

Abstract
Cancer immunotherapy aims to generate long-lived, tumor-specific adaptive immunity to limit dysregulated tumor progression and metastasis. The tumor vasculature has emerged as a critical checkpoint controlling the efficacy of immunotherapy since it is the main access point for cytotoxic T cells to reach tumor cell targets. Therapeutic success has been particularly challenging to achieve because of the local, cytokine-rich inflammatory milieu that drives a pro-tumorigenic program supporting the growth and survival of malignant cells. Here, we focus on recent evidence that systemic thermal therapy can switch the activities of the inflammatory cytokine, interleukin-6 (IL-6), to a predominantly anti-tumorigenic function that promotes anti-tumor immunity by mobilizing T cell trafficking in the recalcitrant tumor microenvironment.
Keywords: Systemic thermal therapy, interleukin-6, immunotherapy, lymphocyte trafficking, tumor vasculature
CD8+ T cell anti-tumor immunity: strength in numbers
Recent advances in the field of immunotherapy have galvanized interest in targeting the immune system for the treatment of cancer. The primary appeal of immune-based treatments stems from the ability of cytotoxic CD8+ T lymphocytes, the major effectors of the adaptive immune response, to specifically recognize and kill tumor targets while sparing normal tissue. T cells can also seek out micrometastatic lesions throughout the body and generate long-term protection against tumor recurrence. Compelling evidence for the relevance of T cells in patient outcome was demonstrated in 2006 when landmark studies in colon cancer revealed that the extent of intratumoral infiltration by CD3+ T cells is more predictive of survival than traditional tumor staging based on lesion size and metastatic spread [1]. The prognostic value of T cells has been recapitulated in several types of cancer, emphasizing that extensive patient populations may benefit from improved T cell responses [1–4].
Adding to the complexity of these studies is the diversity of CD3+ T cell subsets that can have an impact on anti-tumor immunity. The CD4+ T helper 1 subset cell (Th1) has mainly been credited with anti-tumorigenic activity by virtue of their ability to stimulate CD8+ T cell activity [5, 6]. Conversely, CD4+ regulatory T cells (Tregs) and T helper 2 (Th2) subsets primarily elicit pro-tumorigenic functions [3, 5, 7]. Immunosuppressive Tregs are associated with poor patient prognosis and have been shown to inhibit activation, proliferation, and cytotoxic function of CD8+ effector cells [3, 5–8]. The CD4+ T helper 17 subset cells (Th17s) remain a wild card as they have been linked with both pro- and anti-tumorigenic activity in different models [3, 5, 7]. The balance of these opposing T cell subsets within patients can strongly influence the efficacy of the anti-tumor immune response, and numerous studies are underway to understand how to shift the equilibrium to a predominantly CD8+ T cell-driven immune response in patients [5, 7, 9, 10].
Equally evocative in the field of tumor immunology was the discovery that the status of the immune system dictates the anti-tumor efficacy of traditional treatment modalities such as chemotherapy and radiation, whose actions are classically attributed to direct tumor cytotoxicity [11–13]. In this regard, depletion of CD8+ T cells abrogated the ability of the chemotherapeutic doxorubicin to control tumor growth in murine breast cancer models [12]. The radiation response in melanoma tumors was similarly impaired in mice lacking T cells [13]. These studies suggest that improving the magnitude of the endogenous T cell response has the potential to enhance the efficacy of widely-used standard treatments in a broad range of patients.
Two challenges faced in generating anti-tumor immunity are the extremely low frequency of T cells specific for a single tumor antigen [3, 4, 10, 14] and tumor-induced suppression of T cell activity [5–7]. One approach to augment the number of tumor-specific T cells in patients is to exploit the natural process of T cell activation by using dendritic cell (DC) vaccines [14] (Figure 1). Systemic administration of patients’ own DCs that are preloaded ex vivo with tumor antigen stimulates endogenous, tumor-specific naïve CD8+ T cells to differentiate into cytotoxic effectors and proliferate, thus enlarging the available tumor-reactive effector T cell pool [14]. Additional methods to boost the frequency of circulating cytotoxic T cells such as T cell-based adoptive cell transfer (ACT) involve bypassing endogenous T cell activation (which may be suppressed in cancer patients [5–7]) by expanding autologous tumor-specific T cells ex vivo and re-infusing them into the bloodstream at high numbers [10] (Figure 1). These regimens increase the frequency of tumor-specific T cells in peripheral blood up to ~50% [10, 14].
Figure 1. T cell-based immunotherapies share a common requirement for trafficking across vascular checkpoints in tumor tissues.
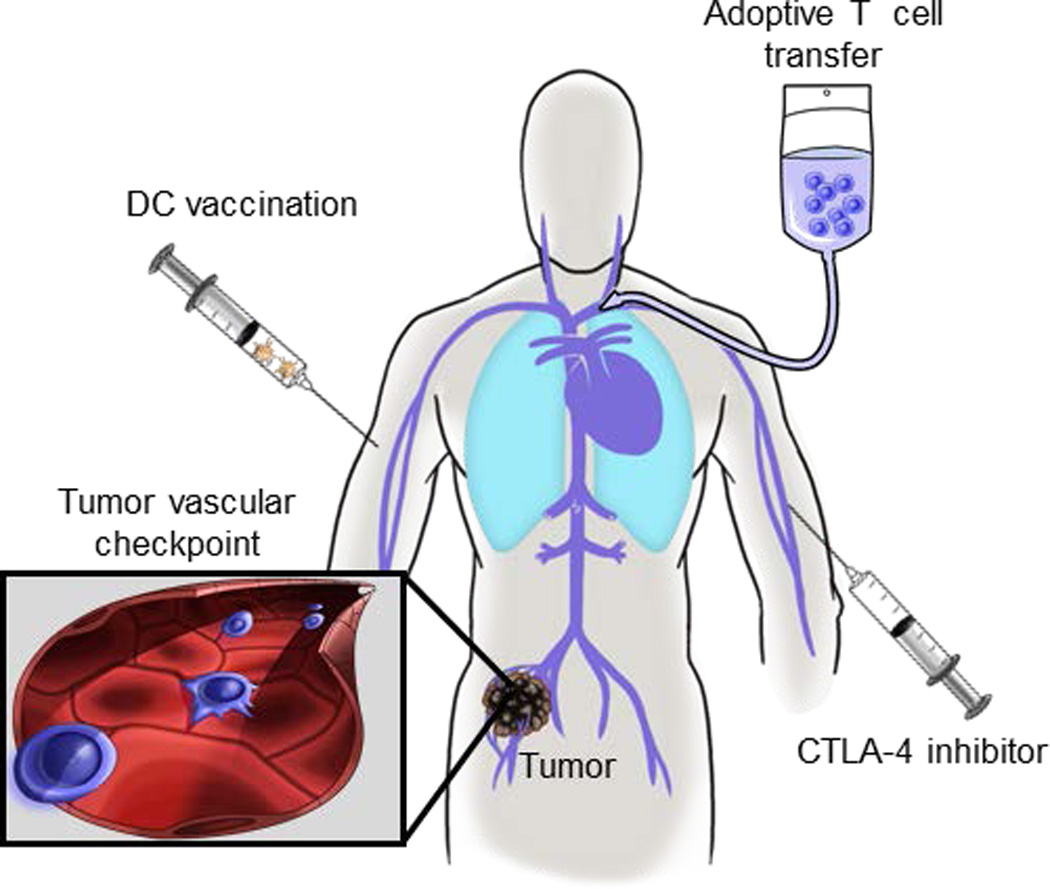
The direct tumoricidal activity of therapeutic interventions including dendritic cell (DC) vaccination, adoptive T cell transfer, and administration of cytotoxic T-lymphocyte antigen-4 (CTLA-4) inhibitors hinge on the ability of cytotoxic CD8+ T cells to traffic across vasculature barriers (blue cells in inset) within the tumor microenvironment.
Alternative strategies seek to relieve the “brakes” on T cell function by dampening inhibitory molecules which repress T cells in vivo. Ipilimumab, an antibody which targets the suppressive molecule, cytotoxic T lymphocyte antigen-4 (CTLA-4), was the first FDA-approved drug to improve the survival of patients with metastatic melanoma over standard therapies [15–17] (Figure 1). Ipilimumab is thought to prolong the activity of tumor-specific T cells in vivo by disrupting CTLA-4-mediated signaling in CD8+ T cells that normally occurs during resolution of immunity [16, 17]. Further clinical trials are underway for combination therapies to extend the survival benefit of ipilimumab treatment and to target other immunosuppressive pathways such as programmed cell death-1 protein (PD-1) and lymphocyte-activation gene-3 (Lag-3) [15, 18, 19].
Despite excitement in the field regarding the promise of immunotherapeutic modalities, there is still substantial room for improvement in terms of patient outcomes. For example, in metastatic melanoma the durable response rates for DC vaccination and ACT are less than 25%, and ipilimumab achieves similarly poor complete responses and only extends patient survival by ~4 months [10, 14, 15]. These poor outcomes are likely due to a combination of multiple mechanisms including loss of tumor antigen, which has been suggested to occur as result of immunoediting [20]. Additionally, the tumor microenvironment often contains several immunosuppressive cells (e.g., Treg, myeloid-derived suppressor cells [MDSC], and tumor-associated macrophage [TAM] of the M2 subset) and soluble factors (e.g., indoleamine 2,3-dioxygenase, TGF-β , and IL-10) that have been implicated in potentiating tumor growth [20, 21]. Notably, a common feature of the immunotherapeutic strategies in use clinically is that they all converge on the requirement for blood-borne cytotoxic T cells to traffic across the tumor vasculature in order to initiate contact-dependent killing of tumor cell targets (Figure 1). The ultimate demand for physical contact between T cells and tumor cells for cancer control is beneficial because it minimizes nonspecific damage of healthy tissues, but it also suggests that limiting T cell access to tumor cells may be an obvious mechanism of tumor escape. One appealing option to improve T cell-based therapies involves identifying methods to drive T cell extravasation across tumor vascular checkpoints; however, surprisingly little is known about the regulation of T cell recruitment into tumor tissue. This review will discuss the bottleneck in CD8+ T cell infiltration at tumor sites as well as recent findings that reveal the potential for thermal modalities to dial up T cell trafficking at the tumor vascular interface through an unexpected mechanism involving the IL-6 proinflammatory cytokine.
Rules of engagement
Insight into effector T cell trafficking can be found in the setting of non-malignant inflammation where T cells must undergo a series of Velcro-like multi-step adhesive interactions with vascular endothelium in order to exit the circulating lymphocyte pool (Figure 2A) [22–25]. This process is initiated by loose tethering and rolling interactions that slow T cells down on the lumenal surface of vessel walls, thereby allowing T cells to sample the local chemokine microenvironment. Chemokine signaling in T cells expressing complimentary chemokine receptors causes the transition to firm adhesion and subsequent extravasation. In the absence of inflammation, endothelial cells do not normally support high rates of effector T cell extravasation. However, during inflammation, production of proinflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), IL-6, and interferon γ (IFN-γ), induce local upregulation of vascular trafficking molecules that function at discrete steps of the adhesion cascade [9, 24, 26]. Depending on the type of insult and location of the inflammatory response, different patterns of trafficking molecules on blood vessels and reciprocal molecules on T cells work in concert to precisely mobilize effector T cells to inflammatory sites.
Figure 2. Thermal therapy primes tumor microvessels to support E/P-selectin– and ICAM-1–dependent trafficking of CD8+ effector T cells.
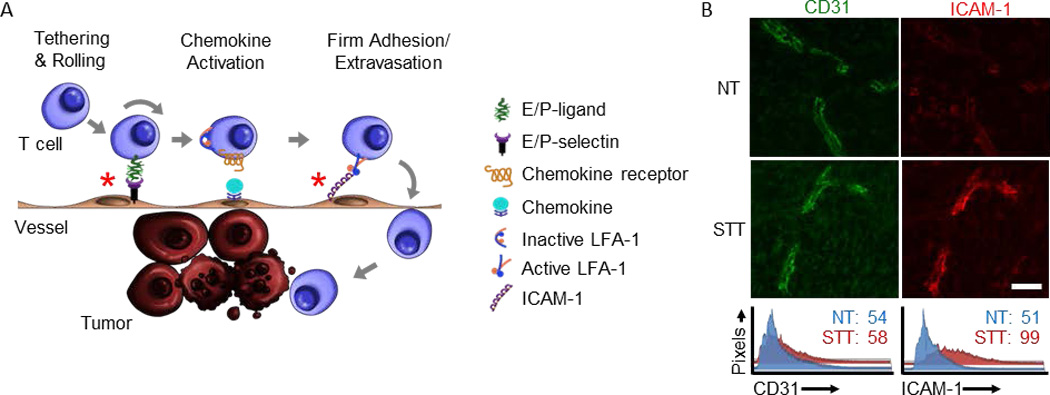
(A) Multistep adhesive interactions direct circulating T cells into tumor sites where they can kill tumor cell targets; thermally responsive adhesion events are demarked by red asterisks (*). (B) STT treatment of B16-OVA tumors increased intravascular expression of ICAM-1 (red) over baseline normothermal control (NT). The landmark position of vessels in photomicrographs is demarked by immunostaining for the CD31 pan-endothelial adhesion molecule (green). Histograms represent quantitative image analysis of relative immunofluorescence staining intensity of CD31 or ICAM-1 on all CD31+ vessels; studies were performed as described [35]. Bar, 50 µm.
While the specifics of T cell trafficking in tumor tissues have been difficult to resolve, histological studies in both mice and humans suggest that tumor vessels may be poor sites of T cell recruitment. Despite the proinflammatory cytokine milieu typically present in malignant lesions (i.e., TNF-α, IL-1β, and IL-6) [5–7, 27], tumor vessels in both mice and humans exhibit limited expression of inflammatory chemokines and adhesion molecules [28–30]. This may provide a partial explanation for why intratumoral infiltration of T cells is often low in a majority of cancer patients [1, 2, 4, 9]. However, infiltration of T cells within patient tumors reflects the culmination of multiple parameters including T cell trafficking across tumor vasculature, T cell frequency in the blood, and T cell fate in situ (i.e., retention, egress, apoptosis, survival, and proliferation). These events downstream of T cell entry can greatly impact overall intratumoral T cell numbers. For example, approximately 40% of T cells in tumors are predicted to proliferate in situ within one day [31], making it difficult to isolate the contribution of T cell entry to anti-tumor immunity in vivo. Although some studies have observed poor leukocyte interactions with tumor vessels [32–34], there has been remarkably little direct investigation of the actual rate of trafficking of effector CD8+ T lymphocytes across tumor vasculature.
To directly examine whether trafficking is an impediment to anti-tumor immunity, we used epifluorescence intravital microscopy to visualize in real-time the stepwise adhesive interactions of fluorescently-tagged CD8+ effector T cells at the tumor vascular checkpoint [35]. For these studies, we utilized the highly aggressive B16-OVA orthotopic melanoma model which expresses the surrogate tumor antigen, ovalbumin (OVA). OT-I CD8+ T cell clones expressing a transgenic T cell receptor specific for OVA were activated and expanded ex vivo prior to use as a source of adoptively transferred effector CD8+ cells. It is noteworthy that despite the presence of chaotic, leaky vasculature within tumor tissue, we did not observe any indiscriminate T cell entry indicating that trafficking is strictly an active process [35]. In this regard, CD8 T cells lacking trafficking molecules necessary for migration to peripheral tissues (e.g., naïve T cells which do not express receptors for E- and P-selectin) did not passively accumulate within tumors. The inability of T cells with a diameter of ~10 µm to passively enter tumors via an intrinsically leaky vasculature is in line with prior studies demonstrating that the permeability of tumor vessels is restricted to molecules less than 400–500 nM in size [36, 37].
Analysis of CD8+ effector T cells expressing a full complement of adhesion molecules and chemokine receptors necessary for trafficking to extralymphoid sites revealed that these cells rarely engage with tumor vessels in various types of murine tumor models including melanoma and colon cancer. Specifically, only ~1% of the transferred effector CD8+ T cells observed within tumor vessels were capable of transitioning to firm arrest in subcutaneous tumors, representing a modest increase above the low frequency interactions (~0.2%) typically detected in normal vessels at the same tissue site. This is in sharp contrast to observations during inflammation in non-malignant settings where blood vessels support a substantially higher frequency of trafficking (e.g., >10% of effector T cells visualized within vessels transition to firm adhesion) [33, 35]. Moreover, despite high numbers of effector T cells in the circulation following adoptive transfer, the level of antigen-specific apoptosis of tumor targets was indistinguishable from that of untreated mice, demonstrating that low constitutive trafficking in tumors limits the therapeutic efficacy of transferred cells [35]. These findings implicate tumor vessels as critical gatekeepers of anti-tumor immunity in vivo and suggest that triggering a more adhesive vasculature may be therapeutically beneficial for immune-based treatment modalities.
Warming up to lymphocyte trafficking in the tumor microenvironment
Those who cannot be cured by medicine can be cured by surgery.
Those who cannot be cured by surgery can be cured by heat.
Those who cannot be cured by heat are to be considered incurable.
Hippocrates, c. 460 BC – c. 370 BC
There has been longstanding interest in the application of exogenous heat as a primary and adjunct therapy since the times of Hippocrates [38]. Evidence that thermal therapy in the febrile range (38–40°C) does not exert direct cytotoxic effects in tumor cells has suggested that anti-tumor activity due to mild temperature hyperthermia reflects microenvironmental changes [35, 39]. Many of the adjuvant activities of fever-range thermal therapy have been attributed to direct effects on vascular permeability which increase blood flow and oxygenation in tumor tissues, thus improving delivery of systemic chemotherapy and the efficacy of radiation therapy [40–44]. Intriguingly, administration of fever-range systemic thermal therapy (STT; core temperature elevated to ~39–40°C for 6 h) also alters the patterns of endogenous leukocyte infiltrates (i.e., natural killer [NK] cells, CD8+ T cells, and neutrophils) in tumor tissues [29, 35, 39, 45–48]. A concomitant decrease in the number of circulating lymphocytes in cancer patients and mice has also been reported following STT [45, 49], supporting the notion that heat mobilizes the redistribution of T lymphocytes out of the circulating pool and into discrete tissue destinations.
We were prompted to examine the effects of STT on the intratumoral trafficking of CD8+ T cells based on prior studies examining the impact of the thermal element of fever on lymphocyte trafficking in an inflammatory setting [9, 22, 49–54]. In non-tumor-bearing mice, inflammatory cues triggered by fever-range STT were shown by intravital imaging to enhance the capacity of specialized cuboidal post-capillary high endothelial venules (HEVs) to direct naïve T cell entry into lymph nodes and Peyer’s patches [9, 22, 49–54]. The trafficking of T cells across HEVs represents a critical checkpoint in the generation of effector T cell responses by regulating the probability that rare T cells specific for a given antigen will encounter an activating DC within lymphoid tissues [25, 55]. T cell entry across HEVs is normally extremely efficient (i.e., ~1 out of 4 blood-borne cells that enter HEVs will go on to extravasate under homeostatic conditions) [55]. Thus, the ability of STT to double lymphocyte trafficking across HEVs can be viewed as a profound boost in trafficking potential.
Intravital microscopy further revealed that not all vascular endothelium within lymphoid organs is responsive to a heat stimulus [50]. In this regard, endothelial cells lining non-HEVs are insensitive to heat-induced stimulation of naïve lymphocyte homing [35, 49–51]. Given evidence that DCs preferentially localize in close proximity to HEVs [25], increasing trafficking of naïve and central memory T cells exclusively across HEVs maximizes the opportunity for productive interactions between these key players during the initiation of adaptive immunity. The restricted action of STT on a select subset of vascular beds (i.e., HEV) within lymphoid tissue despite systemic administration of thermal therapy suggests that heat functions preferentially in the context of immune-relevant sites. These observations raised the question of whether blood vessels in the tumor microenvironment would behave similarly to thermally-sensitive, specialized HEVs or would mirror heat-refractory, non-HEVs during STT.
To investigate the impact of thermal therapy on effector T cell trafficking within an established tumor vasculature in murine tumor models, we again turned to intravital imaging analysis [35]. For these studies we tracked the fate of adoptively transferred, OVA-specific effector OT-I T cells in vessels of B16-OVA tumors as an experimental endpoint that is relevant to clinical immunotherapy. Effector T cell populations were generated through ex vivo activation and expansion and then adoptively transferred post-STT after normal core temperatures were restored and hemodynamic parameters were normalized (e.g., blood flow velocity, vessel diameter and wall shear rate), allowing us to interrogate the intrinsic binding activity of tumor vascular endothelium. Although tumor vessels support few interactions with circulating T cells under homeostatic conditions, they are highly responsive to STT, yielding an ~5-fold induction in effector CD8+ T cell extravasation into the tumor site. The activity of STT on the entry of adoptively transferred cells extends to several implantable and spontaneous tumor models in addition to B16-OVA melanoma including CT26 colorectal tumor, EMT6 mammary tumor, and RIP-Tag5 transgenic pancreatic islet tumor [35].
One notable finding is that STT acts at multiple discrete steps in the adhesion cascade which culminate in improved CD8+ effector T cell trafficking across tumor vascular barriers (Figure 2A) [35]. STT increases the frequency of initial tethering and rolling interactions which are dependent on vascular display of E-selectin and P-selectin. Other molecules known to mediate transient rolling interactions during acute or chronic inflammation (e.g., vascular cell adhesion protein-1 [VCAM-1] and mucosal addressin cell adhesion molecule-1 [MAdCAM-1]) do not appear to be regulated by STT. T cells further require chemokine signaling to transition to firm arrest in the tumor vasculature as demonstrated by the ability of pertussis toxin, an irreversible inhibitor of chemokine-dependent G-protein signaling, to block this event. We also identified an absolute requirement for the prototypical firm arrest molecule intercellular adhesion molecule-1 (ICAM-1) in STT-mediated firm adhesion, where antibody blockade or genetic deficiency in ICAM-1 prevented arrest and extravasation. Quantitative immunofluorescent histology demonstrated that STT strongly upregulates the intravascular density of ICAM-1 on CD31+ tumor vessels in comparison to ICAM-1 expression on normothermal controls (NT) (Figure 2B) [35]. Moreover, STT boosts ICAM-1 induction at both primary and metastatic tissue sites, suggesting that STT takes advantage of common features of tumor vessels regardless of anatomical location. Based on studies in inflammatory scenarios, high ICAM-1 density following STT likely improves the avidity of ICAM-1 for its counter-receptor, lymphocyte function-associated antigen-1 (LFA-1) on T cells, thus supporting shear-resistant adhesion and helping shepherd transmigrating lymphocytes through the endothelium [24, 56].
One concern for strategies that enhance leukocyte trafficking is that therapeutic intervention might “open the floodgates” for recruitment not only of CD8+ cytotoxic T cells but also immunosuppressive subsets. This does not appear to be the case, however, since thermally-induced CD8+ effector T cell trafficking is accompanied by a concomitant decrease in infiltration of CD4+ CD25+ FoxP3+ Tregs in B16 murine melanoma [35]. The inverse relationship between the infiltration of these cell types during STT results in a profound increase in the overall ratio of CD8+ effector T cells to Tregs (i.e., from ~5:1 to ~35:1) which is likely advantageous in view of reports that high CD8+ T cell:Treg ratios correlate with increased patient survival [8]. STT does not change the overall infiltration of other inhibitory leukocytes such as monocytic MDSC (M-MDSC), polymorphonuclear MDSC (PMN-MDSC), or TAM in tumor tissues. However, these studies did not formally exclude the possibility that STT shifts the balance between different TAM subsets, namely pro-tumorigenic M2 and anti-tumorigenic M1 cells.
In light of the beneficial increases in CD8+ T cell trafficking after STT, we performed proof-of-concept studies to determine whether adjuvant STT can enhance the intratumoral entry of tumor-specific T cells during ACT and delay tumor progression (schema for therapeutic treatment regimen shown in Figure 3A) [35]. Adoptive transfer of effector OT-I CD8+ T cells vivo) into mice bearing established B16-OVA tumors is not sufficient to mediate tumor regression as a single therapy [35], even though these T cells are highly cytotoxic for tumor targets in vitro. Additionally, STT treatment alone has no effect on tumor growth control, suggesting that the endogenous T cell pool is insufficient to control tumor growth [35]. However, combination of STT pretreatment with ACT induces entry of tumor-specific T cells, leading to a delay in melanoma tumor growth [35]. Collectively, these findings establish that fever-range STT is an effective preconditioning strategy to boost the efficacy of adoptive T cell transfer immunotherapy.
Figure 3. Thermal therapy induces CD8+ T cell trafficking and target cell apoptosis in the tumor microenvironment.

(A) Schematic for combination systemic thermal therapy (STT) and adoptive cell transfer (ACT) of tumor-specific effector CD8+ T cells. Experimental endpoints for analysis of CD8+ T cell homing and TUNEL staining of apoptotic cells in tumor cryosections as well as tumor growth control are described in a recent report [35]. (B) Administration of IL-6–neutralizing monoclonal antibody (mAb) 30 min prior to STT blocked induction of CD8+ T cell trafficking (red) and apoptosis of intratumoral cellular targets (green). Relative values for normothermal (NT) mice are shown; nucleated cells are labeled with DAPI (blue). Left, representative photomicrographs of tumor cryosections. Bar, 50 μm. Right, quantification of intratumoral CD8+ T cells and TUNEL+ apoptotic cells per unit area of tumor tissue. *, p < 0.001. (C) Thermal induction of ICAM-1-–dependent CD8+ T cell trafficking in tumor vessels involves an IL-6 trans-signaling mechanism whereby intracellular JAK-1/2 and STAT3 activation occurs downstream of ligation of an agonistic, soluble form of the IL-6 receptor α binding subunit (sIL-6Rα) and the gp130 signal transducing subunit.
IL-6: a double agent in the war against cancer
Investigation of the mechanisms underlying thermal regulation of T cell trafficking centered on the contributions of candidate proinflammatory cytokines (i.e., TNF-α, IL-1β, and IL-6) which are abundant in murine and patient tumors and are well-known inducers of vascular trafficking molecules in non-cancerous settings [5–7, 24, 27]. These studies led to the unexpected discovery of opposing roles for IL-6 in the tumor microenvironment. The pro-tumorigenic role of IL-6 has already been intensively studied based on recognition over the past decade of its ability to drive cancer progression and metastasis [5, 6, 27, 35, 57]. IL-6 stimulates tumor cell-intrinsic proliferation and/or survival by inhibiting caspase-3-mediated apoptosis and inducing cyclin D, myc, Bcl-2/Bcl-xL, and survivin [58–61]. IL-6 also promotes tumor growth through extrinsic activities that include fueling angiogenesis via STAT3-dependent induction of vascular endothelial growth factor (VEGF) synthesis [60] and self-seeding of tumors by chemoattraction of aggressive malignant cell subsets [62]. These diverse mechanisms likely contribute to IL-6–induced chemoresistance and radioresistance in cancer [61, 63, 64].
In contrast to the prevailing view of IL-6 as a pro-tumorigenic factor, there is a paucity of information linking IL-6 to anti-tumorigenic activities within the tumor microenvironment. Thus, it came as a surprise that IL-6 was identified as the single cytokine mediating STT stimulation of CD8+ effector T cell trafficking across murine tumor vessels [35]. In this regard, antibody blockade of IL-6, but not TNF-α, IL-1β, or IFN-γ, prevented upregulation of E/P-selectin– and ICAM-1–dependent trafficking of adoptively transferred CD8+ T cells at the vascular interface in multiple tumor models (i.e., B16-OVA melanoma, CT26 colorectal, and RIP-Tag5 pancreatic islet tumors). The IL-6 requirement was further substantiated in IL-6–deficient mice in which STT failed to upregulate ICAM-1 on tumor vessels or induce CD8+ T cell intratumoral accumulation. In sharp contradistinction to evidence that tumor-promoting IL-6 is provided by hematopoietic cells (i.e., macrophages and CD4+ T cells) [5, 27], heat-induced trafficking of CD8+ T cells depends on IL-6 produced by nonhematopoietic stromal cells [35]. Differential sources of IL-6 could feasibly dictate its function within select micro-anatomical niches within the tumor microenvironment. While we were unable to detect an increase in total IL-6 levels within tumor tissue in response to STT, this cytokine was shown to be required for the therapeutic benefit achieved by combining ACT with STT in murine models. In this regard, absence of IL-6 function abrogated the ability of thermal therapy to induce effector T cell trafficking and apoptosis of tumor targets within the initial 12 h window post-ACT (detected by TUNEL assays; Figure 3B) as well as tumor growth delay and prolonged survival [35].
The proinflammatory activities of IL-6 are known to be under tight control by two distinct signaling pathways, i.e., classical versus trans-signaling [22, 27, 65], raising the question as to which mechanism is operative during STT. Classical IL-6 signaling involves the interaction of IL-6 with the membrane-anchored IL-6 receptor α binding subunit (IL-6Rα) and the gp130 signal-transducing component of the IL-6 receptor complex. Under homeostatic conditions only select cell types (i.e., hepatocytes and immune cells) express membrane-bound IL-6Rα, restricting the populations of IL-6–responsive cells [65]. However, ubiquitous gp130 expression confers all cells with the potential to convert to IL-6 responders through trans-signaling in an inflammatory setting. During trans-signaling, dual availability of IL-6 and an agonistic, soluble (s) form of IL-6Rα is required to trigger downstream signaling via activation of JAK-1/2 and STAT3 in cells that are normally refractory to IL-6 (Figure 3C) [22, 27, 65].
Complementary loss-of-function and gain-of-function approaches were taken to distinguish which of these two signaling mechanisms mediate STT responses within an inflammatory tumor microenvironment. Loss-of-function was achieved by systemic administration of soluble gp130 which acts as a decoy to selectively antagonize trans-signaling initiated by the IL-6/sIL-6Rα complex [65]. Our findings demonstrate that blockade of trans-signaling by soluble gp130 abrogates the ability of STT to boost E/P-selectin–dependent rolling and ICAM-1–dependent firm arrest during effector CD8+ T cell trafficking across tumor vessels [35]. Alternatively, systemic administration of a recombinant IL-6/sIL-6Rα fusion protein, hyper-IL-6 (H-IL-6) [66], in a gain-of-function approach, rapidly induces activation of STAT3 directly in endothelial cells lining tumor vessels in mouse tumors [35]. Moreover, H-IL-6 acts as a surrogate for STT by strongly inducing E/P-selectin– and ICAM-1–directed trafficking of CD8+ T cells across murine tumor vessels in addition to achieving tumor growth delay. Interestingly, administration of H-IL-6 did not support increased tumor growth, suggesting that the endogenous IL-6 responsible for growth-promoting activity was already at a saturating level such that the addition of exogenous H-IL-6 is superfluous [35]. The impact of IL-6 trans-signaling on tumor vascular adhesion was also examined in primary patient tumor explants ex vivo and was found to closely mirror observations in mouse models [35]. In this regard, the low baseline levels of ICAM-1 on tumor vessels tumors was profoundly elevated after treatment with H-IL-6 ex vivo in a subset of stage IV colorectal patient samples [35].
Collectively, these data highlight the dichotomy in IL-6 signaling within the tumor microenvironment where pro-tumorigenic activities are counterbalanced by T cell-mediated anti-tumor immunity during STT. Since cohabitating tumor cells and endothelial cells are simultaneously exposed to STT, it is difficult to reconcile how IL-6 trans-signaling during thermal therapy results in an overall anti-tumorigenic effect. Clues into possible mechanisms are provided by the analysis of IL-6 and sIL-6Rα, which are both present in substantial amounts in human and mouse tumors and constitutively drive pro-tumorigenic activities [5, 27, 35]. Endothelial cells embedded in the same microenvironment, on the other hand, are refractory to IL-6 under baseline conditions as evidenced by low STAT3 activation and expression of trafficking molecules [35]. A possible explanation for these observations is that tumor vascular endothelial cells have a higher threshold for IL-6 trans-signaling than tumor cells. Although different cellular sources (hematopoietic versus non-hematopoietic stromal cells) have been linked to pro- versus anti-tumorigenic activity [5, 27, 35], the answer as to how thermal therapy activates trans-signaling in tumor vessels does not appear to lie in the targeting of soluble trans-signaling components. In this regard, STT does not significantly change the intratumoral concentration of IL-6 [35] or sIL-6Rα (i.e., sIL-6Rα concentration in B16 tumor extracts in control mice or after STT, respectively, is 1.2 ± 0.26 and 1.6 ± 0.22 pg/mg total protein; data from ELISA are mean ± SEM for 6 mice). Instead, STT preferentially upregulates the expression of membrane-anchored gp130 on tumor vessels [35], paralleling observations in non-malignant inflammation where gp130 induction lowers the activation threshold for IL-6 trans-signaling [67]. Together, these studies suggest that STT could be a viable strategy to ‘flip the switch’ toward IL-6–driven anti-tumor immunity in a clinical setting in cancer patients.
Perspectives and future directions – the heat is on in the tumor microenvironment
Studies employing mild-temperature STT have revealed the tumor vasculature to be a critical determinant of anti-tumor immunity by controlling the access of cytotoxic T cells to tumor targets. Thermal therapy has emerged as a powerful tool to induce a window of vascular adhesiveness that drives adoptively transferred cytotoxic CD8+ T cells into tumor tissue, thereby controlling tumor growth. An added therapeutic benefit is that STT also acts during the initiation phase of the adaptive immune response to increase naïve T cell entry across HEVs in lymph nodes that are important sites of activation and expansion of tumor-reactive CD8+ T cells [28, 49, 50, 53, 54]. STT unexpectedly unveiled a novel anti-tumorigenic role for IL-6 signaling in the tumor microenvironment, thereby tilting the balance of IL-6 function in tumor tissues away from its pro-tumorigenic role in a clinical setting (Figure 4A). The ability of mild thermal stress to co-opt IL-6 may also extend to higher temperature modalities currently in clinical use such as hyperthermic intraperitoneal chemotherapy (HIPEC), isolated limb perfusion (ILP), radiofrequency ablation (RFA), and high intensity focused ultrasound (HIFU) [68–70]. The contribution of hyperthermia to HIPEC and ILP efficacy has mainly been attributed to increasing vascular permeability which improves the delivery of chemotherapeutics in tumor tissues [39, 43, 71, 72]. Based on recent findings for STT [35], it is tempting to speculate that various localized/regional heat modalities could also act to improve the delivery of endogenous cytotoxic T cells to tumors. It is also possible that heat induces long-term effects in vascular parameters or interstitial fluid pressure which may contribute to patient outcomes [43]. Further investigation into the ability of temperature to shift the outcome of IL-6 signaling in the tumor microenvironment will be needed in order to harness the full potential of thermal treatment as a therapeutic modality.
Figure 4. Collaborative intersection between thermal therapy and IL-6 in the tumor microenvironment.
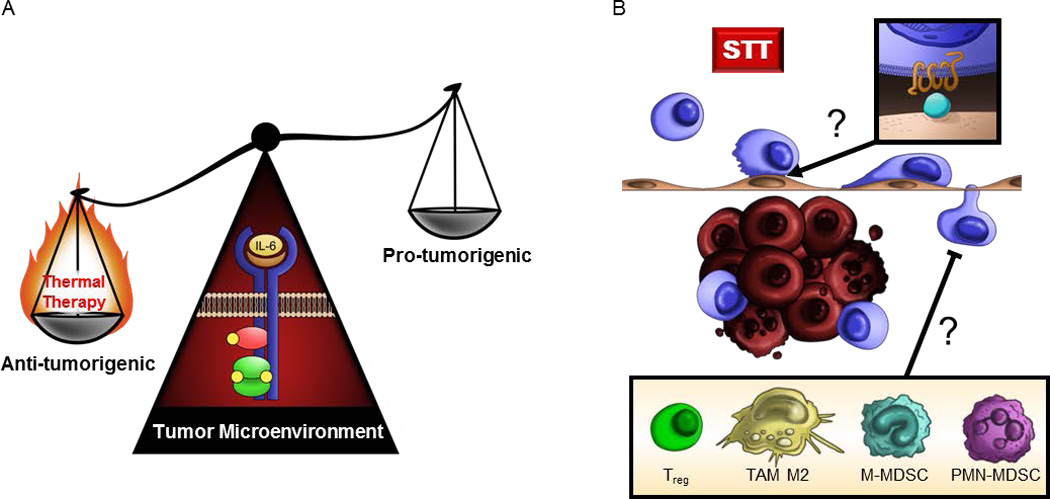
(A) Thermal therapy tips the balance of IL-6 activity in the tumor microenvironment from a tumor-promoting inflammatory role to an anti-tumorigenic role supporting T cell-mediated tumor immunity. (B) Outstanding questions remain regarding which chemokine/chemokine receptor interactions (inset) mediate CD8+ T cell trafficking at the vascular interface during heat therapy and whether clinical responses to preconditioning regimens are predicated on the presence of immunosuppressive cell subsets (inset) within the tumor microenvironment.
A key outstanding question relates to the chemokine/chemokine receptor pairs governing effector T cell trafficking at the tumor vascular interface and whether thermal therapy modifies the chemokine milieu within tumors (Figure 4B). While G-protein coupled chemokine receptor signaling is clearly required during CD8+ effector T cell trafficking in tumors under baseline and STT conditions [35, 73], a major challenge will be to pinpoint the precise molecules involved in view of the diversity of the chemokine network which includes over 50 chemokines and 20 receptors [74–76]. Studies examining homeostatic T cell accumulation in tumor tissues have implicated multiple inflammatory chemokine/chemokine receptor pairs including CXCL9/CXCR3, CCL5/CCR5, CX3CL1/CX3CR1, and CCL28/CCR3 [3, 4, 77, 78]. These studies fall short of extrapolating a direct role for individual chemokines in cytotoxic T cell trafficking, however, since chemokine signaling also contributes to the fate of T cells within tissues (i.e., T cell retention, egress, survival, and proliferation in situ) [74–76]. A related consideration will be whether the IL-6–rich tumor microenvironment modifies chemokine production during STT. Of note, IL-6 has been linked to the expression of lymphocyte-attracting chemokines (i.e., CCL2, CCL5, and CXCL10) during inflammation in a non-tumor setting [79–81], and a recent study in ovarian cancer patients reported a decline in CC L2 and CXCL12 levels after treatment with the anti-IL-6 antibody siltuximab [82]. Additional studies will be needed to evaluate the unique or redundant functions of individual chemokines in the tumor microenvironment in order to gain a more complete picture of how thermal therapy overcomes barriers to anti-tumor immunity.
Another issue warranting investigation is whether the immunosuppressive milieu characteristic of tumor tissues dictates patient responses to preconditioning regimens like thermal therapy (Figure 4B). There is already precedent that suppressive cellular constituents within patient tumors (e.g., Tregs, M2-skewed TAMs, and MDSC) foster resistance to conventional treatments such as radiation and chemotherapy by pro-tumorigenic mechanisms which are not fully understood [5, 7, 61, 63, 64, 83, 84]. In addition, multiple soluble factors commonly produced by these immunosuppressive cell subsets have been implicated in an independent series of studies as negative regulators of vascular adhesion in the tumor microenvironment [33, 34, 85, 86]. For example, VEGF and basic fibroblast growth factor (bFGF) can prevent induction of ICAM-1 on tumor vessels in response to TNF or through signaling by an inflammatory activator known as toll-like receptor 4 (TLR) 4 [34, 85]. Other suppressive cytokines such as IL-10 are reported to interfere with leukocyte adhesion in vitro [86], and blockade of TGF-β has been shown to increase the frequency of leukocyte interactions with tumor vasculature in vivo in a murine model of colorectal cancer [33]. Provocatively, IL-10 and TGF-β have both been shown to suppress IL-6–dependent STAT3 activation in vitro [87, 88] which could potentially antagonize beneficial activities of the STT–IL-6–tumor vessel axis in vivo. Exciting new avenues of research should determine whether the decreased frequency of intratumoral Tregs detected during STT [35] is mechanistically linked to trafficking at the vascular interface or if tumors become refractory to thermal therapy in the settings of high burdens of immunosuppressive cells described in cancer patients [5–8, 89].
In conclusion, studies using thermal stress have been instrumental in identifying the tumor microvasculature as a locus for therapeutic targeting. The development of thermally-sensitive liposomes, in which heat treatment can improve the permeability of tumor vasculature to passive liposomal entry as well as triggered release of encapsulated molecules, also demonstrates the value of thermal effects on tumor vasculature [90, 91]. Adjuvant thermal therapy has already been shown to enhance the efficacy of radiation and chemotherapy, so it is reasonable to predict that it will also be useful in combination with T cell-based immunotherapy [40, 42, 71, 72]. Moreover, it is possible that in addition to thermal therapy, other therapeutic strategies which generate acute inflammation such as the TLR7 agonist imiquimod [92, 93] or radiation [94–96] may also tap into mechanisms which can enhance cytotoxic T cell entry across tumor vessels. While further studies are needed to determine the optimal method to prime tumor vascular adhesiveness prior to T cell-based clinical immunotherapy, preconditioning regimens which hijack IL-6 signaling for anti-tumor immunity in the tumor microenvironment hold great promise to improve patient outcomes in a variety of malignant diseases.
Acknowledgements
We thank our many collaborators for valuable contributions during the development of this work with special appreciation to Q. Chen for originally identifying the role of IL-6 in tumors and HEV. We also thank K. Howard for graphical assistance. Supported by the NIH (CA79765, CA085183, 5T32 CA085183), the Joanna M. Nicolay Melanoma Foundation, the Jennifer Linscott Tietgen Family Foundation, and the Mark Diamond Research Fund.
Footnotes
Conflict of interest statement: The authors have no conflicting financial interests.
References
- 1.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 2.Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011;71:5601–5605. doi: 10.1158/0008-5472.CAN-11-1316. [DOI] [PubMed] [Google Scholar]
- 3.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 4.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 7.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher DT, Chen Q, Appenheimer MM, Skitzki J, Wang WC, Odunsi K, et al. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunol Invest. 2006;35:251–277. doi: 10.1080/08820130600745430. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8:151–160. doi: 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- 12.Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 2011;71:4809–4820. doi: 10.1158/0008-5472.CAN-11-0753. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y, Auh SL, Wang Y, Burnette B, Meng Y, Beckett M, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boon T, Coulie PG, Eynde BJ, Bruggen PV. Human T Cell Responses against Melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 15.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Culver ME, Gatesman ML, Mancl EE, Lowe DK. Ipilimumab: a novel treatment for metastatic melanoma. Ann Pharmacother. 2011;45:510–519. doi: 10.1345/aph.1P651. [DOI] [PubMed] [Google Scholar]
- 17.Sondak VK, Smalley KS, Kudchadkar R, Grippon S, Kirkpatrick P. Ipilimumab. Nat Rev Drug Discov. 2011;10:411–412. doi: 10.1038/nrd3463. [DOI] [PubMed] [Google Scholar]
- 18.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnis ME, Korman AJ, Drake CG, Vignali DA. Combinatorial Immunotherapy: PD-1 may not be LAG-ing behind any more. Oncoimmunology. 2012;1:1172–1174. doi: 10.4161/onci.20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 21.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, et al. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine. 2007;39:84–96. doi: 10.1016/j.cyto.2007.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans SS, Fisher DT, Skitzki JJ, Chen Q. Targeted regulation of a lymphocyte-endothelial-interleukin-6 axis by thermal stress. Int J Hyperthermia. 2008;24:67–78. doi: 10.1080/02656730701772498. [DOI] [PubMed] [Google Scholar]
- 24.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 25.Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol. 2012;12:762–773. doi: 10.1038/nri3298. [DOI] [PubMed] [Google Scholar]
- 26.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 27.Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol. 2010;88:1145–1156. doi: 10.1189/jlb.0410217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q, Evans SS. Thermal regulation of lymphocyte trafficking: hot spots of the immune response. Int J Hyperthermia. 2005;21:723–729. doi: 10.1080/02656730500271734. [DOI] [PubMed] [Google Scholar]
- 29.Chen Q, Wang WC, Evans SS. Tumor microvasculature as a barrier to antitumor immunity. Cancer Immunol Immunother. 2003;52:670–679. doi: 10.1007/s00262-003-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother. 2008;57:1553–1558. doi: 10.1007/s00262-008-0524-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. 2007;204:345–356. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu NZ, Klitzman B, Dodge R, Dewhirst MW. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res. 1992;52:4265–4268. [PubMed] [Google Scholar]
- 33.Bessa X, Elizalde JI, Mitjans F, Pinol V, Miquel R, Panes J, et al. Leukocyte recruitment in colon cancer: role of cell adhesion molecules, nitric oxide, and transforming growth factor beta1. Gastroenterology. 2002;122:1122–1132. doi: 10.1053/gast.2002.32369. [DOI] [PubMed] [Google Scholar]
- 34.Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. Faseb J. 2006;20:621–630. doi: 10.1096/fj.05-4493com. [DOI] [PubMed] [Google Scholar]
- 35.Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. 2011;121:3846–3859. doi: 10.1172/JCI44952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 37.Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res. 2000;60:4440–4445. [PubMed] [Google Scholar]
- 38.Rowe-Horwege RW. Systemic Hyperthermia. Encyclopedia of Medical Devices and Instrumentation. 2006 [Google Scholar]
- 39.Burd R, Dziedzic TS, Xu Y, Caligiuri MA, Subjeck JR, Repasky EA. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J Cell Physiol. 1998;177:137–147. doi: 10.1002/(SICI)1097-4652(199810)177:1<137::AID-JCP15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 40.Jones EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, et al. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol. 2005;23:3079–3085. doi: 10.1200/JCO.2005.05.520. [DOI] [PubMed] [Google Scholar]
- 41.van der Zee J, Gonzalez Gonzalez D, van Rhoon GC, van Dijk JD, van Putten WL, Hart AA. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet. 2000;355:1119–1125. doi: 10.1016/s0140-6736(00)02059-6. [DOI] [PubMed] [Google Scholar]
- 42.Issels RD, Lindner LH, Verweij J, Wust P, Reichardt P, Schem BC, et al. Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncol. 2010;11:561–570. doi: 10.1016/S1470-2045(10)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen A, Capitano ML, Spernyak JA, Schueckler JT, Thomas S, Singh AK, et al. Mild elevation of body temperature reduces tumor interstitial fluid pressure and hypoxia and enhances efficacy of radiotherapy in murine tumor models. Cancer Res. 2011;71:3872–3880. doi: 10.1158/0008-5472.CAN-10-4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sumiyoshi K, Strebel FR, Rowe RW, Bull JM. The effect of whole-body hyperthermia combined with 'metronomic' chemotherapy on rat mammary adenocarcinoma metastases. Int J Hyperthermia. 2003;19:103–118. doi: 10.1080/0265673021000017091. [DOI] [PubMed] [Google Scholar]
- 45.Kraybill WG, Olenki T, Evans SS, Ostberg JR, O'Leary KA, Gibbs JF, et al. A phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: correlation with mouse models. Int J Hyperthermia. 2002;18:253–266. doi: 10.1080/02656730110116704. [DOI] [PubMed] [Google Scholar]
- 46.Ostberg JR, Dayanc BE, Yuan M, Oflazoglu E, Repasky EA. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J Leukoc Biol. 2007 doi: 10.1189/jlb.1106699. [DOI] [PubMed] [Google Scholar]
- 47.Ostberg JR, Ertel BR, Lanphere JA. An important role for granulocytes in the thermal regulation of colon tumor growth. Immunol Invest. 2005;34:259–272. doi: 10.1081/imm-200064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thrall DE, Prescott DM, Samulski TV, Rosner GL, Denman DL, Legorreta RL, et al. Radiation plus local hyperthermia versus radiation plus the combination of local and whole-body hyperthermia in canine sarcomas. Int J Radiat Oncol Biol Phys. 1996;34:1087–1096. doi: 10.1016/0360-3016(95)02260-0. [DOI] [PubMed] [Google Scholar]
- 49.Evans SS, Wang WC, Bain MD, Burd R, Ostberg JR, Repasky EA. Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood. 2001;97:2727–2733. doi: 10.1182/blood.v97.9.2727. [DOI] [PubMed] [Google Scholar]
- 50.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, et al. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol. 2006;7:1299–1308. doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 51.Chen Q, Clancy KA, Wang WC, Evans SS. Inflammatory Cues Controlling Lymphocyte-Endothelial Interactions During Fever-Range Thermal Stress. In: Aird W, editor. Endothelial Biomedicine. Cambridge University Press; 2007. pp. 471–479. [Google Scholar]
- 52.Skitzki JJ, Chen Q, Wang WC, Evans SS. Primary immune surveillance: some like it hot. J Mol Med. 2007;85:1361–1367. doi: 10.1007/s00109-007-0245-7. [DOI] [PubMed] [Google Scholar]
- 53.Evans SS, Bain MD, Wang WC. Fever-range hyperthermia stimulates alpha4beta7 integrin-dependent lymphocyte-endothelial adhesion. Int J Hyperthermia. 2000;16:45–59. doi: 10.1080/026567300285411. [DOI] [PubMed] [Google Scholar]
- 54.Chen Q, Appenheimer MM, Muhitch JB, Fisher DT, Clancy KA, Miecznikowski JC, et al. Thermal facilitation of lymphocyte trafficking involves temporal induction of intravascular ICAM-1. Microcirculation. 2009;16:143–158. doi: 10.1080/10739680802353850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 56.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 58.Tu B, Du L, Fan QM, Tang Z, Tang TT. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012;325:80–88. doi: 10.1016/j.canlet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 59.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 60.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Niu XL, Qu Y, Wu J, Zhu YQ, Sun WJ, et al. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010;295:110–123. doi: 10.1016/j.canlet.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 62.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen CC, Chen WC, Lu CH, Wang WH, Lin PY, Lee KD, et al. Significance of interleukin-6 signaling in the resistance of pharyngeal cancer to irradiation and the epidermal growth factor receptor inhibitor. Int J Radiat Oncol Biol Phys. 2010;76:1214–1224. doi: 10.1016/j.ijrobp.2009.09.059. [DOI] [PubMed] [Google Scholar]
- 64.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 66.Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, et al. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol. 1997;15:142–145. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- 67.Schooltink H, Rose-John S. Cytokines as therapeutic drugs. J Interferon Cytokine Res. 2002;22:505–516. doi: 10.1089/10799900252981981. [DOI] [PubMed] [Google Scholar]
- 68.Seinen JM, Hoekstra HJ. Isolated limb perfusion of soft tissue sarcomas: A comprehensive review of literature. Cancer Treat Rev. 2012 doi: 10.1016/j.ctrv.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Chan DL, Morris DL, Rao A, Chua TC. Intraperitoneal chemotherapy in ovarian cancer: a review of tolerance and efficacy. Cancer Manag Res. 2012;4:413–422. doi: 10.2147/CMAR.S31070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haen SP, Pereira PL, Salih HR, Rammensee HG, Gouttefangeas C. More than just tumor destruction: immunomodulation by thermal ablation of cancer. Clin Dev Immunol. 2011;2011:160250. doi: 10.1155/2011/160250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herzog TJ. The role of heated intraperitoneal chemotherapy (HIPEC) in ovarian cancer: hope or hoax? Ann Surg Oncol. 2012;19:3998–4000. doi: 10.1245/s10434-012-2521-1. [DOI] [PubMed] [Google Scholar]
- 72.Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia. 2005;21:779–790. doi: 10.1080/02656730500271668. [DOI] [PubMed] [Google Scholar]
- 73.Skitzki J, Craig RA, Okuyama R, Knibbs RN, McDonagh K, Chang AE, et al. Donor cell cycling, trafficking, and accumulation during adoptive immunotherapy for murine lung metastases. Cancer Res. 2004;64:2183–2191. doi: 10.1158/0008-5472.can-03-2799. [DOI] [PubMed] [Google Scholar]
- 74.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- 75.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 76.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 77.Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138:1429–1440. doi: 10.1053/j.gastro.2009.10.057. [DOI] [PubMed] [Google Scholar]
- 78.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9589–9594. doi: 10.1073/pnas.0501794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 81.Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, et al. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood. 1998;91:258–265. [PubMed] [Google Scholar]
- 82.Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17:6083–6096. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bouma-ter Steege JC, Baeten CI, Thijssen VL, Satijn SA, Verhoeven IC, Hillen HF, et al. Angiogenic profile of breast carcinoma determines leukocyte infiltration. Clin Cancer Res. 2004;10:7171–7178. doi: 10.1158/1078-0432.CCR-04-0742. [DOI] [PubMed] [Google Scholar]
- 86.Huet O, Laemmel E, Fu Y, Dupic L, Aprico A, Andrews KL, et al. Interleukin 10 antioxidant effect decreases leukocytes/endothelial interaction induced by tumor necrosis factor alpha. Shock. 2013;39:83–88. doi: 10.1097/SHK.0b013e318278ae36. [DOI] [PubMed] [Google Scholar]
- 87.Walia B, Wang L, Merlin D, Sitaraman SV. TGF-beta down-regulates IL-6 signaling in intestinal epithelial cells: critical role of SMAD-2. Faseb J. 2003;17:2130–2132. doi: 10.1096/fj.02-1211fje. [DOI] [PubMed] [Google Scholar]
- 88.Niemand C, Nimmesgern A, Haan S, Fischer P, Schaper F, Rossaint R, et al. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol. 2003;170:3263–3272. doi: 10.4049/jimmunol.170.6.3263. [DOI] [PubMed] [Google Scholar]
- 89.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Manzoor AA, Lindner LH, Landon CD, Park JY, Simnick AJ, Dreher MR, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72:5566–5575. doi: 10.1158/0008-5472.CAN-12-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li L, Ten Hagen TL, Hossann M, Suss R, van Rhoon GC, Eggermont AM, et al. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J Control Release. 2013;168:142–150. doi: 10.1016/j.jconrel.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 92.Urosevic M, Maier T, Benninghoff B, Slade H, Burg G, Dummer R. Mechanisms underlying imiquimod-induced regression of basal cell carcinoma in vivo. Arch Dermatol. 2003;139:1325–1332. doi: 10.1001/archderm.139.10.1325. [DOI] [PubMed] [Google Scholar]
- 93.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 94.Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180:3132–3139. doi: 10.4049/jimmunol.180.5.3132. [DOI] [PubMed] [Google Scholar]
- 95.Cao ZA, Daniel D, Hanahan D. Sub-lethal radiation enhances anti-tumor immunotherapy in a transgenic mouse model of pancreatic cancer. BMC Cancer. 2002;2:11. doi: 10.1186/1471-2407-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–1470. [PubMed] [Google Scholar]