

Abstract
Here we demonstrate a strategy to convert the graded Michaelis-Menten response typical of unregulated enzymes into a sharp, effectively all-or-none response. We do so using an approach analogous to the “branch point effect”, a mechanism observed in naturally occurring metabolic networks in which two or more enzymes compete for the same substrate. As a model system, we used the enzymatic reaction of Glucose Oxidase (GOx) and coupled it to a second, non-signaling reaction catalyzed by the higher affinity enzyme Hexokinase (HK) such that, at low substrate concentrations, the second enzyme outcompetes the first, turning off the latter’s response. Above an arbitrarily selected “threshold” substrate concentration, the non-signaling HK enzyme saturates leading to a “sudden” activation of the first signaling GOx enzyme and a far steeper dose-response curve than that observed for simple Michaelis-Menten kinetics. Using the well-known GOx-based amperometric glucose sensor to validate our strategy, we have steepen the normally graded response of this enzymatic sensor into a discrete yes/no output similar to that of a multimeric cooperative enzyme with a Hill coefficient above 13. We have also shown that, by controlling the HK reaction we can precisely tune the threshold target concentration at which we observe the enzyme output. Finally, we demonstrate the utility of this strategy for achieving effective noise attenuation in enzyme logic gates. In addition to supporting the development of biosensors with digital-like output, we envisage that the use of all-or-none enzymatic responses will also improve our ability to engineer efficient enzyme-based catalysis reactions in synthetic biology applications.
Keywords: cooperativity, enzymatic systems, logic gate, biomolecular filter, synthetic biology
Introduction
Enzymes exhibit extraordinary specificity, selectivity and catalytic activities, attributes that have lead to their widespread use in research, industry and medicine. In synthetic biology, for example, enzyme-catalyzed reactions are used for drug discovery1 and the synthesis of biopharmaceuticals2 and biofuels3-5. Enzymes are also widely employed in biosensors as recognition and signalling elements for the detection of specific molecular analytes and confer to these platforms unprecedented performances in terms of sensitivity and selectivity6-9. In recent years enzymatic reactions have been also used as the basis for novel biomolecular logic systems that could lead to the next generation of diagnostic. An example is the recently proposed use of enzyme logic gates responding to multiple inputs characteristic of specific diseases or injuries (i.e. biomarkers)10-11. The output of these enzyme logic gates can activate counteractions (e.g., drug delivery) against the specific disease, thus aiming to become integrated smart “sense/act” (biosensor-bioactuator) platforms12-13.
Despite all their great attributes, enzymes also display some limitations. For example, the dose-response curve associated with the majority of enzymes follows the well-known Michaelis–Menten equation, producing a fairly shallow, hyperbolic increase in catalytic rate (the “response” or “output”) with increasing substrate concentrations (the “dose” or “input”). Due to the shape of this input-output curve the “dynamic range” of an enzymatic response is generally fixed: an 81-fold increase in substrate concentration is needed to drive most enzymes from 10% to 90% of their maximal product formation rate14-15. This fixed dynamic range limits the utility of enzyme-based technologies in applications for which steeper dose-response curves are required. A steeper, nearly all-or-none “digital” response would be especially desired in biomolecular enzyme “logic gate” applications in order to reduce noise16-19. In such logic operations, the normal physiological level of the biomarker is defined as the logic-0 level, and pathological levels are defined as logic-1 values. The separation between normal and pathological levels, however, is often narrow and thus it is sometimes difficult to strictly define the 0 and 1 logic-values of enzyme logic gates using the shallow input-output curves associated with most enzymes.
Recently, the groups of Wang, Privman and Katz have spent efforts aimed at reducing the “noise” of biological logic gates, proposing the use of filters based on different mechanisms ranging from the use of enzymes with substrates that have self-promoter properties20, redox transformations21 or pH-filters22. Alternatively, the use of a biochemical filter that involves a partial back-conversion of the product generating the output signal was also proposed23 leading to a much better discrimination between the 0 and 1 outputs. Of note, however, this mechanism does not solve the problem related to the definition of the 0- and 1-logic values of the inputs. That is, it would be crucial to find a method to achieve a digital-like output of enzyme systems at specific threshold concentrations of the input. A general strategy to steepen the input/output curves of enzyme-based response coupled with the ability to arbitrarily tune the dynamic range of enzymatic outputs and so the threshold at which the digital-like response is obtained would thus prove of great value.
Several mechanisms have been invented by evolution to solve the problem related to the graded outputs of enzymes. The best known of these is positive allosteric cooperativity, which involves two or more target binding sites that interact such that the first binding events increase the affinity of those that follow. Allostery, however, requires subtle binding-induced conformational and functional changes and thus this approach to generating stepper input-output curves is likely expensive in terms of the number of evolutionary steps required to generate it and certainly quite challenging to engineer in artificial systems. Fortunately for us biomolecular engineers, however, cooperativity is not the only mechanism by which the input-output curves of enzymes can be manipulated. Indeed, several of the many approaches employed by nature in vivo to generate switch-like enzymatic response, such as multistep phosphorylation reactions, partial enzyme saturation and stoichiometric inhibitors appear quite simple and are likely amenable to exploitation in artificial technologies14.
Motivated by the above arguments here we have adapted the metabolic “branch point effect”24-25 to achieve steep input-output curves on enzyme-based systems. In this mechanism, which nature employs to generate ultrasensitive responses in some metabolic networks24-28, two enzymes compete for a single substrate. If one of these has a higher affinity (lower Km) it will sequester the substrate, reducing the response of the other enzyme. If the substrate concentration climbs above the threshold level at which the higher affinity “depletant” enzyme is saturated, then the output of the second enzyme will rise dramatically, producing a nearly all-or-none response. Using this mechanism we demonstrate an efficient and convenient approach to convert the hyperbolic dose-response curve of enzymes to a much steeper input/output response. We use the classic glucose electrochemical enzyme sensor as a model system to demonstrate and validate this approach and then apply this strategy to improve the performance of an enzyme-based AND logic gate.
Materials and Methods
Apparatus, electrodes and reagents
Amperometric measurements were carried out using a Portable Bipotentiostat μ stat 200 (DropSens, Spain). Carbon based screen-printed electrodes (SPEs) were printed with a 245 DEK (Weymouth, UK) screen printing machine using the following inks: graphite-based ink (Elettrodag 421), silver ink (Electrodag 477 SS RFU) and insulating ink (Elettrodag 6018 SS). The substrate was a polyester flexible film (Autostat HT5) obtained from Autotype Italia (Milan, Italy). The printing procedure is already described in previous papers29. Each sensor consists of three printed electrodes, a carbon working electrode, a silver internal pseudoreference electrode and a carbon counter electrode. The diameter of the working electrode was 0.3 cm, resulting in an apparent geometric area of 0.07 cm2. All chemicals from commercial sources were of analytical grade. All solutions were prepared with 0.05 M phosphate buffer + 0.1 M KCl + 0.01 M MgCl2, pH 7.4, unless otherwise specified. The standard solutions were made up daily in the same buffer. Glucose oxidase (GOx) (EC 1.1.3.4, type VII, 185 U/mg), horseradish peroxidase (HRP) (EC 1.11.1.7, 1550 U/mg), hexokinase (HK) (EC 2.7.1.1, 200 U/mg), Adenosine 5’-triphosphate disodium salt (ATP), o-phenylendiamine (OPD), FeCl3, K3Fe(CN)6 were all obtained from Sigma (St. Louis, MO).
Preparation of Prussian Blue (PB) modified screen-printed electrodes
Because of the high overpotential required to detect H2O2, here we have used an electrochemical mediator (Prussian Blue) capable of catalyzing the reduction of H2O2 and thus allowing its detection at low applied potentials30-31. PB modification29 of SPEs was accomplished by placing a drop (10 μL total volume) of a “precursor solution” onto the working electrode area. This solution is a mixture obtained by adding 5 μL of 0.1 M potassium ferricyanide (K3Fe(CN)6) in 10 mM HCl to 5 μL of 0.1 M ferric chloride in 10 mM HCl. The drop is carefully applied exclusively on the working electrode area. The electrodes are shaken gently on an orbital shaker for 10 min and then rinsed with a few millilitres of 10 mM HCl. The electrodes are then left for 90 min in an oven at 100 °C to obtain a more stable and active layer of Prussian blue. The PB modified electrodes are stored dry at room temperature in the dark and are stable for several months.
Preparation of GOx membrane glucose biosensor
Glucose Oxidase (GOx) was immobilized onto PB modified SPEs using a procedure optimized in a previous work29. 10 μL of a mixture of glutaraldehyde, Nafion® and a solution of enzyme+BSA were added onto the working electrode area and the solution was allowed to dry for 45 min at room temperature. 150 μL of the mixture have the following exact composition: • 100 μL of enzymatic solution (4 mg of BSA and 1 mg of GOx in 0.05 M phosphate buffer + 0.1 M KCl, pH 7.4;• 20 μL of glutaraldehyde (2.5 % in water);• 30 μL of Nafion® (5 % in ethanol). The sensors prepared with this procedure are stable and ready for glucose measurement in batch and drop analysis.
Glucose measurements with GOx membrane biosensor
Amperometric batch measurements of glucose were performed in a stirred phosphate buffer solution 0.05 M + KCl 0.1 M + MgCl2 0.01 M + HK 20 U/mL, pH 7.4 (10 mL) with an applied potential of -0.05 V versus internal reference electrode (int. ref.). When a stable baseline current was reached, glucose was added and the current variation was recorded after 3 minutes. In a different embodiment the sensor was tested using “drop” chronoamperometric measurement. In this case a drop (70 μL) of the above buffer solution containing different concentrations of glucose (see text for details) was added onto the electrode held in horizontal position at an applied potential of -0.05 V. The drop was added so as to cover all the three electrodes and to close the circuit. The current signal was measured after 3 minutes before and after three successive additions of ATP (2.5, 5, 7.5 mM). For the sake of clarity in all the figures the relative current (normalized) has been used in the y-axis. The RSD% of the developed sensors is ca. 7% (n=4) and the actual current plateau values obtained are 0.53 (±0.04) and 0.96 (±0.08) μA respectively in drop and batch measurements.
Composition of logic gate and input signals
The AND logic gate consisted of a 2.5 mL phosphate buffer solution 0.05 M + KCl 0.1 M + MgCl2 0.01 M + GOX 10 U/mL + HRP 23 U/mL, pH 7.4. A concentration of 0.17 mM of OPD was added to the solution to achieve a yes/no output signal. Of note, this concentration does not correspond to the actual glucose level (ca. 0.6 mM) at which we observe the steep response for two reasons. The first is related to the fact that the reaction between H2O2 and OPD catalyzed by HRP has a stoichiometric ratio of 3:2 (H2O2:OPD)32. The second is that the real concentration of H2O2 produced by GOx does not correspond to the concentration of glucose present in solution because the enzymatic reaction is already in the plateau region. Also in this case, for graphical purposes, normalized values were used. When GOx was used in solution (i.e. for logic gates applications) the plateau current value obtained in batch measurements was 2.9 (±0.1) μA.
Results
In order to validate and demonstrate our strategy we have selected Glucose Oxidase (GOx), an enzyme widely employed in sensors for the detection of blood glucose levels. This sensor employs a surface-confined Glucose Oxidase (GOx) to catalyze the oxidation of glucose. The resultant production of hydrogen peroxide is detected electrochemically, signalling the presence of the substrate6-9. As expected, the dose-response curve of the glucose sensor obeys the Michaelis-Menten equation, producing a Michaelis-Menten constant (Km) of 0.8 mM (Fig. 1, top). To convert the hyperbolic dose-response curve associated with GOx catalyzed reaction into a steeper, digital-like response, we have created a “branch point effect” using hexokinase (HK) as the competing “depletant” enzyme (Fig. 1, bottom). HK, which possesses a much greater affinity for glucose than that of GOx 33, will sequester glucose by converting it to glucose-6-phosphate, a species that is not recognized by GOx. [Of note, the glucose will also associate much faster to the free HK than to the electrode-bound GOx thus further increasing the HK catalytic rate]. Using this approach we readily compress the hyperbolic dose-response curve of this enzyme-based sensor14-15, 24 by one order of magnitude, pushing the normally 81-fold dynamic range of the sensor to less than 2-fold (Fig. 1, bottom).
Figure 1.
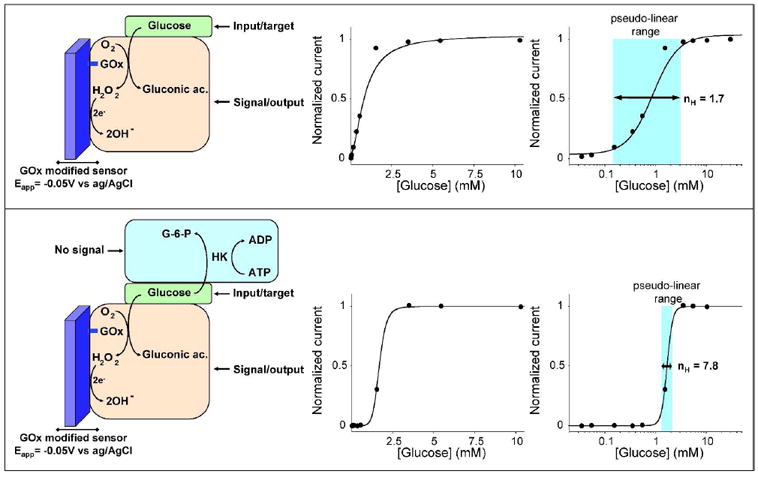
Here we convert the hyperbolic dose-response curve of an enzyme-based sensor into a steep all-or-none digital-like response by employing the “branch point effect”, a mechanism that occurs in some metabolic networks, where two enzymes compete for the same substrate24-26. (top) As our testbed system we have used the well known glucose amperometric sensor, which contains a surface-confined Glucose Oxidase (GOx) that shows a classic Michaelic-Menten response with a Km of 0.8 mM. (bottom) By coupling this enzyme system with another higher affinity competing “depletant” enzyme (here Hexokinase, HK) we can convert the hyperbolic Michaelis-Menten response of GOx into a digital-like output. When the total glucose concentration is equal or lower to the concentration of ATP, all the glucose is converted by HK to glucose-6-phosphate (G-6-P). The glucose is thus sequestered from the GOx, precluding signaling. When the total glucose concentration surpasses the concentration of ATP (the HK catalyzed reaction is saturated), a threshold response is achieved in which further addition of glucose drastically raises its effective concentration. This threshold effect generates a “pseudo-cooperative” dose-response curve in which the output signal arises much more rapidly than would occur in the absence of HK and ATP. In these operative conditions ([ATP]=1.25 mM) the range of glucose concentration at which this sharp transition occurs is compressed to less than 2-fold. The use of the logarithmic scale in the x-axes (right) as opposed to a linear scale (center) renders it easier to evaluate the narrowing of the dose-response curve.
The steepness of the input/output curve can be controlled by altering the ATP concentration. Higher sensitivities (steeper curves) occur when the ATP level is above the saturation limit of GOx ([ATP]/Km > 1). Conversely, when the ATP concentration is in the range where GOx responds linearly to glucose levels the steepness of the resulting dose-response curve is only slightly higher than that in absence of ATP. To show this we have fitted our data to obtain pseudo-Hill coefficients, which, although our system is not classically cooperative, are analogous to the Hill coefficient commonly used to describe cooperative enzymatic systems27, 34. As expected, we observe a pseudo-Hill coefficient near unity (nH=1.7) for a glucose calibration curve obtained in the absence of ATP (Fig. 2). [The slight deviation from the theoretical value of 1 is likely due to the limiting oxygen concentrations present in aqueous solution7]. Upon the addition of ATP the pseudo-Hill coefficient climbs, reaching 2.1 at [ATP] = 0.1 mM ([ATP]/Km = 0.125) before ultimately reaching 13.3 at [ATP] = 3 mM ([ATP]/ Km = 3.75) (Fig. 2).
Figure 2.
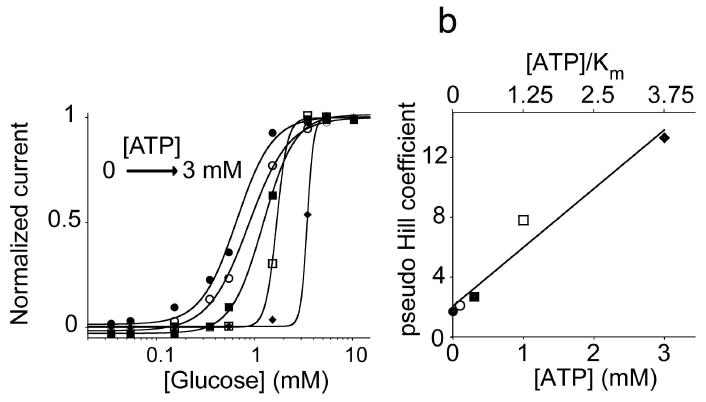
The steepness of the dose-response curve and the substrate concentration at which the threshold response is observed are both strong functions of the activity of the depletant enzyme. (left) Using HK as our depletant we can easily tune these values by varying the ATP concentration in the reaction mix (here we have employed 0.1, 0.3, 1.25, 3 mM ATP). To quantify the steepness of these dose response curves we have fitted them to the Hill equation to define pseudo-Hill coefficients34. (right) The pseudo-Hill coefficient increases monotonically as the [ATP]/Km ratio increases (by increasing the concentration of ATP), reaching 13 when this ratio is at 3.75 (at 3 mM ATP).
In addition to control the steepness of the dose-response curve, we can also control the threshold limit at which HK sequesters glucose (and thus control the threshold at which the sensor signals) by varying the concentration of ATP in the reaction mix. Specifically, HK sequesters glucose only when the ratio of ATP to glucose is more than one (Fig. 2, left). That is, at [ATP] = [glucose] the HK catalyzed reaction is saturated and the injection of any additional glucose produces a large rise in the relative concentration of free glucose which can be now recognized by GOx thus generating an output signal. This provides a valuable tool to tailor the dynamic range over which the enzymatic output is activated.
The ability to achieve digital-like enzymatic responses, together with the possibility to arbitrarily tune the substrate concentration at which this threshold response can occur, should significantly improve the utility of enzymes in many applications ranging from enzymatic sensors to enzyme-logic gates. To demonstrate possible applications, we have employed our testbed glucose sensor and have constructed four glucose sensors, each of which we placed in a solution containing varying concentrations of ATP. Challenging them with injections of increasing glucose concentration we create a set of sensors triggered at different glucose levels (see coloured regions in Fig. 3, left) within the clinically relevant range of glycaemic levels in blood. In a different embodiment of this same strategy we have used a single glucose sensor and have performed three consecutive measurements following the injection of 3 different ATP concentrations (Fig. 3, right). Doing so we were able to easily obtain high precision measurements of the concentration of glucose in the sample. Moreover, the metabolic branch point strategy not only enables a digital-like enzymatic response but also allows us to arbitrarily shift the useful dynamic range of the enzyme to higher, relevant concentrations than those defined by the Km of the enzyme. As normal physiological concentrations of a biomarker (here glucose) can be higher than the Km of the specific enzyme used, this can preclude accurate differentiation between normal and pathological concentrations. And while this problem has been efficiently overcome for glucose sensors by the introduction of diffusion mediators that push the sensor’s dynamic range in line with physiologically relevant levels7, the strategy we propose can be of utility in other specific applications such as those employing enzymes for logic systems.
Figure 3.
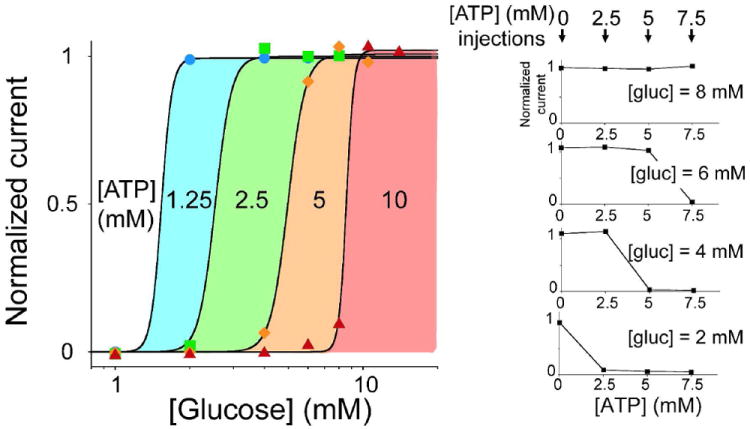
Extending the dynamic range of glucose sensors using multiple all-or-none glucose sensors triggered at different clinically relevant glucose concentrations. (left) Using our metabolic branch point strategy, we engineered four glucose sensors with variable threshold concentration by simply employing different ATP concentrations in the reaction mix (1.25, 2.5, 5, 10 mM). The coloured regions define the glucose level at which each sensor is triggered. (right) In a different embodiment of this strategy, we used a single sensor and tested it after successive additions of ATP in the sample solution. A drop of solution containing different physiologically relevant concentrations of glucose (2, 4, 6 and 8 mM) and HK was placed on the sensor and the current was measured before and after three successive injections of ATP (2.5, 5, 7.5 mM). A yes/no response depending on the glucose level present in the sample was observed.
To demonstrate the utility of the “branch point effect” strategy to logic gate systems, we have applied it to a previously described enzyme-based AND logic gate10-11, 35. Ideally, such logic gates should produce a yes/no (1/0) outcome that corresponds to a “Sense/Act” or “Sense/Diagnose/Treat” response10-11. Historically, however, a limitation of these systems was that the graded response signal observed with most enzymes renders it difficult to unequivocally define the 0 and 1 logic values of the biomolecular gate. Indeed, in the literature the 0 value of the input signal is usually defined as the complete absence of the biomaterial, a situation that rarely occurs under normal physiological conditions. Logic 1 values are likewise typically set to substrate concentrations well above those found even under pathological conditions. The ability to generate steeper, more digital enzymatic input/output curves and the possibility to tune the concentration of the input at which we observe the digital output could thus prove of utility in such applications. For these reasons, several efforts have been recently focused on the development of novel filtering systems to achieve digital output from enzymatic systems16-23. Here we demonstrate for the first time that by using the “branch point effect” strategy, we can activate a model enzyme-logic gate in an all-or-none fashion at arbitrarily fixed input concentrations. To demonstrate this, we have applied our strategy to a classic enzyme-based AND logic gate which was recently described in several works10-11, 35. This logic gate, in its original format, is intended to give an output signal only in presence of two specific inputs (GOx, input A) and glucose (input B). Although this is a very simple and basic example of enzyme logic gate it may give useful insights regarding the possibility to adopt this strategy for other, more complex, examples. As expected, the output signal of this logic gate (at fixed concentrations of GOx -input A- and varying the concentration of glucose -input B) is shallow. In order to define the 0- and 1-logic output values, we then define the threshold level using the branch-point effect. We did do so by employing a biochemical filter composed of horseradish peroxidase (HRP) and o-phenylendiamine (OPD). The enzymatic reaction catalyzed by HRP sequesters the output signal (i.e., H2O2) of the AND logic gates (Fig. 4, top). OPD in this case acts as the threshold level at which this sequestration event can occur. In fact, when the total concentration of H2O2 produced by the logic gate equals or is lower the OPD level the output signal will be effectively 0. Of note, this reaction is characterized by a stoichimoetric ratio of 3:2 (H2O2:OPD)32 so this ratio has to be taken in consideration for a correct evaluation of the threshold level. As soon as the total concentration of H2O2 surpasses the OPD level we will observe a steep threshold response, which can be defined as 1 output (Fig. 4, bottom). Only input values of glucose above a certain threshold (in the presence of GOx) will result in the activation of the logic gate. The digital-like transition between the 0 and 1 output state and the possibility of tuning this transition by varying the concentration of the filter represents an important step towards the design of multienzyme-catalyzed cascades logic gates with strong, unequivocal “sense/act” behaviour.
Figure 4.
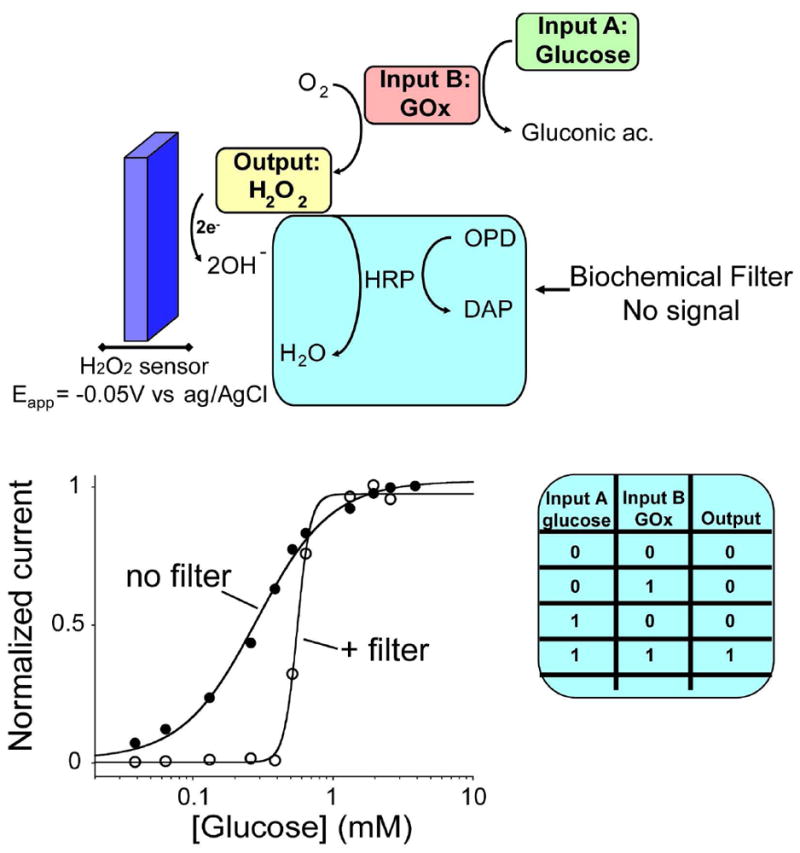
The “branch point effect” mechanism can be used as effective biomolecular filter for enzyme-based logic gates applications. (top) Here we show this by using an enzyme-based AND Boolean logic gate which is activate in an all-or-none fashion only at specific concentration of glucose (Input A) and in the presence of GOx (input B). Here horseradish peroxidase (HRP) sequesters the output signal (H2O2) generated by the logic gate till a threshold level represented by the concentration of the HRP co-substrate (o-phenylendiamine: OPD). (bottom) With this strategy we produce a steep, all-or-none digital output at an arbitrarily selected concentrations of input A, a response far more suitable for enzyme-based logic gate applications than the graded response typically produced by enzymes. Here we used a fixed concentration of GOx (input B) and increasing concentration of glucose (input A) in presence of HRP. When OPD is absent we observe the curve expected for simple, non-cooperative binding (no filter). This converts into an all-or-none curve in presence of OPD (+ filter).
Conclusions
Here we have demonstrated a novel strategy to convert the graded Michaelis-Menten response of a typical enzyme-based system into a sharp all-or-none response. Our approach is inspired by the “branch point effect”, a situation that occurs in some metabolic networks in which two or more enzymes compete for the same substrate24-28. We first used the well-known glucose amperometric sensor as a model system to demonstrate this strategy and its possible applications. Specifically, we coupled the signaling enzymatic reaction of GOx with a second, non-signaling reaction catalyzed by the higher affinity enzyme HK. This latter reaction sequesters the target analyte up to an arbitrarily selected threshold concentration above which the signaling reaction is activated producing a steep dose-response curve. As a result we steepened the normally graded response of GOx until, ultimately, obtaining a discrete yes/no output similar to that of a multimeric cooperative enzyme with a Hill coefficient of greater than 13. The steep dose response curves we achieve open the door to a number of new biosensor applications. Perhaps the most obvious application, as demonstrated here, would be the creation of enzyme logic gates with effectively digital-like outputs, a field that has attracted increasing interest during last years. Additionally, the monitoring of drugs with narrow therapeutic windows, which requires high precision dosage to optimize their therapeutic effect, would be greatly improved with the development of steeper input/output biosensors. Another application provided by “branch point effect” is that it can help to extend the dynamic range of enzyme-based sensor above the saturation level of the enzyme. And while this problem has been cleverly solved for glucose sensors using diffusion mediators7, the demonstration of alternative methods is of utility. Here we showed how this strategy allows us to finely and arbitrarily tune the glucose concentration range over which the sensor is activated by controlling the secondary depleting reaction.
The branch-point effect is also likely versatile: the wide range of enzymatic reactions targeting the same substrate offers the possibility of using this strategy with a wide range of relevant targets. For example, the same digital-like behavior could be obtained by coupling a signaling oxidase enzyme and a non-signaling dehydrogenase enzyme targeting the same substrate. The concentration of the dehydrogenase cofactor (either NAD+ or NADP+) would represent the threshold level at which we will observe the sharp response. We also note that a similar result would be achieved by using non-enzymatic depletant element. For example, a wide variety of periplasmic binding proteins are known to bind with high affinity several enzymatic substrates, including amino acids, peptides, simple and complex sugars, inorganic ions and metals36.
Despite the above advantages, the strategy we propose is not without limitation. For example, the generation of the all-or-none response is achieved at the cost of a reduced affinity as the minimum target concentration giving a detectable signal (detection limit) is shifted towards higher concentrations. Moreover, we also note that the digital-like response is achieved at the cost of additional reagents, a drawback that can limit the applicability of the approach we propose. Finally, a careful control of the concentration of the reagents involved in the depleting reaction must be performed in order to avoid secondary reactions, a problem that can be particularly crucial in complex samples. This is for example true for the specific model system we have employed here. The use of a depleting reaction based on the use of ATP as substrate requires the control of the possible effect that endogenous ATP (or of species that can react with it) can have in the definition of the threshold response. Despite this, the endogenous level of ATP in clinical samples is normally very low (in the micromolar range)37-38 and thus its effect for this specific application is negligible.
To conclude, it is worthwhile to note that the branch point effect is only one of many mechanisms that nature uses to achieve bistability in natural systems, allowing signalling networks to convert continuously graded inputs into discrete outputs39-41. These include positive feedback loops and double- negative feedback loops42-45. Exploitation of these other strategies in the laboratory would likely also give rise to new tools to achieve all-or-none enzymatic systems and would greatly impact our ability to engineer more efficient enzyme-based catalysis reactions in synthetic biology applications.
Acknowledgments
The authors acknowledge members of our research groups for helpful discussions and comments on the manuscript. This work was supported by the Italian Ministry of University and Research (MIUR) through the project FIRB “Futuro in Ricerca” and NIH grant AI076899.
References
- 1.Ro DK, Paradise EM, Quellet M, Fisher KJ, Newman KL, Ndungu JM, et al. Nature. 2006;440(7086):940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 2.Koeller KM, Wong C. Nature. 2001;409(6817):232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 3.Khalil AS, Collins JJ. Nature Rev Genet. 2010;1(5):367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purnick PEM, Weiss R. Nature Rev Mol Cell Biol. 2009;10(6):410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 5.Himmel ME, Ding S, Johnson DK, Adney WS, Nimlos MR, Brady JW, et al. Science. 2007;315(5813):804–807. doi: 10.1126/science.1137016. [DOI] [PubMed] [Google Scholar]
- 6.Wang J. Electroanalysis. 2001;13(12):983–988. [Google Scholar]
- 7.Wang J. Chem Rev. 2008;108(2):814–825. doi: 10.1021/cr068123a. [DOI] [PubMed] [Google Scholar]
- 8.Bakker E. Anal Chem. 2004;76(12):3285–3598. doi: 10.1021/ac049580z. [DOI] [PubMed] [Google Scholar]
- 9.D’Orazio P. Clin Chim Acta. 2003;334(1-2):41–69. doi: 10.1016/s0009-8981(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Katz E. Anal Bioanal Chem. 2010;398(4):1591–1603. doi: 10.1007/s00216-010-3746-0. [DOI] [PubMed] [Google Scholar]
- 11.Katz E, Privman V. Chem Soc Rev. 2010;39(5):1835–1857. doi: 10.1039/b806038j. [DOI] [PubMed] [Google Scholar]
- 12.Manesh KM, Halámek J, Pita M, Zhou J, Tam TK, Santhosh P, et al. Biosens Bioelectron. 2009;24(12):3569–3574. doi: 10.1016/j.bios.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 13.Windmiller JR, Santhosh P, Katz E, Wang J. Sensor Actuat B-Chem. 2010;2010 doi: 10.1016. [Google Scholar]
- 14.Ferrell JE., Jr Trends Biochem Sci. 1996;21(12):460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- 15.Goldbeter A, Koshland DE., Jr Q Rev Biophys. 1982;15:555–591. doi: 10.1017/s0033583500003449. [DOI] [PubMed] [Google Scholar]
- 16.Privman V, Pedrosa V, Melnikov D, Pita M, Simonian A, Katz E. Biosens Bioelectron. 2009;25(4):695. doi: 10.1016/j.bios.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Privman V, Halámek J, Arugula MA, Melnikov D, Bocharova V, Katz E. J Phys Chem B. 2010;114(44):14103. doi: 10.1021/jp108693m. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Katz E. Isr J Chem. 2011;51(1):141. [Google Scholar]
- 19.Privman V. Isr J Chem. 2011;51(1):118. [Google Scholar]
- 20.Doron A, Portnoy M, Lion-Dagan M, Katz E, Willner I. J Am Chem Soc. 1996;118:8937. [Google Scholar]
- 21.Ashton PR, Ballardini R, Balzani V, Credi A, Dress KR, Ishow E, Kleverlaan CJ, Kocian O, Preece JA, Spencer N, Stoddart JF, Venturi M, Wenger S. Chem Eur J. 2000;6:3558. doi: 10.1002/1521-3765(20001002)6:19<3558::aid-chem3558>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 22.Pita M, Privman V, Arugula MA, Melnikov D, Bocharova V, Katz E. Phys Chem Chem Phys. 2011;13(10):4507. doi: 10.1039/c0cp02524k. [DOI] [PubMed] [Google Scholar]
- 23.Halamek J, Zhou J, Halamkova L, Bocharova V, Privman V, Wang J, Katz E. Anal Chem. 2011 doi: 10.1021/ac202139m. in press. [DOI] [PubMed] [Google Scholar]
- 24.Goldbeter A, Koshland DE., Jr J Biol Chem. 1984;259:14441–14447. [PubMed] [Google Scholar]
- 25.Kim SY, Ferrell JE., Jr Cell. 2007;128(6):1133–1145. doi: 10.1016/j.cell.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 26.LaPorte DC, Walsh K, Koshland DE., Jr J Biol Chem. 1984;259(22):14068–14075. [PubMed] [Google Scholar]
- 27.Legewie S, Blüthgen N, Schäfer R, Herzel H. PLoS Comput Biol. 2005;1(5):0405–0414. doi: 10.1371/journal.pcbi.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legewie S, Schoeberl B, Blüthgen N, Herzel H. Biophys J. 2007;93(7):2279–2288. doi: 10.1529/biophysj.107.109132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricci F, Amine A, Palleschi G, Moscone D. Biosens Bioelectron. 2003;18(2-3):165–174. doi: 10.1016/s0956-5663(02)00169-0. [DOI] [PubMed] [Google Scholar]
- 30.Ricci F, Palleschi G. Biosens Bioelectron. 2005;21(3):389–407. doi: 10.1016/j.bios.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Chaubey A, Malhotra BD. Biosens Bioelectron. 2002;17(6-7):441–456. doi: 10.1016/s0956-5663(01)00313-x. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Wang Z, Liu Y, Xiao J, Wang C. Thermochim Acta. 2006;443(2):173. [Google Scholar]
- 33.Wilson JE. J Exp Biol. 2003;206(12):2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 34.Hill AV. J Physiol. 1910;40:iv–vii. doi: 10.1113/jphysiol.1910.sp001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niazov T, Baron R, Katz E, Lioubashevski O, Willner I. Proc Natl Acad Sci U S A. 2006;103(46):17160–17163. doi: 10.1073/pnas.0608319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dwyer MA, Hellinga HW. Curr Opin Struct Biol. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Gorman MW, Feigl EO, Buffington CW. Clin Chem. 2007;53(2):318. doi: 10.1373/clinchem.2006.076364. [DOI] [PubMed] [Google Scholar]
- 38.Bodin P, Burnstock G. J Cardiovasc Pharmacol. 1996;27(6):872. doi: 10.1097/00005344-199606000-00015. [DOI] [PubMed] [Google Scholar]
- 39.Ferrell JE., Jr Current Biology. 2008;18(6):R244–245. doi: 10.1016/j.cub.2008.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchler NE, Louis M. J Mol Biol. 2008;384(5):1106–1119. doi: 10.1016/j.jmb.2008.09.079. [DOI] [PubMed] [Google Scholar]
- 41.Buchler NE, Cross FR. Mol Syst Biol. 2009;5:272. doi: 10.1038/msb.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novick A, Weiner M. Proc Natl Acad Sci USA. 1957;43:553–566. doi: 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monod J, Jacob F. Cold Spring Harb Symp Quant Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 44.Griffith JS. J Theor Biol. 1968;20(2):209–216. doi: 10.1016/0022-5193(68)90190-2. [DOI] [PubMed] [Google Scholar]
- 45.Ferrell JE., Jr Curr Opin Cell Biol. 2002;14(2):140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]