental fellowship. Q.W. was supported by JDRF fellowship. The study was also supported by NIH R01 grants DK053105 (G.L.K). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Non-Standard Abbreviations and Acronyms
- PKC
protein kinase C
- ET-1
endothelin 1
- eNOS
endothelial nitric oxide synthase
- CVD
cardiovascular disease
- apoE
apolipoprotein E
- VCAM-1
vascular cell adhesion molecule-1
- Ach
acetylcholine
- SNP
sodium nitroprusside
- Ang II
angiotensi II
- NO
nitric oxide
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–85. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Semenkovich CF. Insulin resistance and atherosclerosis. J Clin Invest. 2006;116:1813–22. doi: 10.1172/JCI29024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC, Jr, Probstfield JL, Cushman WC, Ginsberg HN, Bigger JT, Grimm RH, Jr, Byington RP, Rosenberg YD, Friedewald WT. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med. 2011;364:818–28. doi: 10.1056/NEJMoa1006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–53. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 6.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, Clermont A, Geraldes P, Dall'Osso C, Wagers AJ, Huang PL, Rekhter M, Scalia R, Kahn CR, King GL. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–89. doi: 10.1016/j.cmet.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–8. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–45. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
- 9.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–54. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 11.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–57. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–8. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 15.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA, Hamburg NM. Protein Kinase C-beta Contributes to Impaired Endothelial Insulin Signaling in Humans With Diabetes Mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeno Y, Li Q, Park K, Rask-Madsen C, Gao B, Matsumoto M, Liu Y, Wu IH, White MF, Feener EP, King GL. Inhibition of insulin signaling in endothelial cells by protein kinase C-induced phosphorylation of p85 subunit of phosphatidylinositol 3-kinase (PI3K) J Biol Chem. 2012;287:4518–30. doi: 10.1074/jbc.M111.286591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, Yan SF. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. FASEB J. 2009;23:1081–91. doi: 10.1096/fj.08-120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mima A, Hiraoka-Yamomoto J, Li Q, Kitada M, Li C, Geraldes P, Matsumoto M, Mizutani K, Park K, Cahill C, Nishikawa S, Rask-Madsen C, King GL. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCbeta activation in diabetes. Diabetes. 2012;61:2967–79. doi: 10.2337/db11-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord. 2013;14:5–12. doi: 10.1007/s11154-012-9229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berk BC. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev. 2001;81:999–1030. doi: 10.1152/physrev.2001.81.3.999. [DOI] [PubMed] [Google Scholar]
- 21.Lavrentyev EN, Estes AM, Malik KU. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ Res. 2007;101:455–64. doi: 10.1161/CIRCRESAHA.107.151852. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K, Ghatei MA, Lam HC, O'Halloran DJ, Bloom SR. Elevated plasma endothelin in patients with diabetes mellitus. Diabetologia. 1990;33:306–10. doi: 10.1007/BF00403325. [DOI] [PubMed] [Google Scholar]
- 23.Yokota T, Ma RC, Park JY, Isshiki K, Sotiropoulos KB, Rauniyar RK, Bornfeldt KE, King GL. Role of protein kinase C on the expression of platelet-derived growth factor and endothelin-1 in the retina of diabetic rats and cultured retinal capillary pericytes. Diabetes. 2003;52:838–45. doi: 10.2337/diabetes.52.3.838. [DOI] [PubMed] [Google Scholar]
- 24.Hemsen A, Ahlborg G, Ottosson-Seeberger A, Lundberg JM. Metabolism of Big endothelin-1 (1-38) and (22-38) in the human circulation in relation to production of endothelin-1 (1-21) Regul Pept. 1995;55:287–97. doi: 10.1016/0167-0115(94)00119-i. [DOI] [PubMed] [Google Scholar]
- 25.Park JY, Takahara N, Gabriele A, Chou E, Naruse K, Suzuma K, Yamauchi T, Ha SW, Meier M, Rhodes CJ, King GL. Induction of endothelin-1 expression by glucose: an effect of protein kinase C activation. Diabetes. 2000;49:1239–48. doi: 10.2337/diabetes.49.7.1239. [DOI] [PubMed] [Google Scholar]
- 26.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–63. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geraldes P, Yagi K, Ohshiro Y, He Z, Maeno Y, Yamamoto-Hiraoka J, Rask-Madsen C, Chung SW, Perrella MA, King GL. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/phosphoinositide 3-kinase/Akt-2 pathway. J Biol Chem. 2008;283:34327–36. doi: 10.1074/jbc.M807036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP, King GL. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation. 2002;105:373–9. doi: 10.1161/hc0302.102143. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami T, Kawakami Y, Kitaura J. Protein kinase C beta (PKC beta): normal functions and diseases. J Biochem. 2002;132:677–82. doi: 10.1093/oxfordjournals.jbchem.a003273. [DOI] [PubMed] [Google Scholar]
- 30.Askari B, Renard CB, Bornfeldt KE. Regulation of smooth muscle cell accumulation in diabetes-accelerated atherosclerosis. Histol Histopathol. 2002;17:1317–28. doi: 10.14670/HH-17.1317. [DOI] [PubMed] [Google Scholar]
- 31.Stout RW, Bierman EL, Ross R. Effect of insulin on the proliferation of cultured primate arterial smooth muscle cells. Circ Res. 1975;36:319–27. doi: 10.1161/01.res.36.2.319. [DOI] [PubMed] [Google Scholar]
- 32.Rask-Madsen C, Buonomo E, Li Q, Park K, Clermont AC, Yerokun O, Rekhter M, King GL. Hyperinsulinemia does not change atherosclerosis development in apolipoprotein E null mice. Arterioscler Thromb Vasc Biol. 2012;32:1124–31. doi: 10.1161/ATVBAHA.111.239558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sirvio ML, Metsarinne K, Saijonmaa O, Fyhrquist F. Tissue distribution and half-life of 125I-endothelin in the rat: importance of pulmonary clearance. Biochem Biophys Res Commun. 1990;167:1191–5. doi: 10.1016/0006-291x(90)90649-8. [DOI] [PubMed] [Google Scholar]
- 34.General findings of the International Atherosclerosis Project. Lab Invest. 1968;18:498–502. [PubMed] [Google Scholar]
- 35.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.